

Abstract
In healthy individuals, sympathetic vasoconstriction is markedly blunted in exercising muscles to optimize blood flow to the metabolically active muscle fibres. This protective mechanism, termed functional sympatholysis, is impaired in rat models of angiotensin-dependent hypertension. However, the relevance of these findings to human hypertension is unknown. Therefore, in 13 hypertensive and 17 normotensive subjects we measured muscle oxygenation and forearm blood flow (FBF) responses to reflex increases in sympathetic nerve activity (SNA) evoked by lower body negative pressure (LBNP) at rest and during moderate-intensity rhythmic handgrip exercise. In the normotensives, LBNP caused decreases in oxygenation and FBF (−16 ± 2% and −23 ± 4%, respectively) in resting forearm but not in exercising forearm (−1 ± 2% and −1 ± 3%, respectively; P < 0.05 vs. rest). In the hypertensives, LBNP evoked decreases in oxygenation and FBF that were similar in the resting and exercising forearm (−14 ± 2%vs.−12 ± 2% and −20 ± 3%vs.−13 ± 2%, respectively; P > 0.05), indicating impaired functional sympatholysis. In the hypertensives, SNA was unexpectedly increased by 54 ± 11% during handgrip alone. However, when SNA was experimentally increased during exercise in the normotensives, sympatholysis was unaffected. Treatment for 4 weeks with the angiotensin receptor blocker irbesartan, but not with the thiazide-type diuretic chlorthalidone, restored sympatholysis in the hypertensives. These data provide the first evidence that functional sympatholysis is impaired in hypertensive humans by a mechanism that appears to involve an angiotensin-dependent increase in sympathetic vasoconstriction in the exercising muscles.
Non-technical summary
In healthy individuals, blunting of the vasoconstriction caused by activation of the sympathetic nervous system is thought to be an important mechanism that optimizes blood flow to the working muscles. We show for the first time that this protective mechanism, called functional sympatholysis, is impaired in middle-aged patients with high blood pressure. We also show that this impairment can be reversed by treatment with an angiotensin receptor blocker, but not with a thiazide-type diuretic. These findings indicate that angiotensin II may augment sympathetic vasoconstriction in the active muscles of hypertensive humans, which may explain the exaggerated rise in blood pressure and blunted decline in systemic vascular resistance during exercise in this population.
Introduction
Essential hypertension is a common disorder that affects 25% of the adult population worldwide and is a major cause of cardiovascular morbidity and mortality. Hypertensive patients are reported to display an exaggerated rise in blood pressure and a blunted decline in systemic vascular resistance during exercise (Glezer & Lediashova, 1975; de Champlain et al. 1991; Lund-Johansen, 1991; Goodman et al. 1992), but the underlying mechanisms are not completely understood. Exercise is a potent stimulus to activate the sympathetic nervous system, which increases cardiac rate and contractility while simultaneously causes vasoconstriction in many vascular beds, including the viscera and inactive skeletal muscles. Sympathetic outflow is also directed to the exercising skeletal muscles (Hansen et al. 1994), but in young healthy humans the vasoconstrictor response is markedly attenuated in part by local metabolic products of contraction (Thomas et al. 1994; Hansen et al. 1996; Rosenmeier et al. 2003; Fadel et al. 2004). Termed functional sympatholysis (Remensnyder et al. 1962), this blunted sympathetic vasoconstriction is thought to be a protective mechanism that optimizes blood flow to the working muscles and prevents an excessive increase in blood pressure during exercise.
We recently showed that functional sympatholysis is impaired in several well-established rat models of hypertension – chronic infusion of angiotensin II and unilateral renal artery stenosis which causes renin release and increases circulating angiotensin II (Zhao et al. 2006). In these anaesthetized animal experiments, we found that the vasoconstrictor responses evoked by electrical stimulation of the lumbar sympathetic chain were attenuated during electrically evoked contractions of the hindlimb muscles of the normotensive rats but not of the hypertensive rats. Despite the novelty of this finding, the relevance for understanding sympathetic control of the skeletal muscle vasculature in human hypertension is not known.
We therefore sought to perform translational experiments to determine if functional sympatholysis is impaired in subjects with primary hypertension. We hypothesized that there would be less attenuation of reflex sympathetic vasoconstriction in the rhythmically contracting forearm muscles of hypertensive compared to normotensive subjects. We further hypothesized that this impairment in functional sympatholysis in the hypertensive subjects would be reversed by treatment with an angiotensin receptor blocker.
Methods
Ethical approval
All experimental protocols were approved by the Institutional Review Board of the University of Texas Southwestern Medical Center and were conducted in accordance with the Declaration of Helsinki principles. Written, informed consent was obtained from all of the subjects.
Subjects
Thirteen subjects with stage 1 essential hypertension and 17 normotensive subjects participated in the study. Subjects had no history of heart disease, diabetes mellitus or evidence of target organ damage such as left ventricular hypertrophy by electrocardiography or chronic kidney disease. Subjects were excluded if they were over the age of 60 or were postmenopausal women because previous studies demonstrated that sympathetic vasoconstriction is enhanced in the exercising muscles of these groups even in the absence of overt cardiovascular disease (Koch et al. 2003; Fadel et al. 2004; Dinenno et al. 2005; Parker et al. 2007). All vasoactive medications were withdrawn from the hypertensive subjects for at least 4 weeks before the study.
Experimental measurements
Subjects were studied in the supine position. Heart rate (HR) was recorded continuously by electrocardiography, and systolic and diastolic blood pressures (BP) were measured by automated oscillometric sphygmomanometry (CE0050, Welch Allyn, Skaneateles Falls, NY, USA). Respiration was monitored with a strain-gauge pneumograph and subjects were instructed to avoid sympathoexcitatory manoeuvers including Valsalvas and prolonged expirations.
Skeletal muscle oxygenation
Near-infrared (NIR) spectroscopy (NIRO-500, Hamamatsu Photonics, Hamamatsu, Japan) was used to measure changes in tissue concentrations of oxygenated haemoglobin and myoglobin (HbO2+MbO2) in the forearm, as previously described (Hansen et al. 1996; Chavoshan et al. 2002; Fadel et al. 2004). To monitor NIR light absorption, two fibre-optic bundles spaced 2 cm apart were placed over the flexor digitorum profundus muscle, which is the main muscle recruited during handgrip (Fleckenstein et al. 1992). NIR signals were sampled at a rate of 1 Hz, converted to chromophore concentrations using established algorithms, output to a personal computer and digitally stored for later analysis. Changes in the NIR signals were quantified as a percentage of the total labile signal (TLS), which was defined in each experiment as the maximal decrease in HbO2+MbO2 achieved during inflation of a pneumatic cuff on the upper arm to 220 mmHg for 3 min. Since blood vessels larger than 1 mm in diameter maximally absorb NIR light, changes in HbO2+MbO2 reflect changes occurring mainly in the microvessels (Mancini et al. 1994).
Forearm blood flow
Brachial artery diameter and mean blood velocity (MBV) were measured by Duplex Doppler ultrasonography (Philips ie33, Bothell, WA, USA) using an 11 MHz probe in the non-dominant arm. The probe was placed in a holder and fixed to the skin over the brachial artery throughout the entire experiment. Diameter measurements were obtained at end-diastole. Blood velocity was acquired with a probe insonation angle of 60 deg. The output of the handgrip dynamometer was transferred into the auxiliary input of the ultrasound system and displayed simultaneously with the ultrasound images during handgrip exercise. Images were stored on DVD discs and were analysed offline, using edge detection software (Brachial Analyzer, Medical Imaging Applications LLC, Coralville, IA, USA). Forearm blood flow (FBF, ml min−1) was calculated as MBV ×π (brachial diameter/2)2× 60. As motion artifact during handgrip produced distortion of the Doppler waveforms, images acquired during muscle contraction were excluded from analysis. Forearm vascular conductance (FVC, ml min−1 (100 mmHg)−1) was calculated as (FBF/MAP) × 100.
Reflex activation of sympathetic nerves
Lower body negative pressure (LBNP) was used to produce reflex sympathetic vasoconstriction in the forearm. The subject's lower body was enclosed to the level of the iliac crest in a negative pressure chamber. LBNP at −20 mmHg or −30 mmHg was used to unload mainly the cardiopulmonary baroreceptors and trigger increases in muscle sympathetic nerve activity (SNA) (Hansen et al. 1996). Multiunit recordings of SNA were obtained with unipolar tungsten microelectrodes inserted into muscle fascicles of the peroneal nerve by microneurography (Vallbo et al. 1979). Neural signals were amplified, filtered (bandwidth 700–2000 Hz), rectified and integrated to obtain mean voltage neurograms. Recordings were considered acceptable based on well-defined criteria that discriminate muscle SNA from other neural signals including skin SNA and muscle spindle activity (Hansen et al. 1996). Muscle SNA was expressed as burst frequency (bursts min−1) and total activity (burst frequency × mean burst amplitude). Changes in SNA (% total activity) during the course of each experimental protocol were expressed as relative increases from the baseline activity at rest. Changes in SNA specifically in response to LBNP were expressed as the relative increases from the pre-LBNP baseline at rest or during handgrip.
Handgrip exercise
Maximal voluntary contraction (MVC) for each subject was designated as the greatest of at least three maximal squeezes of a handgrip dynamometer (Stoelting, Chicago, IL, USA). Subjects performed intermittent handgrip to the rhythm of a metronome (20 handgrips min−1; 50% duty cycle) at 30% MVC for 6 min. Force production was displayed on an oscilloscope to provide subjects with visual feedback. This level of handgrip alone does not increase muscle SNA in healthy subjects (Hansen et al. 1996).
Experimental protocols
Protocol 1. Comparing functional sympatholysis in normotensive (n= 15) and hypertensive (n= 13) subjects
Blood pressure, HR, forearm muscle oxygenation, FBF, FVC and SNA were measured in response to 2 min of LBNP at −20 mmHg applied at rest and during minutes 3–5 of handgrip in both groups of subjects.
Protocol 2. Effect of artificially increasing SNA from the onset of handgrip exercise on functional sympatholysis in normotensive subjects (n= 6), including four subjects from Protocol 1 plus two additional subjects
In our pilot studies for Protocol 1, we observed that SNA unexpectedly increased in hypertensive subjects during handgrip exercise alone. To control for this atypical sympathoexcitatory response to handgrip, normotensive subjects performed a modified version of the sympatholysis protocol in which SNA was artificially elevated by applying LBNP at −20 mmHg during the first 3 min of handgrip. LBNP was then further lowered to −30 mmHg between minutes 3–5 of handgrip to induce an additional rise in SNA. Functional sympatholysis was evaluated by comparing changes in BP, HR, forearm muscle oxygenation, FBF, FVC and SNA during LBNP at −30 mmHg vs. LBNP at −20 mmHg at rest and during handgrip.
Protocol 3. Effect of angiotensin receptor subtype 1 (AT1) blockade on functional sympatholysis in hypertensive subjects (n= 7)
We previously showed that functional sympatholysis is impaired in rats with angiotensin-dependent hypertension (Zhao et al. 2006). To begin to translate this finding from rats and gain potential insight into the mechanism that might be responsible for impairing functional sympatholysis in hypertensive humans, Protocol 1 was performed in a subset of the hypertensive cohort at baseline (no drug), after 4 weeks of daily irbesartan (150 mg), and after 4 weeks of drug washout. As a control for the antihypertensive effect of irbesartan, Protocol 1 was also performed in five hypertensive subjects (including 3 subjects who participated in the irbesartan study plus 2 additional subjects) before and after 4 weeks of daily chlorthalidone (25 mg), a thiazide-type diuretic.
Statistical analysis
Sample sizes were determined based on detecting differences in LBNP-induced decreases in muscle oxygenation and FBF between hypertensive and normotensive subjects. In Protocol 1, a sample size of 24 (12 per group) was required to detect an effect size of 10% TLS for muscle oxygenation and 15% for FBF with 90% power at the alpha level of 0.05. In Protocol 2, a sample size of six was required to detect an effect size of 6% TLS for muscle oxygenation and 11% for FBF within the same group with 80% power at the alpha level of 0.05. In Protocol 3, six subjects were required to detect effect sizes of 10% TLS for muscle oxygenation and 15% for FBF during drug treatment with 80% and 84% power, respectively, at the level of significance of 0.05.
In Protocol 1, comparisons between hypertensive and normotensive groups were made with repeated measures analyses using a mixed linear model approach. The model factors consisted of study group (hypertensive vs. normotensive) and intervention (LBNP, rest, handgrip), with subject modelled as a random effect. The difference in response between the hypertensive and normotensive groups was assessed by evaluating the group by experiment interaction factor from these models. Planned pair-wise comparisons were made from the least-squares means contrasts derived from the linear models. Compound symmetry and unstructured covariance patterns were employed for most models and an autoregressive covariance structure was used for time course models. In Protocol 2, within-group normotensive-adjusted SNA responses were compared using mixed-model repeated-measures analyses with one repeated factor and comparisons of specific experiments were made with the models’ least-square means contrasts. For Protocol 3, the study phases were compared with repeated measures models that included two repeated factors to assess drug treatment and experiment (LBNP, handgrip) and their interaction. In all protocols, variables with skewed distributions were log or rank transformed prior to analysis. Statistical analyses were conducted using SAS version 9.2 (SAS Institute, Cary, NC, USA). All tests were two-sided and a P value < 0.05 was considered statistically significant. Data are presented as means ±s.e.m.
Results
Baseline characteristics of the normotensive and hypertensive subjects are shown in Table 1. The two groups were well matched for all characteristics. Systolic and diastolic BPs were significantly higher by design in the hypertensive vs. normotensive group (P < 0.05). Resting heart rate was also higher in hypertensive vs. normotensive subjects (P < 0.05).
Table 1.
Subject characteristics
| Normotensive | Hypertensive | |
|---|---|---|
| Subjects (n) | 15 | 13 |
| Age (years) | 48 ± 2 | 47 ± 3 |
| Sex (M/F) | 11 / 4 | 9 / 4 |
| Body mass index (kg m−2) | 29 ± 1 | 30 ± 2 |
| Systolic BP (mmHg) | 118 ± 3 | 144 ± 4* |
| Diastolic BP (mmHg) | 72 ± 2 | 87 ± 2* |
| Heart rate (beats min–1) | 57 ± 2 | 65 ± 2* |
| Maximal voluntary contraction (kg) | 31 ± 2 | 31 ± 2 |
| Serum creatinine (mg dl−1) | 1.0 ± 0.07 | 1.0 ± 0.04 |
| Total cholesterol (mg dl−1) | 181 ± 8 | 183 ± 11 |
| Triglycerides (mg dl−1) | 132 ± 44 | 97 ± 14 |
| Fasting plasma glucose (mg dl−1) | 94 ± 3 | 99 ± 4 |
BP, blood pressure.
P < 0.05 vs. normotensive.
Functional sympatholysis is impaired in hypertensive subjects
LBNP at −20 mmHg evoked similar decreases in muscle oxygenation (−16 ± 2%vs.−14 ± 2%), FBF (−23 ± 4%vs.−20 ± 3%) and FVC (−22 ± 4%vs.−19 ± 3%) in the resting forearms of the normotensive and hypertensive subjects, respectively, without affecting MAP or HR (Fig. 1 and Table 2). In the normotensive subjects, this same level of LBNP applied during rhythmic handgrip caused minimal changes in muscle oxygenation (+1 ± 2%), FBF (+1 ± 2%) and FVC (0 ± 2%) (P < 0.05 vs. responses at rest). In contrast, in the hypertensive subjects LBNP evoked decreases in muscle oxygenation (−12 ± 2%), FBF (−13 ± 2%) and FVC (−14 ± 2%) that were not different from the responses to LBNP at rest (P > 0.05).
Figure 1. Functional sympatholysis in normotensive and hypertensive subjects.
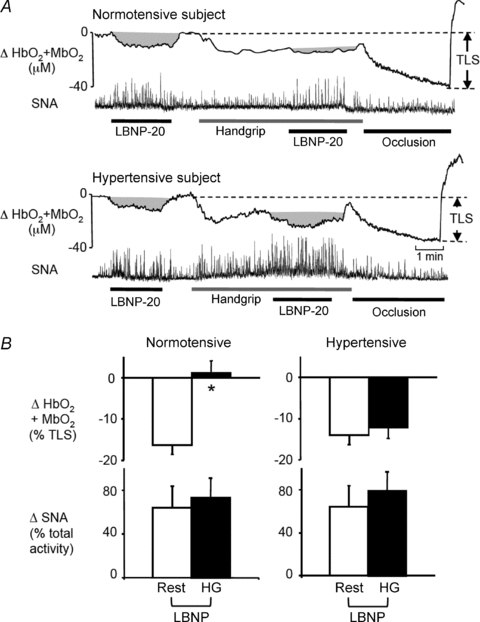
A, original recordings of forearm muscle oxygenation (HbO2+MbO2) and SNA responses to lower body negative pressure (LBNP) applied at rest and during rhythmic handgrip. In both subjects, LBNP evoked similar increases in SNA at rest and during handgrip exercise. In the normotensive subject, this increase in SNA produced a large decrease in muscle oxygenation at rest and an attenuated decrease in oxygenation during handgrip, indicating functional sympatholysis. In the hypertensive subject, the LBNP-induced increases in SNA evoked similar decreases in muscle oxygenation at rest and during handgrip, indicating impaired sympatholysis. Complete forearm vascular occlusion after the exercise produced the maximal decrease in muscle oxygenation that was used to determine the total labile signal (TLS). B, summary data showing the changes in muscle oxygenation and SNA in response to LBNP at rest and during handgrip in normotensive (n= 15) and hypertensive (n= 13) subjects. *P < 0.05 vs. rest.
Table 2.
Haemodynamic and sympathetic responses to LBNP at rest and during handgrip
| Rest | Rest+LBNP | Handgrip | Handgrip+LBNP | |
|---|---|---|---|---|
| Normotensive subjects | ||||
| MAP (mmHg) | 88 ± 2 | 87 ± 2 | 94 ± 2* | 95 ± 2* |
| HR (beats min−1) | 59 ± 2 | 60 ± 2 | 68 ± 3* | 69 ± 3* |
| SNA (bursts min−1) | 31 ± 3 | 39 ± 2* | 31 ± 3 | 41 ± 2*‡ |
| Δ SNA (% total activity) | 0 ± 0 | 63 ± 18* | 5 ± 7 | 78 ± 18*‡ |
| FBF (ml min−1) | 114 ± 15 | 85 ± 10* | 474 ± 41* | 477 ± 39* |
| FVC (units) | 136 ± 17 | 103 ± 12* | 513 ± 50* | 506 ± 44* |
| Hypertensive subjects | ||||
| MAP (mmHg) | 104 ± 2† | 101 ± 3† | 112 ± 3*† | 110 ± 3*† |
| HR (beats min−1) | 66 ± 2† | 66 ± 2 | 73 ± 2* | 75 ± 2* |
| SNA (bursts min−1) | 35 ± 4 | 43 ± 3* | 41 ± 3*† | 47 ± 3*‡ |
| Δ SNA (% total activity) | 0 ± 0 | 64 ± 19* | 54 ± 11*† | 133 ± 23*†‡ |
| FBF (ml min−1) | 98 ± 17 | 76 ± 11* | 447 ± 56* | 384 ± 50*‡ |
| FVC (units) | 93 ± 15 | 73 ± 11* | 397 ± 49* | 343 ± 41*†‡ |
LBNP, lower body negative pressure; MAP, mean arterial pressure; HR, heart rate; SNA, sympathetic nerve activity; FBF, forearm blood flow; FVC, forearm vascular conductance.
P < 0.05 vs. rest;
P < 0.05 vs. normotensive;
P < 0.01 vs. handgrip; Normotensive, n= 15; hypertensive, n= 13.
Sympathoexcitatory responses to LBNP are typical but responses to handgrip are atypical in hypertensive subjects
Baseline SNA at rest and the reflex increases in SNA induced specifically by LBNP applied at rest or during handgrip were similar in the normotensive and hypertensive subjects (P > 0.05; Fig. 1 and Table 2). Responses to handgrip alone at 30% MVC in the normotensive group were characteristic for this moderate level of rhythmic handgrip and included increases in HR, FBF and FVC with no significant changes in MAP or SNA (Fig. 2 and Table 2). Handgrip-induced increases in HR, FBF and FVC were similar in the hypertensive group (P > 0.05 vs. normotensive). However, handgrip alone evoked an unexpected sympathoexcitatory response in the hypertensive subjects: SNA began to increase during the first minute of exercise and was significantly elevated by approximately 50% in the second minute (P < 0.05 vs. rest; Fig. 1A, Fig. 2 and Table 2). This handgrip-induced increase in SNA was accompanied by an augmented increase in MAP in the hypertensive group. MAP increased from 105 ± 2 mmHg at minute 0 to 112 ± 3 mmHg after 3 min of exercise in the hypertensive subjects, while increasing from 90 ± 2 to 94 ± 2 mmHg after 3 min of exercise in the normotensive subjects. Both the absolute and relative increase in MAP during handgrip were greater in the hypertensive than normotensive group (+7 ± 1 vs.+4 ± 1 mmHg and +7.0 ± 0.8%vs. 4.3 ± 0.8%, respectively, both P < 0.05, Fig. 2).
Figure 2. Pressor and sympathoexcitatory responses to rhythmic handgrip alone in normotensive and hypertensive subjects.
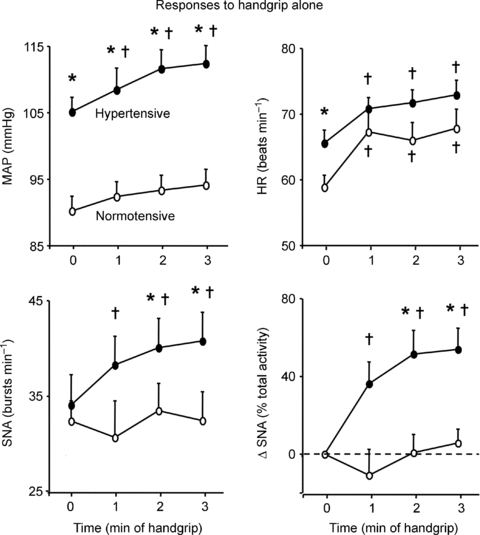
Summary data showing MAP, HR and SNA (expressed as burst frequency and change from resting baseline) during the first 3 min of rhythmic handgrip at 30% MVC. In the normotensive subjects (n= 15), this moderate level of handgrip characteristically increased HR without changing MAP or SNA. In contrast, in the hypertensive subjects (n= 13) handgrip evoked a similar increase in HR that was accompanied by atypical progressive increases in MAP and SNA. *P < 0.05 vs. normotensive; †P < 0.05 vs. min 0 (rest).
Functional sympatholysis is unaffected in normotensive subjects when SNA is atypically increased from the onset of handgrip
At rest, LBNP at −20 mmHg evoked decreases in forearm muscle oxygenation, FBF and FVC that were accompanied by increased SNA (Fig. 3 and Table 3). Further lowering LBNP to −30 mmHg caused additional significant decreases in muscle oxygenation (−7 ± 1%), FBF (−14 ± 3%) and FVC (−15 ± 4%) while further increasing SNA by 55% (P < 0.05 vs. rest plus LBNP −20 mmHg for all variables). Neither level of LBNP affected MAP (P > 0.05 vs. rest) and HR was increased only during LBNP at −30 mmHg (P < 0.05 vs. rest). When LBNP at −20 mmHg was applied at the onset of handgrip, SNA was increased by approximately 70% in the first minute of exercise and remained elevated thereafter (P < 0.05 vs. rest; Fig. 3B and Table 3), successfully reproducing the atypical sympathoexcitatory response to handgrip that was observed in the hypertensive subjects (Fig. 2). MAP, HR, FBF and FVC also were increased during combined handgrip and LBNP at −20 mmHg (P < 0.05 vs. rest; Table 3). When LBNP was further lowered to −30 mmHg during handgrip, SNA increased by an additional 70% but this did not result in a decrease in muscle oxygenation, FBF or FVC (Fig. 3C and Table 3).
Figure 3. Functional sympatholysis in normotensive subjects with artificially elevated SNA from the onset of exercise.
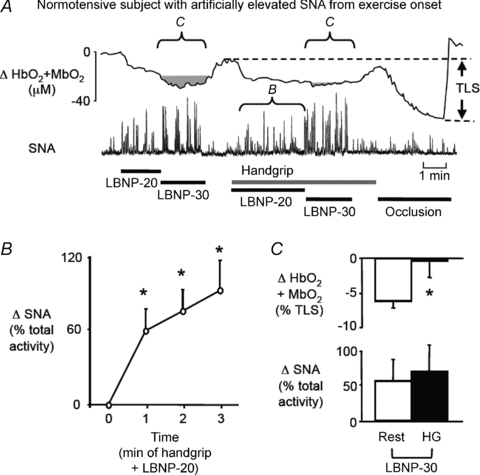
A, an original recording of forearm muscle oxygenation (HbO2+MbO2) and SNA responses to graded LBNP applied at rest and during handgrip in a normotensive subject. At rest, LBNP produced graded increases in SNA and decreases in muscle oxygenation. LBNP at −20 mmHg was applied at the beginning of handgrip to artificially elevate SNA and mimic the exercise-induced sympathoexcitation observed in the hypertensive subjects. When LBNP was then further lowered to −30 mmHg during handgrip, SNA increased further without reducing muscle oxygenation, indicating preserved functional sympatholysis. B, summary data showing the progressive increase in SNA evoked by handgrip plus simultaneous application of LBNP at −20 mmHg in normotensive subjects (n= 6). *P < 0.05 vs. min 0 (rest). C, summary data showing the changes in muscle oxygenation and SNA in response to LBNP at −30 mmHg at rest and during handgrip (n= 6). *P < 0.05 vs. rest.
Table 3.
Haemodynamic and sympathetic responses to LBNP at rest and during handgrip in normotensive subjects with artificially elevated SNA from the onset of exercise
| Rest | Rest+LBNP−20 | Rest+LBNP−30 | Handgrip+LBNP−20 | Handgrip+LBNP−30 | |
|---|---|---|---|---|---|
| MAP (mmHg) | 86 ± 1 | 87 ± 2 | 89 ± 2 | 95 ± 2* | 95 ± 2* |
| HR (beat min−1) | 60 ± 4 | 63 ± 4* | 68 ± 3*† | 70 ± 4* | 72 ± 4* |
| SNA (bursts min−1) | 33 ± 4 | 45 ± 3* | 47 ± 3* | 45 ± 4* | 51 ± 3*‡ |
| Δ SNA (% total activity) | 0 ± 0 | 84 ± 29* | 139 ± 47*† | 108 ± 28* | 176 ± 64*‡ |
| FBF (ml min−1) | 151 ± 30 | 105 ± 21* | 90 ± 18*† | 443 ± 70* | 467 ± 82* |
| FVC (units) | 174 ± 33 | 119 ± 22* | 100 ± 19*† | 463 ± 67* | 491 ± 94* |
P < 0.05 vs. rest;
P < 0.05 vs. rest + LBNP –20;
P < 0.01 vs. handgrip + LBNP –20; n= 6 subjects.
Functional sympatholysis is restored in hypertensive subjects treated with an AT1 receptor blocker
The subset of seven hypertensive subjects studied in this protocol was highly representative of the larger hypertensive cohort as shown by the similarities in baseline haemodynamics and SNA, and by the comparable vasoconstrictor responses evoked by LBNP at −20 mmHg applied at rest or during handgrip (Fig. 4 and Table 4). Treatment of these hypertensive subjects with irbesartan for 4 weeks reduced MAP by 9 ± 3 mmHg and increased SNA at rest by 11 ± 5 bursts min−1 (both P < 0.05 vs. no drug; Table 4) with no effect on resting HR, FBF or FVC. Irbesartan also had no effect on the LBNP-induced increase in SNA (+46 ± 14%) and decreases in muscle oxygenation (−17 ± 3%), FBF (−20 ± 5%) and FVC (−20 ± 5%) in resting forearm. However, these vasoconstrictor responses were abolished when LBNP was applied during handgrip in the irbesartan-treated subjects, despite a comparable effect of LBNP to increase SNA by 58 ± 12% (Fig. 4 and Table 4). Although irbesartan normalized functional sympatholysis in the hypertensive subjects, it did not normalize the sympathoexcitatory response to exercise: SNA still increased by 44 ± 21% in response to handgrip alone (P < 0.05 vs. rest; Table 4). All of the significant effects of irbesartan were reversed following 4 weeks of drug washout (Fig. 4 and Table 4).
Figure 4. Functional sympatholysis in hypertensive subjects treated with an AT1 receptor blocker.
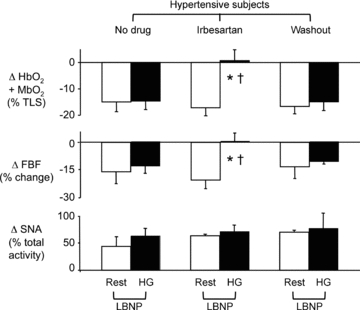
Summary data showing the changes in muscle oxygenation, FBF and SNA in response to LBNP at rest and during handgrip in hypertensive subjects (n= 7) studied at baseline (no drug), after treatment with irbesartan for 4 weeks and after drug washout for 4 weeks. *P < 0.05 vs. rest; †P < 0.05 vs. no drug or washout.
Table 4.
Haemodynamic and sympathetic responses to LBNP at rest and during handgrip in hypertensive subjects treated with an AT1 receptor blocker
| Rest | Rest + LBNP | Handgrip | Handgrip + LBNP | |
|---|---|---|---|---|
| No drug | ||||
| MAP (mmHg) | 106 ± 4 | 103 ± 4 | 112 ± 4* | 112 ± 5* |
| HR (beats min−1) | 63 ± 2 | 65 ± 3 | 72 ± 1* | 74 ± 2* |
| SNA (bursts min−1) | 34 ± 5 | 47 ± 2* | 41 ± 5 | 49 ± 3*† |
| Δ SNA (% total activity) | 0 ± 0 | 62 ± 14* | 44 ± 15* | 109 ± 17 *† |
| FBF (ml min−1 | 113 ± 15 | 89 ± 10* | 627 ± 69* | 551 ± 63*† |
| FVC (units) | 109 ± 16 | 88 ± 12* | 570 ± 73* | 498 ± 63*† |
| Irbesartan | ||||
| MAP (mmHg) | 97 ± 4‡ | 97 ± 5‡ | 102 ± 5‡ | 104 ± 5*‡ |
| HR (beats min−1) | 68 ± 3 | 72 ± 4‡ | 74 ± 3* | 80 ± 4* |
| SNA (bursts min−1) | 46 ± 4‡‖ | 58 ± 3*‡ | 53 ± 4*‡‖ | 61 ± 5*‡‖ |
| Δ SNA (% total activity) | 0 ± 0 | 46 ± 14* | 44 ± 21* | 102 ± 16*† |
| FBF (ml min−1) | 122 ± 14 | 97 ± 13* | 631 ± 105* | 631 ± 101* |
| FVC (units) | 129 ± 19 | 102 ± 16* | 626 ± 99* | 618 ± 98*‡ |
| Washout | ||||
| MAP (mmHg) | 105 ± 3 | 102 ± 3 | 112 ± 4 | 112 ± 4 |
| HR (beats min−1) | 65 ± 2 | 67 ± 3 | 70 ± 2* | 72 ± 4* |
| SNA (bursts min−1) | 32 ± 4 | 49 ± 6* | 40 ± 5* | 46 ± 7* |
| Δ SNA (% total activity) | 0 ± 0 | 74 ± 25* | 46 ± 13* | 174 ± 44*† |
| FBF (ml min−1) | 166 ± 30 | 136 ± 16* | 560 ± 131* | 511 ± 124*† |
| FVC (units) | 158 ± 35 | 123 ± 20* | 496 ± 111* | 452 ± 105*† |
P < 0.05 vs. rest;
P < 0.05 vs. handgrip
P < 0.05 vs. no drug,
P < 0.05 vs. washout; n= 7 subjects.
In the five hypertensive subjects treated with chlorthalidone, MAP decreased by 6 ± 3 mmHg, which was similar to the effect of irbesartan (P > 0.05). In addition, chlorthalidone increased resting SNA by 9 ± 6 bursts min−1 with no effect on resting HR, FBF or FVC. Chlorthalidone had no effect on the LBNP-induced increases in SNA and decreases in muscle oxygenation, FBF and FVC in either resting or exercising forearm (Fig. 5).
Figure 5. Functional sympatholysis in hypertensive subjects treated with a thiazide-type diuretic.
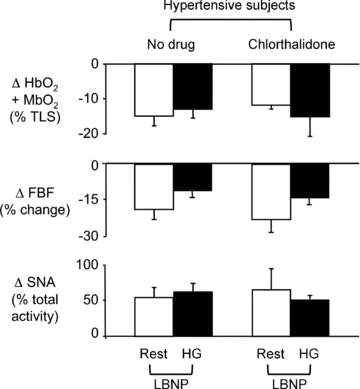
Summary data showing the changes in muscle oxygenation, FBF and SNA in response to LBNP at rest and during handgrip in 5 hypertensive subjects studied before and after treatment with chlorthalidone for 4 weeks.
Discussion
The major new findings of the present study are three-fold. First, sympathetic vasoconstriction was not attenuated in the exercising forearm muscles of individuals with mild, uncomplicated hypertension in contrast to the robust functional sympatholysis observed in the exercising muscles of normotensive individuals. Second, muscle SNA was uncharacteristically increased within the first minute of moderate-intensity rhythmic handgrip exercise in the hypertensive subjects. However, this exercise-induced sympathoexcitation is unlikely to be the principal cause of the impaired functional sympatholysis in the hypertensive subjects. When muscle SNA was experimentally increased from the beginning of exercise in normotensive subjects, sympatholysis was unaffected. Third, functional sympatholysis was readily restored in the hypertensive subjects during short-term treatment with an ARB but not with a thiazide-type diuretic, despite equivalent antihypertensive effects of the two drugs. Collectively these findings provide the first evidence that functional sympatholysis is impaired in humans with a common cardiovascular disease and suggest that the underlying mechanism involves an angiotensin-dependent increase in sympathetic vasoconstriction in the exercising muscles. These data also support the emerging hypothesis that the sympathoexcitatory response to exercise is exaggerated in hypertensive humans, and show for the first time that this occurs even during a relatively low level of exercise that would not be expected to cause muscle ischaemia and activate the muscle metaboreflex.
In young healthy individuals, we and others have found that moderate vasoconstrictor responses evoked by reflex sympathetic activation (i.e. simulated orthostatic stress) or infusion of sympathomimetic drugs in resting muscle are largely abolished during a low to moderate intensity of rhythmic exercise (Hansen et al. 2000; Rosenmeier et al. 2003; Wray et al. 2004). Our current study now extends this finding of robust functional sympatholysis to middle-aged normotensive men and women. We specifically excluded individuals over the age of 60 and postmenopausal women from our study because of previous reports that sympathetic vasoconstriction is enhanced in exercising muscle in this population even in the absence of overt cardiovascular disease (Koch et al. 2003; Fadel et al. 2004; Dinenno et al. 2005; Parker et al. 2007).
Although our normotensive and hypertensive subjects were carefully matched for physical and metabolic characteristics, functional sympatholysis was clearly impaired only in the hypertensive group as shown by the large decreases in muscle oxygenation and blood flow evoked by LBNP during forearm exercise. The impaired sympatholysis could not be explained by a greater sympathetic stimulus because LBNP evoked similar increases in SNA at rest and during exercise in both normotensive and hypertensive subjects. Nor could it be explained by a general increase in sympathetic vasoconstrictor responsiveness as LBNP evoked similar decreases in muscle oxygenation and blood flow in the resting forearms of both groups of subjects. However, one factor that we could not immediately exclude was the increase in SNA that occurred during exercise alone in the hypertensive subjects. This exercise-induced sympathoexcitation was unexpected because rhythmic handgrip exercise performed at 30% MVC does not reflexively activate muscle SNA in healthy subjects (Hansen et al. 1996). Our concern was that the combined effect of handgrip exercise plus LBNP resulted in a 50% larger increase in SNA in the hypertensive vs. normotensive subjects, which could potentially explain the impaired sympatholysis in the hypertensive group. We addressed this concern by performing additional experiments in normotensive subjects using graded LBNP to achieve increments in SNA that mimicked the pattern of sympathoexcitation observed during exercise alone and exercise plus LBNP in the hypertensive group. Even in this setting of augmented sympathetic outflow, the normotensive subjects displayed robust functional sympatholysis. Taken together, these findings indicate that the impaired sympatholysis observed in the hypertensive subjects is most probably mediated by an increased vascular responsiveness to sympathetic activation which occurs only in the exercising muscles.
To begin to understand the specific mechanism underlying the impaired functional sympatholysis in our hypertensive subjects, we chose to initially focus on a potential role for angiotensin II because we previously found that functional sympatholysis was impaired in angiotensin-dependent rat models of hypertension (Zhao et al. 2006). While the pathogenesis of primary human hypertension is thought to be heterogeneous, a large body of experimental and clinical research implicates a pivotal role for angiotensin II in the genesis and progression of human hypertension (Simon, 2004; Dzau, 2005; Shafiq et al. 2008). Our novel finding that functional sympatholysis was readily normalized by a short 4 week treatment with an ARB lends credence to the hypothesis that angiotensin II is a key mediator of the excessive sympathetic vasoconstriction in the exercising muscles of the hypertensive subjects. This is further supported by the lack of an effect of the thiazide-type diuretic chlorthalidone to restore functional sympatholysis in hypertensive subjects despite achieving a reduction in blood pressure that was similar to irbesartan.
We do not know the mechanism by which angiotensin II impairs functional sympatholysis in human hypertension, but the peptide has effects on both neural and vascular tissue that could augment sympathetic vasoconstriction. In animal studies, angiotensin II has been shown to act centrally to increase SNA and peripherally to enhance release of noradrenaline (norepinephrine, NA) from sympathetic nerve terminals (Phillips, 1987). However, the findings from our study confirm previous reports that resting SNA is not decreased, but is either increased or unaffected by ARBs (Esler, 2002; Fu et al. 2005; Krum et al. 2006). We also think that it is unlikely that a presynaptic effect of AT1 receptor blockade to reduce NA release explains our findings because the LBNP-induced vasoconstriction in resting muscle was unchanged during irbesartan treatment. Furthermore, in contrast to the effect of ARBs in animals, studies in humans have not shown that NA release is reduced by blockade of these receptors (Esler, 2002; Fu et al. 2005; Krum et al. 2006). We speculate that a vascular effect of angiotensin II is more likely to explain the impaired functional sympatholysis. Angiotensin II can act postjunctionally to potentiate NA-induced vasoconstriction, especially when it involves α2 adrenergic receptors (Dunn et al. 1991a,b; Ikeoka & Faber, 1993). We and others have previously shown that functional sympatholysis is mediated in part by an attenuation of α2 adrenergic vasoconstriction (Thomas et al. 1994; Rosenmeier et al. 2003; Wray et al. 2004). Finally, angiotensin II also can increase the production of reactive oxygen species, which we have previously shown to impair functional sympatholysis in rat models of heart failure and hypertension (Thomas et al. 2001; Zhao et al. 2006).
Although incidental, our novel finding that a moderate level (30% MVC) of rhythmic forearm exercise increased SNA in the hypertensive subjects merits further investigation. In healthy individuals, sympathoexcitation typically only occurs during higher intensities (45% MVC or greater) of rhythmic handgrip exercise and has been attributed mainly to activation of muscle metaboreceptors (Victor et al. 1988). However, little is currently known about exercise-induced sympathoexcitation in hypertensive subjects with previous studies reporting either an attenuated or exaggerated SNA response to static handgrip exercise compared to normotensive controls (Rondon et al. 2006; Delaney et al. 2010). The mechanisms underlying these abnormal sympathoexcitatory responses remain obscure and could potentially involve changes in central command, muscle mechano- or metaboreflexes, or baroreflexes (Rondon et al. 2006; Smith et al. 2006; Delaney et al. 2010). While our study was not specifically designed to examine mechanisms of exercise-induced sympathoexcitation in hypertension, we can rule out a role for AT1 receptors because irbesartan treatment did not prevent SNA from increasing during handgrip exercise.
There are several limitations to our study. First, the exercise protocol that we used engages only a small muscle mass in the forearm and we do not know if the results are applicable to exercise involving larger muscle masses. However, functional sympatholysis is present in the thigh muscles during dynamic exercise in normotensive humans and is mediated by attenuation of α1 and α2 adrenergic vasoconstriction as in forearm muscles (Wray et al. 2004). Second, our drug treatment study in the hypertensive subjects was not randomized or placebo-controlled. However, it is unlikely that restoration of sympatholysis with irbesartan was the result of random variation or subject familiarization because this effect was reversed after drug washout. Third, we only evaluated the effect of two antihypertensive drugs and do not know if the beneficial effect of an ARB to restore sympatholysis is specific only to this class of drug. However, we do know that decreases in blood pressure alone are not sufficient to restore sympatholysis as chlorthalidone failed to do so despite its antihypertensive effect.
Even with these limitations, our study may have important clinical implications. Impaired functional sympatholysis in hypertensive patients may contribute to an exaggerated pressor response to exercise and impaired exercise tolerance even in the absence of coronary artery disease or left ventricular dysfunction (Fagard et al. 1988; Lim et al. 1996). Interestingly, treatment with ARBs has been shown to attenuate the pressor response to exercise and improve exercise capacity in hypertensive patients (Nashar et al. 2004; Omvik et al. 2000), effects which were not observed with other antihypertensive drugs such as beta blockers (Hamada et al. 1987; Cleroux et al. 1994) or calcium channel blockers (Taylor et al. 1982; Dvorak et al. 1991). Further studies are needed to determine if restoration of functional sympatholysis can improve functional capacity in hypertensive subjects.
Acknowledgments
Sources of funding: supported by grants to W.V. from the National Institute of Health (R01 HL-078782) and the O’Brien Kidney Center and to B.A.-H. from the Clinical and Translational Sciences Award (UL1RR-024982). There are no conflicts of interest.
Glossary
Abbreviations
- ARB
AT1 receptor blocker
- AT1
angiotensin II type 1 receptor
- BP
blood pressure
- FBF
forearm blood flow
- FVC
forearm vascular conductance
- HR
heart rate
- LBNP
lower body negative pressure
- MAP
mean arterial pressure
- MBV
mean blood velocity
- MVC
maximal voluntary contraction
- NA
noradrenaline
- NIR
near-infrared
- RHG
rhythmic handgrip
- SNA
sympathetic nerve activity
- TLS
total labile signal
Author contributions
W.V., G.D.T. and R.G.V. conceived and designed the experiments. W.V., Z.W., D.A., G.A., J.H.M. and B.A.-H. collected, analysed and interpreted the data. W.V. and G.D.T. drafted the manuscript. All authors contributed to the revision of the manuscript and approved the final version.
References
- Chavoshan B, Sander M, Sybert TE, Hansen J, Victor RG, Thomas GD. Nitric oxide-dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J Physiol. 2002;540:377–386. doi: 10.1113/jphysiol.2001.013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleroux J, Beaulieu M, Kouame N, Lacourciere Y. Comparative effects of quinapril, atenolol, and verapamil on blood pressure and forearm hemodynamics during handgrip exercise. Am J Hypertens. 1994;7:566–570. doi: 10.1093/ajh/7.6.566. [DOI] [PubMed] [Google Scholar]
- de Champlain J, Petrovich M, Gonzalez M, Lebeau R, Nadeau R. Abnormal cardiovascular reactivity in borderline and mild essential hypertension. Hypertension. 1991;17:III22–III28. doi: 10.1161/01.hyp.17.4_suppl.iii22. [DOI] [PubMed] [Google Scholar]
- Delaney EP, Greaney JL, Edwards DG, Rose WC, Fadel PJ, Farquhar WB. Exaggerated sympathetic and pressor responses to handgrip exercise in older hypertensive humans: role of the muscle metaboreflex. Am J Physiol Heart Circ Physiol. 2010;299:H1318–H1327. doi: 10.1152/ajpheart.00556.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA, Masuki S, Joyner MJ. Impaired modulation of sympathetic α-adrenergic vasoconstriction in contracting forearm muscle of ageing men. J Physiol. 2005;567:311–321. doi: 10.1113/jphysiol.2005.087668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WR, McGrath JC, Wilson VG. Influence of angiotensin II on the α-adrenoceptors involved in mediating the response to sympathetic nerve stimulation in the rabbit isolated distal saphenous artery. Br J Pharmacol. 1991a;102:10–12. doi: 10.1111/j.1476-5381.1991.tb12123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WR, McGrath JC, Wilson VG. Postjunctional α-adrenoceptors in the rabbit isolated distal saphenous artery: indirect sensitivity to prazosin of responses to noradrenaline mediated via postjunctional α2-adrenoceptors. Br J Pharmacol. 1991b;103:1484–1492. doi: 10.1111/j.1476-5381.1991.tb09815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak I, Blaha M, Nemcova H. Antihypertensive effect of isradipine on blood pressure at rest and during exercise. Am J Hypertens. 1991;4:144S–146S. doi: 10.1093/ajh/4.2.144s. [DOI] [PubMed] [Google Scholar]
- Dzau V. The cardiovascular continuum and renin-angiotensin-aldosterone system blockade. J Hypertens Suppl. 2005;23:S9–S17. [PubMed] [Google Scholar]
- Esler M. Differentiation in the effects of the angiotensin II receptor blocker class on autonomic function. J Hypertens Suppl. 2002;20:S13–S19. [PubMed] [Google Scholar]
- Fadel PJ, Wang Z, Watanabe H, Arbique D, Vongpatanasin W, Thomas GD. Augmented sympathetic vasoconstriction in exercising forearms of postmenopausal women is reversed by oestrogen therapy. J Physiol. 2004;561:893–901. doi: 10.1113/jphysiol.2004.073619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagard R, Staessen J, Amery A. Maximal aerobic power in essential hypertension. J Hypertens. 1988;6:859–865. doi: 10.1097/00004872-198811000-00003. [DOI] [PubMed] [Google Scholar]
- Fleckenstein JL, Watumull D, Bertocci LA, Parkey RW, Peshock RM. Finger-specific flexor recruitment in humans: depiction by exercise-enhanced MRI. J Appl Physiol. 1992;72:1974–1977. doi: 10.1152/jappl.1992.72.5.1974. [DOI] [PubMed] [Google Scholar]
- Fu Q, Zhang R, Witkowski S, Arbab-Zadeh A, Prasad A, Okazaki K, Levine BD. Persistent sympathetic activation during chronic antihypertensive therapy: a potential mechanism for long term morbidity? Hypertension. 2005;45:513–521. doi: 10.1161/01.HYP.0000158312.63381.c1. [DOI] [PubMed] [Google Scholar]
- Glezer GA, Lediashova GA. Changes in general haemodynamics and renal function during exercise in patients with arterial hypertension. Cor Vasa. 1975;17:1–13. [PubMed] [Google Scholar]
- Goodman JM, McLaughlin PR, Plyley MJ, Holloway RM, Fell D, Logan AG, Liu PP. Impaired cardiopulmonary response to exercise in moderate hypertension. Can J Cardiol. 1992;8:363–371. [PubMed] [Google Scholar]
- Hamada M, Kazatani Y, Shigematsu Y, Ito T, Kokubu T, Ishise S. Enhanced blood pressure response to isometric handgrip exercise in patients with essential hypertension: effects of propranolol and prazosin. J Hypertens. 1987;5:305–309. doi: 10.1097/00004872-198706000-00007. [DOI] [PubMed] [Google Scholar]
- Hansen J, Sander M, Thomas GD. Metabolic modulation of sympathetic vasoconstriction in exercising skeletal muscle. Acta Physiol Scand. 2000;168:489–503. doi: 10.1046/j.1365-201x.2000.00701.x. [DOI] [PubMed] [Google Scholar]
- Hansen J, Thomas GD, Harris SA, Parsons WJ, Victor RG. Differential sympathetic neural control of oxygenation in resting and exercising human skeletal muscle. J Clin Invest. 1996;98:584–596. doi: 10.1172/JCI118826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J, Thomas GD, Jacobsen TN, Victor RG. Muscle metaboreflex triggers parallel sympathetic activation in exercising and resting human skeletal muscle. Am J Physiol Heart Circ Physiol. 1994;266:H2508–H2514. doi: 10.1152/ajpheart.1994.266.6.H2508. [DOI] [PubMed] [Google Scholar]
- Ikeoka K, Faber JE. ANG II reverses selective inhibition of alpha 2-adrenoceptor sensitivity after in vitro isolation of arterioles. Am J Physiol Heart Circ Physiol. 1993;265:H1988–H1995. doi: 10.1152/ajpheart.1993.265.6.H1988. [DOI] [PubMed] [Google Scholar]
- Koch DW, Leuenberger UA, Proctor DN. Augmented leg vasoconstriction in dynamically exercising older men during acute sympathetic stimulation. J Physiol. 2003;551:337–344. doi: 10.1113/jphysiol.2003.042747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum H, Lambert E, Windebank E, Campbell DJ, Esler M. Effect of angiotensin II receptor blockade on autonomic nervous system function in patients with essential hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H1706–H1712. doi: 10.1152/ajpheart.00885.2005. [DOI] [PubMed] [Google Scholar]
- Lim PO, MacFadyen RJ, Clarkson PB, MacDonald TM. Impaired exercise tolerance in hypertensive patients. Ann Intern Med. 1996;124:41–55. doi: 10.7326/0003-4819-124-1_part_1-199601010-00008. [DOI] [PubMed] [Google Scholar]
- Lund-Johansen P. Twenty-year follow-up of hemodynamics in essential hypertension during rest and exercise. Hypertension. 1991;18:III54–61. doi: 10.1161/01.hyp.18.5_suppl.iii54. [DOI] [PubMed] [Google Scholar]
- Mancini DM, Bolinger L, Li H, Kendrick K, Chance B, Wilson JR. Validation of near-infrared spectroscopy in humans. J Appl Physiol. 1994;77:2740–2747. doi: 10.1152/jappl.1994.77.6.2740. [DOI] [PubMed] [Google Scholar]
- Nashar K, Nguyen JP, Jesri A, Morrow JD, Egan BM. Angiotensin receptor blockade improves arterial distensibility and reduces exercise-induced pressor responses in obese hypertensive patients with the metabolic syndrome. Am J Hypertens. 2004;17:477–482. doi: 10.1016/j.amjhyper.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Omvik P, Gerdts E, Myking OL, Lund-Johansen P. Long-term central hemodynamic effects at rest and during exercise of losartan in essential hypertension. Am Heart J. 2000;140:624–630. doi: 10.1067/mhj.2000.109919. [DOI] [PubMed] [Google Scholar]
- Parker BA, Smithmyer SL, Jarvis SS, Ridout SJ, Pawelczyk JA, Proctor DN. Evidence for reduced sympatholysis in leg resistance vasculature of healthy older women. Am J Physiol Heart Circ Physiol. 2007;292:H1148–H1156. doi: 10.1152/ajpheart.00729.2006. [DOI] [PubMed] [Google Scholar]
- Phillips MI. Functions of angiotensin in the central nervous system. Annu Rev Physiol. 1987;49:413–435. doi: 10.1146/annurev.ph.49.030187.002213. [DOI] [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- Rondon MU, Laterza MC, de Matos LD, Trombetta IC, Braga AM, Roveda F, Alves MJ, Krieger EM, Negrao CE. Abnormal muscle metaboreflex control of sympathetic activity in never-treated hypertensive subjects. Am J Hypertens. 2006;19:951–957. doi: 10.1016/j.amjhyper.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Rosenmeier JB, Dinenno FA, Fritzlar SJ, Joyner MJ. α1- and α2-adrenergic vasoconstriction is blunted in contracting human muscle. J Physiol. 2003;547:971–976. doi: 10.1113/jphysiol.2002.037937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiq MM, Menon DV, Victor RG. Oral direct renin inhibition: premise, promise, and potential limitations of a new antihypertensive drug. Am J Med. 2008;121:265–271. doi: 10.1016/j.amjmed.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon G. Pathogenesis of structural vascular changes in hypertension. J Hypertens. 2004;22:3–10. doi: 10.1097/00004872-200401000-00002. [DOI] [PubMed] [Google Scholar]
- Smith SA, Williams MA, Leal AK, Mitchell JH, Garry MG. Exercise pressor reflex function is altered in spontaneously hypertensive rats. J Physiol. 2006;577:1009–1020. doi: 10.1113/jphysiol.2006.121558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SH, Silke B, Ahuja RC, Okoli R. Influence of nicardipine on the blood pressure at rest and on the pressor responses to cold, isometric exertion, and dynamic exercise in hypertensive patients. J Cardiovasc Pharmacol. 1982;4:803–807. doi: 10.1097/00005344-198209000-00016. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Hansen J, Victor RG. Inhibition of α2-adrenergic vasoconstriction during contraction of glycolytic, not oxidative, rat hindlimb muscle. Am J Physiol Heart Circ Physiol. 1994;266:H920–H929. doi: 10.1152/ajpheart.1994.266.3.H920. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Zhang W, Victor RG. Impaired modulation of sympathetic vasoconstriction in contracting skeletal muscle of rats with chronic myocardial infarctions: role of oxidative stress. Circ Res. 2001;88:816–823. doi: 10.1161/hh0801.089341. [DOI] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Victor RG, Bertocci LA, Pryor SL, Nunnally RL. Sympathetic nerve discharge is coupled to muscle cell pH during exercise in humans. J Clin Invest. 1988;82:1301–1305. doi: 10.1172/JCI113730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray DW, Fadel PJ, Smith ML, Raven P, Sander M. Inhibition of α-adrenergic vasoconstriction in exercising human thigh muscles. J Physiol. 2004;555:545–563. doi: 10.1113/jphysiol.2003.054650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Swanson SA, Ye J, Li X, Shelton JM, Zhang W, Thomas GD. Reactive oxygen species impair sympathetic vasoregulation in skeletal muscle in angiotensin II-dependent hypertension. Hypertension. 2006;48:637–643. doi: 10.1161/01.HYP.0000240347.51386.ea. [DOI] [PubMed] [Google Scholar]