

Abstract
Synaptic latency at cortical synapses is determined by the presynaptic release probability (Pr). Short- and long-term presynaptic plasticity is associated with modulation of synaptic delay. We show here that the duration and amplitude of the presynaptic action potential also determine synaptic latency at neocortical and hippocampal excitatory synapses. Blockade of voltage-gated potassium (Kv) channels with 4-aminopyridine or dendrotoxin-I, but not tetraethylammonium, induced a 1–2 ms shift in latency at excitatory synaptic connections formed by pairs of neocortical pyramidal neurons. 4-Aminopyridine or dendrotoxin-I, but not tetraethylammonium, increased the duration of the action potential recorded in the axon, suggesting that presynaptic spike duration is controlled by axonal Kv1 potassium channels. Spike width-dependent changes in latency have been identified at the mossy fibre–CA3 cell synapses and contribute to stabilization of synaptic timing during repetitive stimulation. The effects of presynaptic spike amplitude on synaptic latency were also examined. Decreasing the amplitude of the presynaptic action potential with 15–30 nm TTX reduced synaptic latency by ∼0.5 ms. The regulation of synaptic timing by potassium and sodium channel blockers could not be attributed to modulation of axonal conduction. Rather, these effects are compatible with modifications of the kinetics of the presynaptic calcium current. We conclude that synaptic latency at cortical neurons is not constant but dynamically regulated by presynaptic action potential waveform.
Non-technical summary
Synaptic delay at cortical synapses is determined by the presynaptic release probability. We show here that the duration and amplitude of the presynaptic action potential also determine synaptic latency at neocortical and hippocampal excitatory synapses. Broadening the presynaptic spike with blockers of potassium channels increased latency by 1–2 ms. Decreasing the amplitude of the presynaptic action potential by partly blocking sodium channels reduced synaptic latency by ∼0.5 ms. These changes may contribute to stabilization of synaptic timing during repetitive stimulation. The regulation of synaptic timing by these pharmacological agents could not be attributed to modulation of axonal conduction. Rather, the effects are compatible with modifications of the kinetics of the presynaptic calcium current. We conclude that synaptic latency at cortical neurons is not constant but dynamically regulated by presynaptic action potential waveform.
Introduction
Nerve cells transmit information not only by their firing rate, but also by the fine temporal organization of their discharge (Rieke et al. 1997). In simple neuronal networks, the timing between connected neurons is usually described by the synaptic latency, which is the sum of the conduction time and the synaptic delay (Sabatini & Regehr, 1999). We have recently shown that EPSC latency at monosynaptically connected pairs of layer 5 (L5) or CA3 pyramidal neurons is not fixed, but rather determined by the presynaptic release probability (Pr; Boudkkazi et al. 2007). Synaptic latency was found to be inversely correlated with synaptic strength and sensitive to changes in the extracellular Ca2+ to Mg2+ ratio. Changes in synaptic latency were also observed during paired-pulse synaptic plasticity and presynaptic long-term potentiation and depression. We furthermore established that the observed covariation in latency and synaptic strength was a synergistic combination that significantly affects postsynaptic spiking (Boudkkazi et al. 2007), suggesting that it might be a putative code for short- and long-term synaptic dynamics in cortical networks.
Classical studies on giant synapses (Katz & Miledi, 1967; Llinas et al. 1981; Augustine et al. 1985) showed that the duration of the presynaptic waveform strongly determines synaptic latency because calcium entry essentially occurs during the repolarization of the action potential. However, the precise role of the presynaptic action potential waveform is still unclear at central synapses of the mammalian brain. This question is potentially of great importance, since the shape of the presynaptic action potential (AP) is subject to considerable variation in mammalian axons in physiological conditions. In mossy fibre terminals, the AP duration is extended following repetitive stimulation (Geiger & Jonas, 2000). Likewise, in L5 pyramidal neurons the duration of the axonal spike increases when voltage-gated K+-selective (Kv) channels are inactivated by sustained depolarization of the presynaptic neuron (Shu et al. 2006, 2007; Kole et al. 2007). Finally, the shortening of AP duration observed during postnatal development in the calyx of Held terminal is associated with a decrease in the delay of the presynaptic calcium current and a shortening of synaptic latency (Taschenberger & von Gersdorff, 2000), suggesting that presynaptic AP waveform could be a major determinant of synaptic latency.
We show here that extension of the axonal spike duration with Kv1 channel blockers (20 μm 4-aminopyridine (4-AP) or dendrotoxin-I (DTx-I)) increased synaptic latency in the millisecond range. This spike-dependent latency is independent of a presynaptic release because release-dependent variation in latency was still observed in the presence of Kv channel blockers. In addition, it does not result from a modification of the conduction properties because the latency of antidromically evoked APs was not affected. However, AP waveform-dependent regulation of synaptic latency is compatible with modification of the kinetics of the presynaptic calcium current. Functionally, this spike width-dependent variation in latency is operant during repetitive stimulation to stabilize synaptic latency. In parallel, we also show that the reduction of AP amplitude with TTX shortens latency. Our study therefore demonstrates that synaptic latency at excitatory contacts is finely tuned by both Pr and presynaptic spike waveform.
Methods
Slices and slice cultures
Cortical slices (350–400 μm thick) were obtained from 13- to 20-day-old Wistar rats. All experiments were carried out according to the European and Institutional guidelines for the care and use of laboratory animals (Council Directive 86/609/EEC and French National Research Council). In addition, our experiments comply with The Journal of Physiology policy on animal experimentation described by Drummond (2009). Rats were deeply anaesthetized with chloral hydrate (intraperitoneal, 200 mg kg−1) and killed by decapitation. Slices were cut in an ice-cold solution containing (mm): 280 sucrose, 26 NaHCO3, 10 d-glucose, 10 MgCl2, 1.3 KCl and 1 CaCl2, and were bubbled with 95% O–5% CO2, pH 7.4. Slices recovered (1 h) in a solution containing (mm): 125 NaCl, 26 NaHCO3, 3 CaCl2, 2.5 KCl, 2 MgCl2, 0.8 NaH2PO4 and 10 d-glucose, and were equilibrated with 95% O2–5% CO2.
Interface hippocampal slice cultures were prepared as described previously (Stoppini et al. 1991; Debanne et al. 2008). In brief, hippocampal slices (250 μm) were obtained from 6- to 10-day-old Wistar rats and were grown on culture inserts. Culture medium was replaced three times per week. Slice cultures were maintained at 35°C for at least 4 days in vitro before experiments.
Each slice was transferred to a submerged chamber mounted on an upright microscope (Olympus, equipped with a ×40 water-immersion objective). Layer 5 pyramidal neurons were visualized using differential interference contrast infrared videomicroscopy.
Recording and data analysis
Dual whole-cell recordings were obtained as detailed previously (Boudkkazi et al. 2007; Debanne et al. 2008). The external solution contained (mm): 125 NaCl, 26 NaHCO3, 3 CaCl2, 2.5 KCl, 2 MgCl2, 0.8 NaH2PO4 and 10 d-glucose, and was equilibrated with 95% O2–5% CO2. Patch pipettes (5–10 MΩ) were filled with a solution containing (mm): 120 potassium gluconate, 20 KCl, 0.5 EGTA, 10 Hepes, 2 Na2ATP, 0.3 NaGTP and 2 MgCl2, pH 7.4. Some experiments were performed with another presynaptic pipette solution containing (mm): 140 CsMeSO4, 10 Hepes, 0.5 EGTA, 4 MgATP and 0.3 NaATP, pH 7.3. Recordings were made at 34°C in a temperature-controlled recording chamber (Luigs & Neumann, Ratingen, Germany). Classically, the presynaptic neuron was recorded in current clamp with an Axoclamp 2B amplifier (Axon Instruments) and the postsynaptic cell in voltage clamp with an Axopatch 200B amplifier (Axon Instruments). Pre- and postsynaptic cells were held at their resting membrane potential (∼–65 mV). Presynaptic APs were generated by injecting brief (5–10 ms) depolarizing pulses of current at a frequency of 0.3 Hz. Short-term facilitation was assessed with two presynaptic stimulations delivered at a frequency of 20 Hz (Debanne et al. 1996; Boudkkazi et al. 2007). The voltage and current signals were low-pass filtered (3 kHz), and acquisition of 500 ms sequences was performed at 10–15 kHz with Acquis1 (G. Sadoc, CNRS, Gif-sur-Yvette, France) or DAAD software (N. Ankri, INSERM UMR 641, Marseille, France).
Mossy fibre EPSCs recorded in CA3 neurons were evoked by minimal stimulation in the dentate gyrus of organotypic slice cultures and were characterized by a marked frequency facilitation and by their sensitivity to group II metabotropic glutamate receptor agonist DCG-IV ((2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl) glycine; Nicoll & Schmitz, 2005). For these particular experiments, external calcium and magnesium concentrations were 1 and 3 mm, respectively, in control condition. When 4-AP was added, the calcium to magnesium ratio was decreased to 0.5/9 to avoid epileptiform activity. However, in all the other experiments (i.e. paired recordings of L5–L5 and CA3–CA3 neurons), the calcium to magnesium ratio remained constant (3/2) before and after application of 4-AP or DTX-I.
Synaptic responses were averaged following alignment of the presynaptic action potentials using automatic peak detection (Detectivent 4.0, N. Ankri, INSERM). The presence or absence of a synaptic connection between two neurons was determined on the basis of averages of 30–50 individual traces, including failures (Debanne et al. 2008). With this technique, even very small responses (<0.2 mV or <10 pA) could easily be detected. In practice, the smaller synaptic responses were 0.1 mV and 4 pA (Boudkkazi et al. 2007). The analysis was restricted to a corpus of connections with mean amplitude larger than 10 pA. The latency of individual EPSCs was measured from the peak of the presynaptic AP measured in the cell body to 5% of the EPSC amplitude (Boudkkazi et al. 2007).
In a few experiments, simultaneous recordings were obtained from the soma in whole-cell configuration and the axon in ‘loose whole-cell’ configuration. Typically, the axon was recorded 10–15 min after whole-cell access of the somatic compartment. The axon was visualized with differential interference contrast infrared videomicroscopy, and negative pressure was applied to the pipette to obtain a high-resistance seal on the axon. Occasionally, a short length of the axon was pulled into the lumen of the patch pipette. In order to obtain a loose whole-cell patch recording, a brief suction of negative pressure was applied to the pipette that was occasionally maintained during the recording. In these conditions, the spike measured in the axon had a positive polarity (amplitude 0.3–10.1 mV). Its waveform was comparable to the spike waveform recorded in whole-cell configuration in the cell body (Scott et al. 2007; Boudkkazi et al. 2007), indicating that the time course of the recorded spike is minimally distorted in this recording configuration. The estimated conduction velocity between the axon and the soma (∼0.1 m s−1) was compatible with previous values measured in neocortical and pyramidal cell axons (Meeks et al. 2005; Palmer & Stuart, 2006). Axonal APs were averaged following alignment of the somatic APs using automatic peak detection (Detectivent 4.0). Axonal APs were analysed on the basis of acquisition of 300–2000 individual traces. All values shown in the figures correspond to averages over at least 2 min (i.e. >20 trials).
High-frequency components in the signals were filtered with a median filter (rank 1 or 2) and in some cases de-noising filtering (wavelet methods) was used. Special care was taken to verify that filtering of signals did not affect the original signals by superposition of the raw and filtered signals.
Drugs and statistical analysis
Tetraethylammonium (TEA) and 4-AP were obtained from Sigma, DCG-IV from Tocris and DTx-I from Latoxan. Data are presented as means ±s.e.m.
Results
Prolonged latency by extended spike duration with potassium channel blockers
To explore the consequences of AP broadening on EPSC latency, the latency of L5–L5 EPSCs was compared when the presynaptic cell was recorded with a control pipette solution (potassium gluconate) or Cs+ as the main cation. As Cs+ applied intracellularly is poorly permeable, this experimental approach allows inhibition of voltage-gated K+ channels selectively in the presynaptic compartment. In this case, the presynaptic cell was voltage clamped to avoid uncontrolled presynaptic spikes. Interestingly, EPSP latency was significantly longer in caesium gluconate (4.0 ± 0.2 ms, n= 6) compared with potassium gluconate (1.4 ± 0.1 ms, n= 9, Student's unpaired t test P < 0.001; Fig. 1). To further confirm that the prolonged latency was the result of the broadening of the presynaptic spike, we studied the effects of extracellular Kv channel blockers on synaptic latency in the same synaptic connections.
Figure 1. Delayed synaptic latency with Cs+ in the presynaptic pipette.

A, comparison of synaptic latency (Lat) at L5–L5 connections when the presynaptic neuron is recorded with potassium gluconate (black traces) or caesium gluconate (grey traces) in the intracellular solution. Note the longer EPSP latency in the presence of Cs+. B, analysis of EPSP latency in potassium gluconate and caesium gluconate. Note that EPSP latency was on average significantly longer with Cs+ as the main intracellular cation.
Bath application of TEA (2 mm) increased somatic AP amplitude (from 85.4 ± 4.5 to 89.3 ± 4.2 mV, n= 6, Student's paired t test P < 0.05) as well as somatic AP duration (from 1.6 ± 0.1 to 3.0 ± 0.3 ms, n= 6, Student's paired t test P < 0.05; Fig. 2Aa and b). Compared with experiments using presynaptic Cs+, TEA slightly increased EPSC latency (from 1.3 ± 0.2 to 1.5 ± 0.2 ms, n= 6, Student's paired t test P < 0.05; Fig. 2Ac and d). Moreover, no significant change in paired-pulse ratio (PPR) was observed (from 50 ± 8 to 50 ± 8% in TEA, n= 6, Student's paired t test P > 0.10). To further explore the effect of TEA on the presynaptic AP, simultaneous soma and axon recordings were performed (Fig. 2Ba). While TEA increased the somatic AP width by almost a factor of 2 (from 1.7 ± 0.1 to 2.9 ± 0.4 ms, i.e. 175 ± 14%, n= 7, Student's paired t test P < 0.01), its effect on axonal AP duration diminished with the axonal distance (Fig. 2Bc). In fact, in the proximal part of the axon (<45 μm), the spike increased (from 1.4 ± 0.2 to 2.5 ± 0.4 ms, n= 4, Student's paired t test P < 0.01) but not in its more distal part (>45 μm, from 1.0 ± 0.1 to 1.0 ± 0.1 ms, n= 3, Student's paired t test P > 0.10; Fig. 2Bc). These results indicate that TEA-sensitive Kv channels are expressed in the soma and the proximal part of L5 pyramidal axons, but not in more distal axonal membranes.
Figure 2. Tetraethylammonium-sensitive Kv channels do not control synaptic latency and axonal spike duration.

Aa, effect of 2 mm TEA (grey traces) on synaptic transmission at a connected pair of L5 neurons. Ab, time course of AP duration (filled diamonds; top panel) and EPSC latency (open diamonds; bottom) in control conditions (black symbols) and in the presence of TEA (grey symbols). Averages in control conditions are indicated by dotted lines. Ac, plot of EPSC latencies versus amplitudes measured on individual currents in control conditions (open circles) and in TEA (grey open diamonds). Note that control and TEA distributions are almost superimposed. Black curve, logarithmic fit (y=−0.46ln(x) + 3.19; r2= 0.36) over the control distribution. Mean values of EPSCs and latencies are symbolized by dotted lines in control conditions and by grey arrows in TEA. Ad, group data corresponding to 6 pairs tested. Ba, left panel, differential interference contrast infrared videomicrograph of an L5 neuron recorded simultaneously at the soma in whole-cell configuration and at the axon with the loose whole-cell patch recording technique. Right panel, effect of TEA on somatic and axonal APs. Note the prolongation in the soma but not in the axon. Bb, time course of spike duration in the soma (filled diamonds) and in the axon (open diamonds) following bath application of TEA. Mean values in control conditions are indicated by dotted lines. Bc, normalized AP width as a function of distance from the soma after application of TEA (7 experiments). Note the large effect in the soma and the proximal part of the axon. The effect of TEA disappears in the distal part of the axon.
Next, the effect of blocking Kv channels with 4-AP was tested on synaptic latency. Bath application of 4-AP (2 mm) increased both somatic AP amplitude (from 79 ± 4 to 85 ± 3 mV, n= 6, Student's paired t test P < 0.10; data not shown) and somatic AP duration (from 1.3 ± 0.1 to 1.6 ± 0.1 ms, n= 6, Student's paired t test P < 0.05). In contrast with TEA, 4-AP produced a large increase in both EPSC amplitude (from 27 ± 5 to 34 ± 5 pA, n= 6, Student's paired t test P < 0.01) and EPSC latency (1.4 ± 0.3 to 2.6 ± 0.3 ms, n= 6, Student's paired t test P < 0.01; Fig. 3Aa–c). Interestingly, the enhanced transmission was associated with a reduced PPR (from 68 ± 7 to 37 ± 2%, n= 6, Student's paired t test P < 0.01), indicating that it was mediated by an increase in Pr. As a consequence of Pr-dependent latency (Boudkkazi et al. 2007), the 4-AP-induced effect could be underestimated, because increasing Pr leads to reduced latency, which tends to counterbalance the effect of 4-AP. For this reason, latencies were measured on EPSCs of similar amplitudes (average from 37 ± 3 to 37 ± 3 pA, n= 6, Student's paired t test P > 0.10; Fig. 3Ac, inset). In these conditions, the latency increased from 1.3 ± 0.3 to 3.1 ± 0.4 ms in the presence of 4-AP (n= 6, Student's paired t test P < 0.01; Fig. 3Ad). To examine the effects of 4-AP on spike duration along the axo-somatic axis, simultaneous somatic and axonal recordings were obtained from L5 neurons (Fig. 3Ba). 4-Aminopyridine produced a larger prolongation of the spike duration in the axon (from 1.2 ± 0.1 to 2.8 ± 0.4 ms n= 10, Student's paired t test P < 0.01) than in the soma (from 1.6 ± 0.1 to 1.9 ± 0.1 ms, n= 10, Student's paired t test P < 0.01; Fig. 3Bb–c). In conclusion, these data demonstrate that 4-AP-sensitive Kv channels control latency at excitatory cortical synapses.
Figure 3. 4-Aminopyridine-sensitive Kv channels control synaptic latency and axonal spike duration.

Aa, effect of 2 mm 4-AP (blue traces) on spike duration and synaptic transmission at a connected pair of L5 neurons. Note the large increase in EPSC latency. Ab, time course of presynaptic AP duration recorded in the cell body (filled circles; top panel) and EPSC latency (open circles; bottom panels) in control conditions (black symbols) and in the presence of 4-AP (blue symbols). Ac, plot of EPSC latencies versus amplitudes measured on individual currents in control conditions (black open circles) and in 4-AP (blue filled circles). Note a rightward shift of the 4-AP distribution. Each distribution is fitted by a logarithmic curve (y=−0.56ln(x) + 4.56, r2= 0.37 in control conditions; y=−0.92ln(x) + 7.55, r2= 0.18 in 4-AP). The Pr-independent shift in EPSC latency induced by 4-AP was determined for EPSCs of the same amplitude (dotted box). Ad, group data corresponding to 6 pairs tested. Ba, effect of 4-AP on somatic and axonal APs during simultaneous soma and axon recordings. Note the prolongation of the axonal spike. Bb, time course of spike duration in the soma (filled circles) and in the axon (open circles) following bath application of 4-AP. Bc, normalized AP width as a function of recording distance after application of 2 mm 4-AP (10 experiments). Note the dramatic extension in the distal part of the axon.
Axonal Kv1 channels control synaptic latency
We next identified the type of axonal Kv channel involved in the spike width-dependent shift in latency. Prolonged latency was observed with 2 mm 4-AP but not with 2 mm TEA, suggesting the involvement of A- or D-type Kv channels (Storm, 1990). To distinguish between these two possibilities, additional experiments were performed with a low concentration of 4-AP (20 μm) that preferentially blocks D-type Kv current. In these conditions, EPSC amplitude increased (from 17 ± 3 to 20 ± 4 pA, n= 6, Student's paired t test P < 0.01), the PPR decreased (from 62 ± 7 to 41 ± 4%, n= 6, Student's paired t test P < 0.01), the Pr-independent shift in latency (i.e. measured on similar EPSC amplitudes) increased from 1.0 ± 0.2 to 1.7 ± 0.2 ms (n= 6, Student's paired t test, P < 0.01) and axonal spike duration increased (from 0.8 ± 0.1 to 1.6 ± 0.1 ms, Student's paired t test, P < 0.01, n= 9; Fig. 4A). D-type potassium channels (Kv1) located in the axon control spike width and are highly sensitive to dendrotoxin (Shu et al. 2006, 2007; Kole et al. 2007). We therefore tested whether DTx-I, which specifically blocks Kv1.1, Kv1.2 and Kv1.6 subunits, also prolonged the synaptic latency at L5–L5 synapses. As previously shown (Kole et al. 2007), DTx-I (200 nm) was found to increase the amplitude of EPSCs (from 28 ± 5 to 33 ± 6 pA, n= 7), prolong the axonal spike (from 0.9 ± 0.1 to 1.5 ± 0.2 ms, n= 3, distance >45 μm), and reduce PPR (from 67 ± 6 to 44 ± 5%, n= 7, Student's paired t test P < 0.01). In addition, DTx-I was found to prolong the latency of EPSCs of comparable amplitude (23 pA; from 0.9 ± 0.2 to 1.4 ± 0.3 ms, n= 7; Student's paired t test, P < 0.01; Fig. 4B). Thus, axonal Kv1 channels that mediate the D-type current determine synaptic latency.
Figure 4. D-type Kv channels control synaptic latency and axonal spike duration.

A, top left, effect of 20 μm 4-AP (blue traces) on EPSC latency at an L5–L5 connection. Top right, group data of EPSC latency changes measured at constant amplitude (compensated for Pr-dependent latency changes). Bottom left, 20 μm 4-AP extends axonal but not somatic AP. Bottom right, normalized effects of 20 μm 4-AP on AP width as a function of recording distance. B, top left, effect of DTx-I on EPSC latency. Top right, group data of EPSC latency changes measured at constant amplitude. Bottom left, DTx-I extends axonal but not somatic AP. Bottom right, normalized effects of DTx-I on AP width as a function of recording distance.
Axonal spike duration determines synaptic latency
The latency between the presynaptic AP in the somatic compartment and the EPSC onset depends, in theory, on the following factors: (1) the conduction velocity along the axon; (2) the back-propagating time from the spike initiation zone to the soma; (3) the coupling between the spike in the presynaptic terminal and the calcium influx; and (4) the kinetics of Ca2+ binding to the Ca2+ sensor. We first tested the effect of 4-AP on axonal propagation. Antidromic APs were evoked in L5 neurons with an extracellular stimulating electrode located in the white matter (at approximately 200 μm from the soma). The prolongation of the spike produced by 4-AP (2 mm) was associated with a slight reduction in latency (from 1.2 ± 0.4 to 1.1 ± 0.3 ms, n= 4, Student's paired t test P < 0.01; Fig. S1A and B). Therefore, the prolonged latency is not due to modification of the conduction velocity, and other mechanisms should be envisaged.
At invertebrate synapses, latency is largely determined by the duration of the presynaptic depolarization (Katz & Miledi, 1967; Augustine et al. 1985). To further determine the role of axonal spike duration in the synaptic latency at L5–L5 synapses, the variation in latency (ΔLat) produced by the Kv channel blockers was plotted as a function of the induced variation in AP duration measured in the axon. Overall, the increase in synaptic latency produced by the blockers was well predicted by the extension of the AP width in the axon (linear regression: y= 0.72x+ 0.11, r2= 0.95; Fig. 5), indicating that AP extension induced by Kv channel blockers is a major determinant of synaptic latency at L5–L5 synapses.
Figure 5. Axonal AP duration controls synaptic latency.
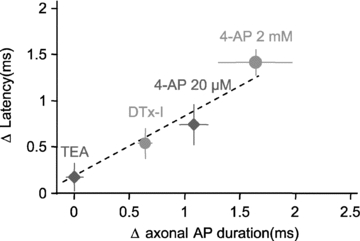
Summary of the changes in EPSC latency and the corresponding effects on axonal AP duration induced by TEA, DTx-I and 4-AP (20 μm and 2 mm). Note the linear relation between latency and AP duration in the axon.
Does AP amplitude determine latency?
In parallel with the prolongation of the axonal spike duration, 4-AP (2 mm) or DTx-I also slightly increased its amplitude in the axon (respectively 9 ± 2%, n= 10 and 2 ± 2%, n= 8). To explore the consequences of AP reduction on EPSC latency, low concentrations of TTX (15–30 nm) were bath applied. In these conditions, action potentials and synaptic transmission were not totally abolished. In fact, the somatic AP amplitude decreased from 79 ± 3 to 63 ± 4 mV (i.e. 21% reduction, n= 12, Student's paired t test P < 0.01) and EPSC amplitude was decreased (from 32 ± 4 to 26 ± 4 pA, n= 12, Student's paired t test P < 0.01). This reduction in synaptic transmission was followed by a significant increase in PPR (from 61 ± 8% in control conditions to 80 ± 9% in TTX, n= 12, Student's paired t test P < 0.01; Fig. 6A), suggesting that the TTX-induced reduction in EPSC is due to an apparent decrease in Pr. The reduction in EPSC amplitude was surprisingly associated with a shortening of EPSC latency (Fig. 6B). Postsynaptic current latency decreased on average from 1.2 ± 0.1 to 0.9 ± 0.1 ms (n= 12, Student's paired t test P < 0.01). Since Pr was apparently reduced in the presence of TTX, the change in EPSC latency was probably underestimated. Therefore, for each synaptic connection, a restricted range of EPSC amplitude (width 10 pA) was defined, and EPSC latencies were compared before and after TTX application (inset in Fig. 6Bb). In these conditions, EPSC latency decreased from 1.3 ± 0.1 to 0.8 ± 0.1 ms in TTX (n= 12, Student's paired t test P < 0.01; Fig. 6Bc). This effect could not be attributed to an increase in conduction along the axon, because the latency of antidromic AP was slightly delayed (from 1.1 ± 0.3 ms in control conditions to 1.4 ± 0.3 ms in TTX, n= 4, Student's paired t test P < 0.01; Fig. S1C and D). To estimate to what extent the axonal spike was decreased, we also measured the effect of TTX in the axon. On average, the amplitude decreased by 37 ± 3% in the axon (n= 9). We conclude that presynaptic AP amplitude also determines synaptic latency. The moderate increase in the axonal spike amplitude induced by Kv channel blockers may also account for 0.13 ms of the observed effect if the amplitude-dependent change in latency is linear.
Figure 6. Reduced latency induced by low concentration of TTX.

A, TTX (15–30 nm) reduces synaptic transmission and increases paired-pulse ratio at L5–L5 connections. A low concentration of TTX reduces the presynaptic spikes but does not block synaptic transmission. Representative example (Aa) and group data on 12 connections (Ab). Note the significant increase in paired-pulse ratio (***P < 0.01, Student's paired t test). B, a low concentration of TTX shortens synaptic latency evoked by the first AP. Example (Ba) and analysis of the EPSC latency vs. amplitude relation in the same pair (Bb). The Pr-independent shift in latency induced by TTX was determined with EPSCs of the same amplitude (dotted box and right inset). Bc, group data of the change in latency after compensation for Pr. C, effect of TTX on somatic and axonal APs during simultaneous soma and axon recordings.
Generalization to other excitatory synapses
To determine whether Kv-dependent latency was a general feature of central synapses, we examined whether 4-AP also produced a shift in the latency at other neocortical or hippocampal synapses. Pairs of connected L5 pyramidal neurons and putative GABAergic interneurons were recorded. In contrast with L5 pyramidal neurons, these neurons displayed a bipolar shape, a brief AP (<1 ms) and deep fast after-hyperpolarization (usually >15 mV). 4-Aminopyridine enhanced synaptic transmission (237 ± 78% of the control amplitude, n= 3). After compensation for the increased release probability, the latency was found to be prolonged in the presence of 2 mm 4-AP (from 1.0 ± 0.1 to 2.4 ± 0.1 ms, n= 3, Student's paired t test P < 0.01; Fig. 7A). Likewise, 4-AP also produced a shift in the synaptic latency at connected CA3 pyramidal neurons recorded in hippocampal slice cultures (from 3.4 ± 1.0 to 5.9 ± 1.4 ms, n= 4, Student's paired t test P < 0.01; Fig. 7B). We conclude that spike width-dependent latency is a general principal at central excitatory synapses.
Figure 7. Generalization of Kv-dependent latency to other excitatory synapses.
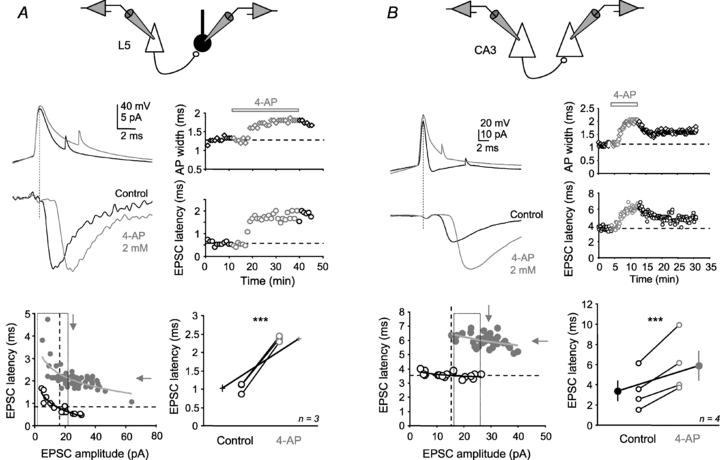
A, effect of 2 mm 4-AP on EPSC latency at connections between L5 pyramidal neuron and inhibitory interneuron. Scheme shows experimental configuration. Top left, 4-AP (2 mm) increased EPSC latency. Top right, time courses of the EPSC latency and AP width. Bottom left, latency vs. amplitude distributions in control conditions (black open circles) and in the presence of 4-AP (grey filled circles). Bottom right, group data of EPSC latency changes measured at constant amplitude. B, effect of 2 mm 4-AP on EPSC latency at a CA3–CA3 connection. Scheme shows experimental configuration. Top left, raw data. Top right, time courses of the EPSC latency and AP width. Bottom left, latency vs. amplitude distributions in each set of experimental conditions. Note the large increase in latency in the presence of 4-AP.
Evidence for Pr-independent prolongation of synaptic latency
Action potentials in presynaptic mossy fibre boutons from granule cell axons are broadened following repetitive high-frequency (50 Hz) stimulation as a result of inactivation of A-type K+ current (Geiger & Jonas, 2000). If the presynaptic spike width determines the latency, repetitive stimulation of granule cell axons should reveal an activity-dependent increase in synaptic latency. CA3 pyramidal neurons were recorded in hippocampal slice cultures (day in vitro 8–15), and trains of 25 EPSCs were evoked at 20 Hz with minimal stimulation in the dentate gyrus. A large facilitation was observed (ratio of the 25th EPSC to the 1st EPSC: 322 ± 30%, n= 12; Fig. 8A). The evoked response was fully antagonized by the type 2/3 metabotropic glutamate receptor agonist DCG-IV (10 μm), confirming that it was mediated by mossy fibre inputs (data not shown). As expected from our previous work (Boudkkazi et al. 2007), the synaptic latency decreased during paired-pulse stimulation, but interestingly, synaptic latency reached a plateau and increased during subsequent stimulations (Fig. 8B). This shift out of the Pr-law suggests that a second mechanism may determine synaptic latency during repetitive activity (Fig. 8C). This change probably results from a broadening of the presynaptic spike. In fact, the width of APs recorded in the soma of granule cells was increased by 77 ± 6% (from 0.9 ± 0.1 to 1.9 ± 0.2 ms; n= 26, data not shown). Although the presynaptic spike width has not been directly measured in the terminal, additional broadening would be observed in the mossy fibre bouton (see Geiger & Jonas, 2000). It is noteworthy that the data points followed the Pr law in the presence of 40 μm 4-AP (Fig. 8B, inset), indicating that increased latency during repetitive stimulation depends on Kv channels. We conclude that presynaptic Kv channels stabilize synaptic latency during repetitive stimulation of mossy fibres.
Figure 8. Pr-independent modulation of latency at DG–CA3 synapse.

A, scheme shows experimental configuration. A CA3 pyramidal cell was recorded in voltage clamp in the whole-cell configuration of the patch-clamp technique, and a train of 25 mossy fibre EPSCs was evoked by stimulating a granular cell of the dentate gyrus in organotypic hippocampal slices. Bottom, example of short-term facilitation of the mossy fibre synapse stimulated at 20 Hz. B, plot of EPSC latencies vs. normalized amplitude for pooled data (n= 12). Note that amplitude-dependent latency variation (fitted by a logarithmic curve in orange; y=−1.4ln(x) + 12.5, r2= 0.92) was only present for the first EPSCs, whereas the last EPSCs diverge from this law (red arrow). Inset, amplitude-dependent latency distribution in the presence of 4-AP (40 μm) is fitted by a logarithmic curve (y=−0.8ln(x) + 14.1, r2= 0.75). C, summary of the latency stabilization during repetitive stimulation at the mossy fibre–CA3 cell synapse. From stimulation 1 to 5, latency decreases because of enhanced Pr (orange traces and arrowhead), whereas latency increases from stimulation 5 to 25 because of AP broadening (red traces and arrowhead). Abbreviations: Mfb, mossy fibre bouton; and Pyr, CA3 pyramidal cell.
Discussion
We show here that besides the Pr-dependent latency reported previously (Boudkkazi et al. 2007; Saez & Friedlander, 2009), the axonal spike duration also determines synaptic latency at cortical synapses. In fact, pharmacological inactivation of Kv channels with 4-AP (2 mm or 20 μm) or DTx-I (200 nm), but not TEA (2 mm), prolonged synaptic latency by ∼2 ms. These two mechanisms are independent, because Pr-dependent latency was still observed before and after blocking Kv channels with 4-AP. The fact that the shift in latency was linearly related to the induced change in axonal spike duration strongly suggests that the underlying mechanism principally involves the shift of the presynaptic calcium current resulting from the prolonged axonal spike. The Kv-dependent shift in latency was not only observed at L5–L5 synapses but also at L5–interneuron synapses and at unitary CA3–CA3 synapses, indicating that it may be a general feature of many central excitatory synapses. The Kv-dependent latency shift is functionally relevant in at least one way. It may stabilize synaptic latency during repetitive stimulation at mossy fibre–CA3 cell synapses (Fig. 8). Importantly, we also demonstrated that reducing the spike amplitude by ∼15 mV with a low concentration of TTX shortened synaptic delay (Fig. 6). In agreement with previous findings (Augustine et al. 1985), the reduction in presynaptic spike amplitude may produce an advance of the presynaptic calcium current, and subsequently a reduction in synaptic delay.
Axonal Kv1 channels determine the synaptic latency at L5–L5 connections
We report here that synaptic latency at L5–L5 connections is increased by Cs+ in the presynaptic neuron, 4-AP (20 μm or 2 mm) or DTx-I (200 nm), but not by TEA (2 mm). In fact, TEA markedly prolonged the spike in the cell body but had no effect on either synaptic latency or the duration of the AP measured in the axon. In contrast, 4-AP or DTx-I significantly extended the axonal AP and delayed synaptic latency, but had little effect on the somatic spike waveform. These data indicate that 4-AP-sensitive Kv channels are selectively expressed in the axon, whereas TEA-sensitive Kv channels are mainly located in the cell body compartment and/or in the proximal part of the axon. In fact, cell-attached recordings from axons of rat L5 pyramidal neurons revealed that the axon initial segment and the axon proper express DTx-sensitive, slowly inactivating D-type potassium currents that are respectively 10 and seven times larger in these compartments than in the cell body (Kole et al. 2007). Supporting this view, another study has indicated that the DTx-sensitive K+ currents are three to four times larger in the axon of L5 cortical neurons of the ferret (Shu et al. 2007). In hippocampal and cortical pyramidal neurons, the slowly inactivating D-type K+ current is principally mediated by Kv1 channels (Stühmer et al. 1989; Storm, 1990; Kole et al. 2007). In fact, immunohistochemistry of Kv1.1 and Kv1.2 proteins has indicated that Kv1 channels are highly expressed in the axon initial segment and axon terminals (Sheng et al. 1993; Veh et al. 1995; Inda et al. 2006; Goldberg et al. 2008; Lorincz & Nusser, 2008).
Tetraethylammonium broadens action potentials in the soma, but not in the axon. Several TEA-sensitive channels controlling spike width are specifically located in the soma of cortical neurons. Spike broadening might be caused by the blockade of the large conductance, calcium-activated (IC) K+ current (Storm, 1987; Lancaster & Nicoll, 1987; Marrion & Tavalin, 1998) or by the blockade of Kv2.1 channels carrying IK (Murakoshi & Trimmer, 1999; Du et al. 2000; Mitterdorfer & Bean, 2002). In addition, TEA blocks Kv7 channels located in the axon initial segment that mediate the M-type current (Devaux et al. 2004). However, these channels do not strongly influence the action potential waveform (Vervaeke et al. 2006; Shah et al. 2008).
The effects of DTx-I on EPSC amplitude were found to be smaller (26%) than those initially reported by Kole et al. (2007; ∼200% of the control amplitude). This discrepancy may result from the high release probability in our experiments (PPR close to 100% in Kole's paper vs. 60% in the present study), due to both the experimental conditions (2 mm external Ca2+vs. 3 mm in our case) and the age of the preparation (2- to 5-week-old rats vs. 13- to 20-day-old rats in our case; see Angulo et al. 1999; Reyes & Sakmann, 1999). Thus, spike prolongation caused by DTx-I only slightly enhances synaptic strength.
Prolonged synaptic latency has been observed at CA3–CA3 synaptic connections when the D-type potassium current was inactivated with 4-AP (Saviane et al. 2003). However, our study is the first that clearly establishes a relation between the change in synaptic latency and the modification in AP waveform measured in the axon of mammalian central neurons. Increased synaptic latency in the millisecond range has been reported at connected pairs of hippocampal neurons when the presynaptic A-type current was partly inactivated (Debanne et al. 1997). In these experiments, the prolonged latency cannot be attributed to the mechanism described here. Rather, it may result from delayed axonal conduction caused by the combination of a change in local geometry and an increase in conductance (Segev, 1990; Manor et al. 1991).
Delayed presynaptic calcium current
Modification of axonal spike duration strongly determined the magnitude of the shift in latency. For instance, TEA (2 mm) had no effect on both axonal spike duration and synaptic latency, whereas 4-AP (2 mm or 20 μm) or DTx-I produced a major effect on both parameters (Fig. 5). Thus, axonal spike prolongation represents a key parameter in the 4-AP-dependent prolongation of synaptic latency. However, conduction velocity does not play a critical role in this mechanism. In fact, 4-AP slightly decreased the latency of antidromically evoked AP, indicating that conduction velocity was not reduced.
What is the mechanism underlying axonal spike width-dependent prolongation of synaptic latency? Although direct recording from presynaptic terminals of L5 neurons is almost impossible because of their small size, our data are consistent with the fact that prolonged axonal duration delays the presynaptic calcium current. As the presynaptic spike overshoots at approximately +50 mV, the peak of the calcium current develops essentially during the repolarization phase of the presynaptic spike (Katz & Miledi, 1967; Llinas et al. 1981; Augustine et al. 1985; Sabatini & Regehr, 1996, 1997; Bischofberger et al. 2002; Lin & Faber, 2002). Thus, any prolongation in the duration of the presynaptic spike, such as that produced by 4-AP, will delay the calcium current (see Bischofberger et al. 2002) and will subsequently shift release towards longer latencies. The precise characterization of the underlying mechanisms is the matter of a separate study.
Amplitude of the presynaptic AP
An increase in AP amplitude in the axon constitutes the third mechanism that may account for the prolongation of EPSC latency observed when Kv1 channels are inactivated. In fact, the axonal spike was increased by 9% in the presence of 4-AP. A similar increase has been previously reported in the cell body (Storm, 1987) and results from the blockade of Kv channels that normally interrupt the rising phase of the AP. To study the effect of the amplitude of the presynaptic spike we applied a low concentration of TTX that efficiently reduced the amplitude of the AP but did not block synaptic transmission. We showed that reducing presynaptic AP amplitude with 15–30 nm TTX shortened EPSC latency by 0.5 ms. This effect is also compatible with the modification of coupling between the presynaptic waveform amplitude and the activation of calcium current reported at giant synapses (Katz & Miledi, 1967; Augustine et al. 1985; Bischofberger et al. 2002). Here, the reduction in amplitude of the presynaptic spike probably shortens the latency of the presynaptic calcium tail current. Our data indicate that the modification in amplitude accounts only for a relatively small portion of the induced change in latency. The 37% reduction of the axonal spike amplitude induced by TTX prolongs the latency by 0.5 ms, suggesting that the 9% increase in axonal spike amplitude would produce a delay in the range of 0.1–0.2 ms if this relation is linear. The amplitude-dependent reduction in synaptic delay might be functionally important. The prolongation of the axonal spike observed during repetitive stimulation (Geiger & Jonas, 2000) or somatic depolarization (Shu et al. 2006) is associated with a reduction in amplitude. Thus, it may partly counterbalance the increase in latency produced by the prolongation of the spike.
Functional implications
The presynaptic spike width-dependent latency is potentially implicated in the stabilization of synaptic timing during repetitive stimulation. We show that at the mossy fibre–CA3 cell synapse the change in synaptic latency during facilitation initially follows the Pr-dependent rule (i.e. facilitation with a reduction in latency; Boudkkazi et al. 2007), but progressively diverges from this law after the fifth stimulation and finally returns to high values despite a very large EPSC facilitation. This observation is consistent with the extension of the AP at the mossy fibre terminal due to the inactivation of A-type Kv channels during repetitive stimulation (Geiger & Jonas, 2000). Although we did not measure directly the spike extension in the mossy fibre terminal, our results indicate that repetitive firing in granule cells increased the duration of the spike width in the soma. Interestingly, this behaviour was absent in the presence of 4-AP.
At the calyx of Held, a parallel reduction in the AP duration and the synaptic delay is observed during development (Taschenberger & von Gersdorff, 2000). This presynaptic acceleration is thought to result from the maturation of presynaptic Nav and Kv channels. Thus, our study provides a causal link between the speeding up of synaptic transmission and the changes in presynaptic AP waveform during development.
Activity-dependent changes in the presynaptic AP waveform represent a powerful means for precise control of the timing of synaptic transmission. We show here that inactivation of presynaptic Kv1 channels is a major mechanism for stabilizing synaptic latency. We may also speculate that the timing of synaptic transmission may be controlled through the activity-dependent regulation of Kv1 (Grosse et al. 2000; Raab-Graham et al. 2006; Cudmore et al. 2010) or Nav channel activity (Cantrell & Catterall, 2001; Carlier et al. 2006).
Acknowledgments
We thank P. Jonas and M. Seagar for constructive criticisms of the manuscript, P. Giraud and E. Campanac for the help with the slice cultures, N. Ankri for help with the analysis and E. Carlier, O. Caillard and J. M. Goaillard for discussion. This work was supported by CNRS, INSERM, Ministry of Research (doctoral grant to S.B.), European Community (LSHM-CT-2004-511995, Synaptic Scaffolding Proteins Orchestrating Cortical Synapse Organization during Development to D.D.), Région PACA (APO 2009, ‘Plexin’ to D.D.) and Agence Nationale de la Recherche (ANR-06-Neuro-014-01 to D.D.).
Glossary
Abbreviations
- AP
action potential
- 4-AP
4-aminopyridine
- DCG-IV
(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine
- DTx-I
dendrotoxin-I
- IR
infrared
- Kv channel
voltage-gated K+-selective channel
- PPR
paired-pulse ratio
- Pr
release probability
Author contributions
S.B., L.F.M. and D.D. conceived and designed the experiments. S.B., L.F.M. and D.D. collected, analysed and interpreted the data. S.B. and D.D. drafted the manuscript. The experiments were performed in the laboratory of D.D. at INSERM U641, Marseille, France. All authors approved the final version of the manuscript.
Supplementary material
Figure S1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Angulo MC, Staiger JF, Rossier J, Audinat E. Developmental synaptic changes increase the range of integrative capabilities of an identified excitatory neocortical connection. J Neurosci. 1999;19:1566–1576. doi: 10.1523/JNEUROSCI.19-05-01566.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J Physiol. 1985;369:163–181. doi: 10.1113/jphysiol.1985.sp015819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JRP, Jonas P. Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J Neurosci. 2002;22:10593–10602. doi: 10.1523/JNEUROSCI.22-24-10593.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudkkazi S, Carlier E, Ankri N, Caillard O, Giraud P, Fronzaroli-Molinieres L, Debanne D. Release-dependent variations in synaptic latency: a putative code for short- and long-term synaptic dynamics. Neuron. 2007;56:1048–1060. doi: 10.1016/j.neuron.2007.10.037. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Carlier E, Sourdet V, Boudkkazi S, Déglise P, Ankri N, Fronzaroli-Molinieres L, Debanne D. Metabotropic glutamate receptor subtype 1 regulates sodium currents in rat neocortical pyramidal neurons. J Physiol. 2006;577:141–154. doi: 10.1113/jphysiol.2006.118026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore RH, Fronzaroli-Molinieres L, Giraud P, Debanne D. Spike-time precision and network synchrony are controlled by the homeostatic regulation of the D-type potassium current. J Neurosci. 2010;30:12885–12895. doi: 10.1523/JNEUROSCI.0740-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Boudkkazi S, Campanac E, Cudmore R, Giraud P, Fronzaroli-Molinieres L, Carlier E, Caillard O. Paired-recordings from synaptically-coupled cortical and hippocampal neurons in acute and cultured brain slices. Nat Protoc. 2008;3:1559–1568. doi: 10.1038/nprot.2008.147. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Action-potential propagation gated by an axonal IA-like K+ conductance in hippocampus. Nature. 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol. 2000;522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JRP, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse G, Draguhn A, Höhne L, Tapp R, Veh RW, Ahnert-Hilger G. Expression of Kv1 potassium channels in mouse hippocampal primary cultures: development and activity-dependent regulation. J Neurosci. 2000;20:1869–1882. doi: 10.1523/JNEUROSCI.20-05-01869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, DeFelipe J, Muñoz A. Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc Natl Acad Sci U S A. 2006;103:2920–2925. doi: 10.1073/pnas.0511197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. A study of synaptic transmission in the absence of nerve impulses. J Physiol. 1967;192:407–436. doi: 10.1113/jphysiol.1967.sp008307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MHP, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurons. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Faber DS. Modulation of synaptic delay during synaptic plasticity. Trends Neurosci. 2002;25:449–455. doi: 10.1016/s0166-2236(02)02212-9. [DOI] [PubMed] [Google Scholar]
- Llinas R, Steinberg IZ, Walton K. Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys J. 1981;33:323–352. doi: 10.1016/S0006-3495(81)84899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor Y, Koch C, Segev I. Effect of geometrical irregularities on propagation delay in axonal trees. Biophys J. 1991;60:1424–1437. doi: 10.1016/S0006-3495(91)82179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Jiang X, Mennerick S. Action potential fidelity during normal and epileptiform activity in paired soma–axon recordings from rat hippocampus. J Physiol. 2005;566:425–441. doi: 10.1113/jphysiol.2005.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitterdorfer J, Bean BP. Potassium currents during the action potential of hippocampal CA3 neurons. J Neurosci. 2002;22:10106–10115. doi: 10.1523/JNEUROSCI.22-23-10106.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Palmer LM, Stuart GJ. Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci. 2006;26:1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab-Graham KF, Haddick PCG, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation to dendrites. Science. 2006;314:144–148. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- Reyes A, Sakmann B. Developmental switch in the short-term modification of unitary EPSPs evoked in layer 2/3 and layer 5 pyramidal neurons of rat neocortex. J Neurosci. 1999;19:3827–3835. doi: 10.1523/JNEUROSCI.19-10-03827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Warland D, de Ruyter van Steveninck R, Bialek W. Spikes: Exploring the Neural Code. Cambridge, MA: MIT Press; 1997. [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of synaptic transmission. Annu Rev Physiol. 1999;61:521–542. doi: 10.1146/annurev.physiol.61.1.521. [DOI] [PubMed] [Google Scholar]
- Saez I, Friedlander MJ. Synaptic output of individual layer 4 neurons in guinea pig visual cortex. J Neurosci. 2009;29:4930–4944. doi: 10.1523/JNEUROSCI.0046-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, Mohajerani MH, Cherubini E. An ID-like current that is downregulated by Ca2+ modulates coding at CA3–CA3 synapses in the rat hippocampus. J Physiol. 2003;552:513–524. doi: 10.1113/jphysiol.2003.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LL, Hage TA, Golding NL. Weak action potential backpropagation is associated with high-frequency axonal firing capability in principal neurons of the gerbil medial superior olive. J Physiol. 2007;583:647–661. doi: 10.1113/jphysiol.2007.136366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev I. Computer study of presynaptic inhibition controlling the spread of action potentials into axonal terminals. J Neurophysiol. 1990;63:987–998. doi: 10.1152/jn.1990.63.5.987. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Liao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. 1993;365:72–75. doi: 10.1038/365072a0. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Shu Y, Yu Y, Yang J, McCormick DA. Selective control of cortical axonal spikes by a slowly inactivating K+ current. Proc Natl Acad Sci U S A. 2007;104:11453–11458. doi: 10.1073/pnas.0702041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Müller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Stühmer W, Ruppersberg JP, Schröter KH, Sakmann B, Stocker M, Giese KP, Perschke A, Baumann A, Pongs O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989;8:3235–3244. doi: 10.1002/j.1460-2075.1989.tb08483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veh RW, Lichtinghagen R, Sewing S, Wunder F, Grumbach IM, Pongs O. Immunohistochemical localization of five members of the Kv1 channel subunits: contrasting subcellular locations and neuron-specific co-localizations in rat brain. Eur J Neurosci. 1995;7:2189–2205. doi: 10.1111/j.1460-9568.1995.tb00641.x. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.