

Abstract
Multifocal multiphoton microscopy (MMM) enhances imaging speed by parallelization. It is not well understood why the imaging depth of MMM is significantly shorter than conventional single-focus multiphoton microscopy (SMM). In this report, we show that the need for spatially resolved detectors in MMM results in a system that is more sensitive to the scattering of emission photons with reduced imaging depth. For imaging depths down to twice the scattering mean free path length of emission photons (), the emission point spread function (PSFem) is found to consist of a narrow, diffraction limited distribution from ballistic emission photons and a broad, relatively low amplitude distribution from scattered photons. Since the scattered photon distribution is approximately 100 times wider than that of the unscattered photons at , image contrast and depth are degraded without compromising resolution. To overcome the imaging depth limitation of MMM, we present a new design that replaces CCD cameras with multi-anode photomultiplier tubes (MAPMTs) allowing more efficient collection of scattered emission photons. We demonstrate that MAPMT-based MMM has imaging depth comparable to SMM with equivalent sensitivity by imaging tissue phantoms, ex vivo human skin specimens based on endogenous fluorophores, and green fluorescent protein (GFP) expressing neurons in mouse brain slices.
1. Introduction
Multiphoton microscopy is a three dimensional (3D) imaging technique based on nonlinear excitation of fluorophores [1]. Nonlinear excitation results in fluorescence generation only at the focus of the microscope objective. Because of its inherent 3D resolution, minimal phototoxicity, and excellent imaging depth, multiphoton microscopy is commonly used for in vivo tissue studies. It has become an important tool for various biomedical applications such as studying neuronal plasticity [2-4], monitoring angiogenesis in solid tumors [5], and performing non-invasive optical biopsy [6, 7]. A practical limitation of multiphoton microscopy is its imaging speed which typically ranges from 0.1 to 2 frames per second. While this imaging speed is sufficient in some cases, two classes of problems demand higher speeds. First, high-speed multiphoton microscopy can be applied to study kinetic processes in biological systems. For example, high-speed imaging can map calcium wave propagation inside cells, the rolling dynamics of leukocytes along blood vessel walls located inside solid tumors [5], and the release of neurotransmitter at synaptic terminals [8]. Second, the imaging volume of conventional multiphoton microscopy is limited to about several hundred microns on a side using typical high numerical aperture objectives [1, 4, 6]. While this volume is sufficient for cellular imaging, many tissues have physiologically relevant structures that are significantly larger. For example, a neuron with its extensive dendritic tree structure can span a volume over 1 mm3. Furthermore, many organs have hierarchical structures spanning length scales from sub-microns to several millimeters. A recent study showed that cardiac structures of a whole mouse heart, approximately 1 cm3 in volume, can be imaged with micron level resolution providing morphological information of physiological structures over multiple scales [9]. Equally important, traditional 3D microscopes with frame rate on the second scale can realistically study only a few hundreds of cells. These microscopes cannot hope to provide comparable statistical accuracy and precision of quantitative assays such as flow cytometry. High-speed imaging can circumvent this difficulty by increasing the number of cells that can be efficiently sampled in tissue specimens. This approach opens the possibility of extending image cytometry into 3D. A study in transdermal drug transport demonstrated this application where high-speed microscopy provided statistically meaningful transport coefficients for hydrophilic and hydrophobic chemicals across the stratum corneum by imaging sufficiently large tissue sections [10]. Large volume imaging has also been applied to elucidate receptor dependent cancer metastasis processes [11].
Given the biomedical utilities of high-speed multiphoton microscopy, several methods have been implemented. These methods can be categorized into three classes. The first method increases imaging speed by using higher speed scanners such as polygonal mirror scanners [12], or resonant mirror scanners [13] instead of galvanometric mirror scanners used in conventional multiphoton microscopes. This method typically achieves the scanning speed of about 10 frames per second in tissues with a comparable imaging depth as conventional multiphoton microscopy. For these microscopes, an increase in frame rate results in a reduction of pixel dwell time which eventually decreases the image signal-to-noise ratio (SNR). This SNR decrease can be partially compensated by increasing excitation power. For the same SNR, an imaging speed increase by a factor of n requires the excitation power to increase by a factor of for two-photon excitation. However, the maximum excitation power cannot be increased arbitrary due to specimen photodamage as well as excitation saturation [1, 14, 15]. Excitation saturation results from the finite fluorescence lifetime of fluorophores. When a fluorophore is excited, it stays in the excitation state for a few nanoseconds before returning to the ground state. The residence time of the fluorophore in the excitation state is called the fluorescence lifetime. Since the pulse width of multiphoton excitation light sources is orders of magnitude shorter than the fluorescence lifetime of typical fluorophores, the fluorophores can be excited at most once per excitation pulse. Excitation saturation results from trapping fluorophores in the excitation state and depleting them in the ground state at high input power. With sufficient depletion in the ground state, the excitation efficiency is no longer proportional to the nth power of the laser intensity for an n-photon excitation process. Since saturation is more severe at the center of the excitation point spread function (PSFex), where the intensity is the highest, this results in resolution degradation. Given saturation limitation, the maximum photon emission rate from the focal volume is only a function of fluorophore concentration and cannot be adjusted in most biological experiments. It can be shown that for a specimen containing 10 μM fluorophores requires at least 450 ns pixel residence time.
The second method to improve speed is to scan selected regions of interest in the sample rather than the whole volume. This method uses acousto-optical deflectors (AODs), for fast point-to-point scanning. Since AODs cause significant dispersion of ultra-short pulses and introduce spherical aberration, high-speed multiphoton microscopes utilizing AODs have been implemented with fairly complex laser pulse width and shape compensation subsystems [16-18]. This type of microscopes has been applied to studies such as monitoring neuronal communication and signal propagation. Other methods to improve imaging speed by optimizing scanning trajectory have been introduced [19].
The third method increases imaging speed by parallelizing the multiphoton imaging process and has been called multifocal multiphoton microscopy (MMM) [20-24]. In MMM, a specimen is scanned with multiple excitation foci instead of a single excitation focus. The emission light generated from the foci is collected simultaneously by a spatially-resolved detector such as a charge coupled device (CCD) camera. The detector integrates the emission photons from the specimen during 2D scanning of multiple excitation foci in the sample plane. The imaging speed enhancement of MMM is proportional to the number of excitation foci. MMM achieves a high frame rate without the need to increase the excitation power at each excitation focus, thus circumventing the excitation power limitation as well as reducing photo-toxicity. MMM can always achieve higher imaging speed than single-focus approach for a given SNR. One practical speed limitation of MMM is that more input power is needed to generate multiple excitation foci. For imaging relatively thin specimens, approximately 10 mW is needed for each focus, thus requiring 640 mW for 64 independent excitation foci. For imaging deeper into tissues, higher total laser power is needed to compensate signal loss in tissues. Improvements in laser technology have recently led to Ti-Sapphire lasers with the mode-locked output power up to almost 5W making MMM deep tissue imaging with more foci possible. Therefore, MMM is the most promising high-speed multiphoton imaging approach. Unfortunately, for thick tissue applications, MMM has also an important disadvantage. Through many private communications with researchers, it has been noted that the imaging depth of MMM in tissues is significantly less than that of conventional single-focus multiphoton microscopy (SMM). This report aims to understand and quantify the tissue optics origin of this limitation both theoretically and experimentally. We further propose a novel instrumental solution based on multianode photomultiplier tubes to improve imaging depth in tissues.
2. Effects of emission photon scattering on MMM
2.1 Sensitivity comparison of SMM and MMM to emission photon scattering
Conventional MMM uses a spatially resolved detector, such as a charge coupled device (CCD) camera, for simultaneous signal collection from multiple excitation foci. Spatial information of emission light in the MMM image is encoded by its distribution over the detector pixels. If emission photons deviate from the original paths due to scattering, they are not collected at correct pixels of the detector causing image degradation. Therefore, conventional MMM is sensitive to emission photon scattering. Conventional single-focus multiphoton microscopy (SMM) uses a single pixel detector with a large active area such as a photomultiplier tube (PMT). The detector collects emission photons without spatial information and the spatial information of emission photons is obtained by temporally correlating the amplitude of the optical signal with the trajectory of the scanning excitation focus in the sample plane. The difference becomes clear by examining the formulation of total point spread functions (PSFtot’s) of two systems: SMM [25] and MMM [26]
PSFex and PSFem are intensity point spread functions of excitation and emission light respectively. (xs ,ys ,zs) is the coordinate in the sample volume (plane) and (xi, yi, zi) is the coordinate in the image volume (plane). For convenience, it is assumed that the image plane is telecentric to the sample plane with 1x magnification. For SMM, PSFem is spatially integrated over the active area of the detector, and the microscope’s spatial resolution is determined only by PSFex. SMM is not sensitive to PSFem variation due to photon scattering and it has excellent imaging depth in tissues. For MMM, the emission signals from the excitation foci are collected simultaneously by a spatially resolved detector. The detector integrates the emission light during the 2D scanning of excitation foci on the sample plane. Therefore, PSFex has to be integrated over the sample plane for MMM. PSFem determines the spatial resolution of MMM in the transverse direction and the product of PSFem and PSFex determines the axial resolution. As a consequence, MMM is sensitive to PSFem variation due to photon scattering.
The effects of photon scattering on image resolution and contrast in SMM have been investigated extensively by both Monte Carlo simulations [27-29] and experiments [30-33]. These studies examined the scattering effects on PSFex [27, 28, 30-32] and on the collection efficiency of emission light [29, 31, 33]. These studies focused on idealized specimens with homogenous fluorophore distribution and ignored pulse dispersion of excitation light with depth. Results of these studies can be summarized as following. (1) Unscattered ballistic photons are mostly responsible for multiphoton excitation generation within an imaging depth less than a few times the mean free path length () of excitation light. The contribution of scattered excitation photons is suppressed by the nonlinear relationship of excitation probability to excitation photon flux. The amount of multiphoton fluorescence generation (F(z)) shows exponential decay with imaging depth (z) as , where z is normalized by and c is the decay coefficient. The decay coefficient is at least 2 for two-photon excitation. In practice, the decay coefficient is slightly higher than 2, because the average path length of excitation photons traveling to the focus is longer than the imaging depth for high NA objectives [31]. Within the depth range of a few times , (2) image resolution is not degraded, because the full-width-at-half-maximum intensity (FWHM) of PSFex is not affected by photon scattering [28, 31, 32], and (3) image contrast is degraded mainly due to the decrease of excitation power as photon scattering effect. (4) Emission light becomes diffusive due to multiple scattering in the specimen. The collection efficiency of emission light can be enhanced by maximizing the solid angles of all the optical components in the detection path [29, 33].
While PSFem variations are not crucial in SMM configuration, its variations due to photon scattering are important in MMM configuration. It should be noted that PSFem is more sensitive to scattering than PSFex in nonlinear microscopy for two reasons. First, the contribution of low amplitude scattered excitation photons to PSFex is suppressed in the nonlinear excitation process. On the other hand, PSFem responds linearly and is more sensitive to scattered emission photon distribution. Second, emission light in nonlinear microscopy has shorter wavelengths than excitation light. Since the scattering cross section increases at shorter wavelengths, PSFem is more affected by scattering than PSFex. Effect of emission photon scattering may be minimized by using the fluorophores of longer emission wavelengths and the detectors sensitive to the wavelengths.
2.2 Effect of emission photon scattering on image resolution
The effect of emission photon scattering on image resolution was analyzed by measuring variations in the lateral and axial FWHMs of PSFem with imaging depth in tissue phantoms. PSFem FWHM can be evaluated by measuring the FWHM of , because PSFex FWHM is known to be invariant at the imaging depth of less than [31, 32]. Therefore, the dependence of the profile on scattering is determined solely by the response of PSFem. The FWHM of was measured by imaging 0.22 μm diameter fluorescent microspheres (yellow-green 505/515, F8811, Molecular Probes, Eugene, OR) as effective point sources with a CCD camera (Pentamax, Princeton Instrument, Trenton, NJ). The experimental apparatus is depicted in Fig. 1. A single excitation focus scans the specimen and emission light is collected by the CCD camera. We chose to perform single focus scanning to simplify data interpretation by removing channel crosstalk effects in the MMM configuration. Excitation light is sourced from a pulsed Ti:sapphire laser (Mira 900, Coherent, Santa Clara, CA) pumped by a continuous wave (CW) Nd:YVO4 laser (Verdi, Coherent, Santa Clara, CA). The laser has the pulse width of a few hundred femtoseconds and the pulse repetition rate of 78 MHz. Its wavelength is set at 800 nm. The excitation beam is reflected by galvanometric x-y mirror scanners (6350, Cambridge Technology, Watertown, MA) and is directed into a microscope (Axiovert 100TV, Zeiss, Thorwood, NY). A lens pair (a scan lens and a tube lens) expands the excitation beam to over-fill the back aperture of an objective. Proper over-filling of the back aperture is required for the objective to achieve its maximal numerical aperture. The expanded excitation beam is reflected by a dichroic mirror (675DCSX, Chroma Technology, Brattleboro, VT) toward the objective. The objective focuses excitation light into the specimen to produce fluorescence at the focus. The focus of the excitation light raster scans across the sample plane via the x-y scanners. Scanning in the depth direction is performed by a piezo-objective translator (P-721.00, Physik Instrumente, Auburn, MA). Emission light is collected by the same objective and is transmitted through the dichroic mirror. An additional barrier filter (ESP650, Chroma Technology, Brattleboro, VT) is used to further attenuate scattered excitation light. Emission light is collected by the CCD camera in the image plane. An image is formed by integrating emission light from the specimen during 2D scanning of the excitation focus.
Fig. 1.

An experimental setup for quantifying photon scattering effects on PSFem. Excitation beam path is very similar to conventional multiphoton microscopy. For measuring the PSFem FWHM variation, a CCD camera is used as a detector. For measuring the PSFem spatial distribution, a pinhole aperture of various sizes is positioned in the image plane and a PMT behind the aperture collects the transmitted emission light.
The specimens were 3D blocks of sparsely distributed fluorescent latex microspheres immobilized in a medium containing 2 % agarose gel (UltraPure Low Melting Point Argarose, Invitrogen, Carlsbad, CA). Intralipid emulsion (Liposyn III, Abbott Laboratories, North Chicago, IL) was added as scatterer at various concentrations from 0 %, 0.5 %, 1 %, to 2 %. The specimen of 2 % intralipid concentration has scattering properties similar to typical biological tissues [31, 32]. We measured the variation in FWHM as a function of imaging depth from the surface down to 125 μm deep at each scatterer concentration. The objective used in the experiment was a 40× water immersion lens with 1.2 N.A. (C-Apochromat 40× W Korr, Zeiss, Thornwood, NY). The images had a lateral field of view of 25 μm × 25 μm with 245 × 245 CCD pixels resulting in approximately 0.1 μm pixel resolution. An image cube was acquired with 0.25 μm depth increment throughout the total imaging depth, resulting in 500 frames total. The acquisition time was approximately 6.5 seconds per frame. For data analysis, the image cube was divided into 5 sections each of which was 25 μm thick. Average values of FWHMs were obtained from more than 10 microsphere images in each image section. Since the optical properties of intralipid emulsion vary (depending on stocks), the scattering properties of these samples were independently measured using the multi-distance frequency-domain method [34]. The reduced scattering coefficient, was obtained by measuring the amplitude and the phase of the scattered light as a function of the source-detector separation distance. The mean free path length, ls was calculated by using the anisotropy (g) value reported in the literature [31], . Optical properties of the 2 % intralipid emulsion used in the experiment were similar to literature-reported parameters [31], (, ). The measured FWHMs as a function of imaging depth are presented in Fig. 2. The imaging depths of different scatterer concentration samples were normalized by . The theoretical values for FWHMs are 0.26 μm and 0.56 μm in the transverse and axial directions respectively in the absence of scattering and taking into account the finite size of the microspheres. In the sample of 0 % scatterer concentration, the measured values were within 30 % of the theoretical values. These variations from the theoretical values may be due to Gaussian intensity distribution of light in the objective back focal plane. The measurement showed no significant change of FWHM in either transverse or axial direction in this depth range. Since image resolution is determined by FWHM, this result leaded us to conclude that emission photon scattering has no effect on image resolution of MMM at the imaging depth from surface down to .
Fig. 2.

Variations in FWHM of as a function of imaging depth due to photon scattering. FWHMs in (a) transverse direction and (b) axial direction were measured from surface down to the imaging depth of at emission wavelength (515 nm). There was no significant change of FWHM in either direction.
2.3 Effect of emission photon scattering on image contrast
In order to study the effect of emission photon scattering on the signal decay, PSFem peak intensity was measured as a function of imaging depth. This experiment was carried out in fluorescent dye solutions mixed with intralipid emulsion. The fluorescent dye solution was 233 μM fluorescein (F1300, Molecular Probes, Eugene, OR) dissolved in water and its pH was adjusted to 8.0 with sodium hydroxide. Intralipid emulsion was added as scatterer up to 2 % in concentration. The measurement was performed using the instrument described in Section 2.2. In this measurement, the excitation focus was kept stationary in the center of the field in the specimen and emission light from the focus was collected by the CCD camera. Images were acquired by moving the excitation focus from surface down to . In contrast to the imaging of the sub-diffraction sized microsphere specimens, the acquired image (Ispot) of the fluorescence signal was a convolution of the PSFex and PSFem, Ispot = PSFex ⊗ PSFem. The scattering effect on the PSFem peak intensity was easily extracted from Ispot, because its effect on PSFex was known [31, 32]. The decay of PSFex peak intensity (F(z)) was expressed as with its decay coefficient (c) of 2.5 for the 40× objective used (1.2 NA). The emission photons collected only at the ‘correct’ pixels of the CCD camera were considered as signal, whereas the ones collected at the ‘incorrect’ pixels due to scattering were counted as background. With 40× magnification, each pixel of the CCD camera covered the area of 0.56 μm × 0.56 μm in the sample plane so that the signal within Ispot FWHM should be collected at a single pixel in the absence of emission photon scattering. The PSFem peak intensity was obtained by dividing the peak intensity of Ispot with that of PSFex. For comparison, the spatially integrated PSFem intensity over the image plane was also measured by using a photomultiplier tube (PMT) in the image plane instead of the CCD camera. The use of the single pixel detector with a large active area such as the PMT was equivalent to the spatial integration of PSFem. The PMT (R7400p, Hamamatsu, Bridgewater, NJ), used in this study, had a circular 8 mm diameter active area. With 40× magnification, the active area of the PMT corresponded to a circular area of 200 μm diameter in the sample plane. The decays of PSFem peak and integrated intensities with imaging depth are presented in Fig. 3(a). The decay of PSFem peak intensity was faster than that of the integrated intensity. The intensity decays (I(z)) were fitted to an exponential decay formula, where c was the decay coefficient with respect to the normalized imaging depth by . The decay coefficients (c’s) of the peak and integrated intensity were 0.94 and 0.18, respectively. Since the peak intensity decay had its coefficient of approximately unity, it could be concluded that only unscattered emission photons contributed to the PSFem peak intensity. Further, the decay coefficient of 0.18, close to zero, in the integrated PSFem indicated that a majority of the scattered emission photons were effectively collected by the PMT. The integrated intensity of the PSFem initially showed a slight increase rather than a decrease with imaging depth. This initial increase could be explained also by the scattering of emission photons. Since fluorescence is isotropic, a fraction of the emission photons is going forward into the specimen in the absence of scattering. In the presence of scattering, a portion of these forward emission photons may be back-scattered and collected by the detector [29].
Fig. 3.

Effects of emission photon scattering on PSFem peak intensity and shape. (a) Decay profiles of PSFem peak intensity and integrated intensity (over a 200 μm diameter circular area) as a function of normalized imaging depth down to . The lines denote best exponent fit of the data points. (b) Variations in the PSFem lateral profile (PSFem(r, φ) in cylindrical coordinates) with the imaging depth. The PSFem profile was measured up to 100 μm in radius from peak location. Data points are connected by lines for eye guidance.
Since PSFem peak intensity decreased rapidly with imaging depth, we could conclude that there must be minimal contribution to the PSFem peak intensity by the scattered emission photons. Further since the PSFem FWHM showed no significant change with imaging depth, we concluded that the scattered emission photons must have a broad spatial distribution with low amplitude relative to the peak intensity of PSFem, which is formed by unscattered ballistic emission photons. This distribution has not been previously measured experimentally. In principle, the image of the multiphoton induced fluorescence focus in the fluorescent solution sample (Ispot) contained this information. However, the relatively low SNR of the CCD pixel signal precluded direct measurement. A higher sensitivity measurement was required to quantify the low amplitude distribution of these scattered emission photons. For this measurement, the same apparatus, as described in Section 2.2, was used, but a high SNR PMT (R7400p) with a pinhole aperture replaced the CCD camera. The pinhole aperture was placed in the image plane and the PMT collected emission light transmitted through the aperture. A series of pinhole apertures with radius ranging from 0.7 μm to 100 μm in the sample plane (PxxS, SM1D12R, Thorlabs, Newton, NJ) were used. In this configuration, we measured the integrated intensity deposited within a specific radius (r0) from the center of Ispot as I(r0, z). The mathematical relationship between Ispot and I(r0, z) was . Therefore, Ispot could be obtained by step-wise differentiation of I(r0, z) in the radial direction. Since the shape of PSFex was known to be invariant, the shape of PSFem could be obtained by deconvoluting PSFex from Ispot. The normalized PSFem profiles at the imaging depths of 0×, 1×, and are presented in Fig. 3(b). Within the resolution of this measurement, the PSFem profiles were similar for all three depths within 2 μm radius from the excitation focus corresponding to approximately 10 times the FWHM of PSFem. This result confirmed that PSFem close to the peak was mainly contributed by the unscattered emission photons. However, the PSFem intensity was observed to increase with imaging depth at distances exceeding 2 μm in radius from the excitation focus. This spatially broadened intensity distribution was due to scattered emission photons. FWHM of the scattered emission photon distribution was approximately 25 μm at the depth of below surface which was 100 times larger than the FWHM of PSFem and the width of the distribution increased at the rate of approximately 12.5 μm per . The wide distribution of scattered emission photons implied that most of the scattered emission photons became background noise by being collected at the ‘incorrect’ detector pixels for MMM imaging. One may note that at , the amplitude of the scattered emission photon distribution was very low corresponding to only about one thousandth of the PSFem peak amplitude.
2.4 Evaluation of emission photon scattering effect on MMM and SMM image contrast by numerical simulation
Is the low amplitude scattered emission photon distribution responsible for the reduced imaging depth of MMM? We assert that in MMM, these scattered emission photons degrade the image contrast in two important ways: (1), The signal decreases very fast in highly scattering specimens, because the scattered emission photons are not collected at the ‘correct’ pixels. (2) The background noise increases as the emission photons are scattered broadly throughout the image.
We verified this assertion regarding the effect of emission photon scattering on contrast reduction by a numerical simulation (Fig. 4). The simulated specimen was a 5 × 5 square array of 10 μm diameter fluorescent microspheres separated by 40 μm. An MMM based on a CCD camera was assumed to be used for imaging. The effect of scattering was modeled by convoluting the known object distribution with . Figures 4(a) and 4(b) show the simulated images at the surface and at a depth of respectively. Figure 4(b) shows the relatively high background noise which is contributed by widely distributed scattered emission photons. The center region of the image has the highest background noise level, because the simulation image has a small size and the scattered emission photons from the surrounding microspheres were accumulated. Furthermore, the background noise distribution had a square-tiled appearance, because of the symmetric distribution of the microspheres used in this simulation. Figure 4(c) shows the SNR decay in the center region of the image with imaging depth. For comparison, the SNR decay of SMM is also included in this plot. The SNR decay of MMM is faster than that of SMM, because MMM experiences both faster signal reduction and background noise increase which is contributed by the low amplitude scattered emission photon distribution.
Fig. 4.

A simulation of photon scattering effect on image contrast in MMM tissue imaging. An original image comprising 5 × 5 microspheres of 10 μm diameter equally spaced with 40 μm separation distance and the experimentally measured PSFtot was used in the simulation. (a) MMM image at the sample surface, (b) MMM image at imaging depth of , where increased background noise is observed in the center of the image. (c) The decay of SNR of the MMM image at its center location as a function of the imaging depth.
The high sensitivity of MMM to emission photon scattering can be explained partially by the effective detection area which is defined as the area in the sample plane covered by each detector pixel. The larger the effective detection area of a detector, the better the detector can collect scattered emission photons. Since the microscope is a telecentric system, the effective detection area in the sample plane is linearly related with the detector size in the image plane. With a magnification (M), and a detector with the linear dimension (LD), the linear dimension of effective detection area (LE) is LE=LD/M. For the PMT with a circular active area of 8 mm diameter and the 40× magnification microscope system, LE is approximately 200 μm. This dimension is significantly larger than the FWHM of the scattered emission photon distribution at the imaging depth of which is approximately 25 μm. On the other hand, for the CCD camera which is used in conventional MMM, the effective area of each CCD pixel is very small. For a CCD camera with 20 μm × 20 μm pixel size and at 40× magnification, LE is only 0.5 μm. Therefore, conventional MMM is very sensitive to emission photon scattering, because the effective detection area of CCD pixels is much small compared to the FWHM of the scattered emission photon distribution.
3. A new MMM based on a multianode multiplier tube (MAPMT)
3.1 Design of MAPMT-based MMM
In order to improve the imaging depth of MMM in turbid tissues, scattered emission photons need to be collected more efficiently and into the correct detector pixels. A detector with a small number of pixels is advantageous, because each pixel has a large effective detection area for a given field of view. The large effective detection area ensures that more of the scattered emission photons are collected in the correct pixels. Since each excitation focus can be mapped to the corresponding detector pixel in a de-scanned configuration, it is optimal to use a detector which has equal number of pixels as the excitation foci. The detector collects signals synchronously with the scanning of excitation foci. Given the laser power limitation, about 10-100 excitation foci are typically feasible for MMM imaging.
A MAPMT is an excellent choice for this purpose. The MAPMT is composed of a cathode, a dynode chain and a segmented anode. Its structure is optimized to ensure the spatial distribution of photons incident upon the cathode to be reproduced faithfully as the signal at the anode. Similar to the conventional PMT, the MAPMT has a good quantum efficiency (over 20% in the blue/green spectral range), negligible read-out noise, and minimal dark noise with cooling. The anode of the MAPMT used in this instrument is a segmented, 8 × 8 pixel array (H7546, Hamamatsu, Bridgewater, NJ).
A schematic of the new MMM design is depicted in Fig. 5. The light source used is a Ti-Sapphire (Ti-Sa) laser (Tsunami, Spectra-Physics, Mountain View, CA) pumped by a continuous wave Nd:YVO4 laser (Millenia, Spectra-Physics, Mountain View, CA). It outputs approximately 2 W of mode locked pulsed light at 800 nm wavelength. Excitation beam from the laser is expanded and illuminates a square micro-lens array with 12×12 micro-lenses. The micro-lenses are of 1 mm × 1 mm in size with 17 mm focal length (1000-17-S-A, Adaptive Optics, Cambridge, MA). The degree of beam expansion is set such that 8 × 8 beam-lets are produced after the micro-lens array. The beam-lets are collimated after a lens (L1) and reflected onto x-y galvanometric mirror scanners (6220, Cambridge Technology, Cambridge MA) which is positioned in the focal plane of L1. In this configuration, the beam-lets overlap each other on the scanner mirror. The beam-lets, reflected by the scanner, incident upon the epi-illumination light path of a microscope at different entrance angles (BX51, Olympus, Melville, NY). A lens combination of L2 and L3 expands the beam-lets to slightly under-fill the back aperture of an objective (approximately 0.75 of the aperture diameter) balancing the need to maximize excitation power at each foci and to utilize the high numerical aperture of the objective. The expanded beam-lets are reflected by either a dichroic mirror (for CCD setup) or a mirror (for MAPMT setup) toward the objective. The objective used is a 20 × water immersion lens with 0.95 NA (XLUMPLFL20XW, Olympus, Melville, NY). The effective NA due to under-filling is approximately 0.71. The objective generates an array of 8 × 8 excitation foci in the sample plane of the specimen. The image is formed by raster scanning the excitation foci across the specimen using the scanner mirrors. The excitation foci are separated each other by 45 μm and each focus scans the area of 45 μm × 45 μm in the sample plane. The frame size covered by the 8 × 8 array of foci is 360 μm × 360 μm.
Fig. 5.

A schematic of the multifocal multiphoton microscope based on a MAPMT. Excitation beams are depicted in red/orange colors and emission beams are in green color. In this figure, only two beam-lets are ray-traced. The excitation beam is splitted into 8 × 8 beam-lets via a microlens array. Multiple excitation foci (8 × 8) scan the specimen. The emission beam-lets are collected either by a CCD camera or a MAPMT which has 8 × 8 pixels. L1, L2, and L3 are lenses. The transmission of light through the microlens array and high NA objective causes appreciable pulse dispersion which can be corrected by pre-chirping using a pair of prisms (not shown).
Emission light is collected by the same objective forming an array of emission beam-lets. For signal collection using the CCD camera, the emission beam-lets are transmitted through a short-pass dichroic mirror (650dcxxr, Chroma Technology, Brattleboro, VT) and focused on the CCD camera via a lens. The CCD camera integrates the emission signal during 2D scanning of the foci in the sample plane. For the MAPMT, the emission beam-lets are reflected on the mirror toward the scanner mirror retracing the excitation paths and are descanned via the scanner mirrors. The de-scanned emission beam-lets become stationary irrespective of the scanner motion. The emission beam-lets are reflected by a long-pass dichroic mirror (Chroma Technology, Brattleboro, VT) toward the MAPMT and are focused onto the MAPMT by a lens (L4). A short-pass filter (E700SP, Chroma Technology, Brattleboro, VT) blocks any strayed excitation light. The 8 × 8 emission beam-lets are focused at the centers of the corresponding MAPMT pixels. Since the emission beam-lets are detected after de-scanning, a loss of emission light (approximately 30 %) is expected compared to non-de-scanning systems. Both the MAPMT and the CCD detection light paths are implemented on the same system so that accurate comparison of these two detection methods is possible.
Signals from the MAPMT pixels are collected synchronously with the scanning of excitation foci in the specimen. They are collected in photon counting mode by a multichannel photon counter card (mCPhC) system which has 64 channels in total for the simultaneous signal collection from the 8 × 8 MAPMT. Detailed electronic design of the mCPhC has been previously reported [35]. Each mCPhC has 18 channels of the photon-counting circuits and a 32-bit parallel interface to the computer for high-speed data transfer. The mCPhC is designed to be expandable so that 64 channels can be readily implemented by using 4 mCPhC cards in parallel. The signal from each focus is assigned to the appropriate location in the image based on the known raster scan pattern of the foci array. The frame rate for 320 × 320 pixel images is approximately 19 frames per second with the pixel dwell time of 33 μs. The pixel dwell time can be varied to optimize image SNR depending on the specimen.
For performance comparison, SMM was implemented to the MAPMT-based MMM setup. The configuration can be easily converted to the SMM which is based on single-focus scanning and signal collection with a conventional PMT. In this case, the microlens – L1 combination is removed from the excitation beam path and the excitation beam is not expanded and is directly projected on the galvanometric scanner. Specimens are scanned by a single excitation focus via the scanner mirror. The emission light collected by the objective lens transmits a dichroic mirror (short-pass) and is collected by a conventional PMT (R7400p). The emission light forms an image by correlating the PMT signals with the known raster pattern of the excitation focus.
In the current design, each MAPMT pixels have the effective detection area of 45 μm × 45 μm. This detection area is comparable with the FWHM of the scattered emission photon distribution which is approximately 25 μm at an imaging depth of . Therefore each pixel of the MAPMT fairly effectively collects most of the scattered emission photons originating from its corresponding excitation focus. For the CCD camera, each CCD pixel has the effective detection area of 1.1 μm × 1.1 μm in the sample plane with 20× magnification. For the SMM setup, the PMT, which has 8 mm diameter active area, has the effective detection area of 400 μm diameter providing more efficient collection of scattered emission photons.
3.2 Quantitative comparison of SMM, conventional MMM, and MAPMT-based MMM in a tissue phantom
We quantitatively compared MAPMT-based MMM and the CCD-based system for the imaging of multiple scattering specimens. Using both type of detectors in the MMM configuration, we measured the signal decay rate with imaging depth which accounts for the scattering effect of both excitation photons and emission photons. These systems were further compared with SMM. The signal decay was measured by imaging 4 μm diameter fluorescent latex microspheres (F8858, Molecular Probes, Eugene, OR) immobilized in 3D by 2 % agarose gel. Large-size microspheres were chosen for their higher intensity allowing pixel crosstalk to be quantified with better precision. Intralipid emulsion was added as scatterer in various concentrations from 0.5 to 2 %. Intralipid emulsion of 2 % volume concentration has scattering mean free path lengths (ls) of 85 μm and 167 μm at the emission wavelength (605 nm), and the excitation wavelength (800 nm) respectively. The peak intensity of each sphere image was recorded as a function of imaging depth. Signal decays in the three systems were measured down to the imaging depth of 190 μm which is equivalent to and are shown in Fig. 6. The signal decay (I(z)) was fitted to an exponential function, where c is the decay coefficient with respect to the normalized imaging depth by . The decay coefficient (c) was measured as 1.22, 1.87, 2.30 for SMM, MAPMT-based MMM, and CCD-based MMM respectively. The signal decay is the combined effect of both excitation and emission photon scattering. Since the effect of the excitation photon scattering is the same for all the systems, the difference in decay coefficients is due to emission photon scattering. The decay rate in SMM was the lowest, as expected. The decay coefficient, c in MAPMT-based MMM (1.87) was lower than for the CCD-based MMM (2.30), but still higher than that of SMM (1.22). It is consistent with the fact that the spatial distribution of scattered emission photons has 25 μm FWHM and the MAPMT pixels, with an area of 45 μm × 45 μm, cannot contain all the scattered photons and thus a portion of the scattered emission photons are collected in the neighboring pixels. Also, a MAPMT pixel has electronic crosstalk with its neighbors resulting in incorrect registering of some photons.
Fig. 6.
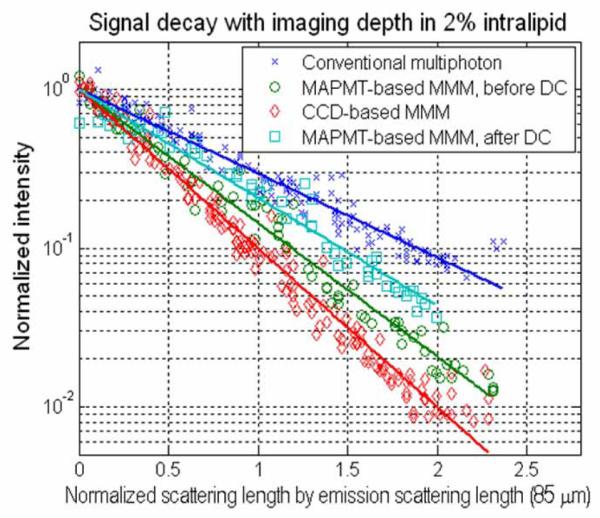
Quantifying signal decay with the increase of imaging depth. The intensity decay coefficient of a MAPMT-based MMM stands between that of a CCD-based MMM and that of a conventional SMM. A simple deconvolution algorithm (DC) is applied to MAPMT-based MMM. The decay coefficient of DC stands close to the that of SMM.
3.3 Effect of crosstalk and its minimization by image deconvolution
Since the distribution of scattered emission photons is broad, some of these photons invariably scattered away from the correct MAPMT pixels and into their neighboring pixels. The effect of crosstalk due to emission photon scattering is shown in Fig 7(a). This is an image of microspheres (4 μm diameter) at 150 μm deep from the surface in 2 % intralipid emulsion concentration (). Microspheres appear as repeated patterns in the image, similar to ghost images in a defective television. The origin of this ghost image requires further explanation. The MMM image is generated by combining 8 × 8 sub-images from signals collected by the 8 × 8 pixels of the MAPMT. Each sub-image is generated by temporally correlating the signal detected at a specific MAPMT pixel with the known raster scan position of the corresponding excitation focus. The emission photons scattered into the adjacent pixels are similarly correlated resulting in the formation of these ghost images. It is important to note that there is no ghost image in CCD-based MMM, because the image is formed purely based on the spatial registration of emission photons on the 2D imager and no information of the raster scanning pattern is used in image formation. We will shortly show that the spatial information contained in these ghost images allows reassignment of these photons back to their correct pixels using a deconvolution approach that is impossible with CCD-based MMM where the scattered photons just contribute to an uniform background.
Fig. 7.

The effect of emission photon scattering on images acquired with MAPMT-based MMM. (a) An image of microspheres at deep from the surface in the tissue phantom sample. Ghost images appeared due to the scattered emission photons collected in neighboring pixels of the MAPMT. (b) Image after the deconvolution process. (c) Variation of PSFem due to emission photon scattering. The increase of intensity in the tail region was due to emission photon scattering. (d) Crosstalk in the MAPMT pixels. It was calculated by integrating the PSFem over the effective detection area of individual MAPMT pixels.
The spatial broadening of the PSFem (Fig. 7(c)) shows an increased number of emission photons scattered out of their assigned pixels with scattering length. The level of this crosstalk can be determined by the spatial integration of the PSFem over the detection areas of individual MAPMT pixels. Fig. 7(d) shows result of this spatial integration. Horizontal axis is the MAPMT pixel location and vertical axis is normalized intensity with respect to the intensity at the correct pixel (pixel 0). It is noted that the relative intensity in the neighboring pixels (pixels 1 and 2) increases with the scattering length and can be on the order of 10 to 20 %. The signals from the 8 × 8 image pixels are collected simultaneously and these signals can be expressed as an acquired signal vector ({Sacq} , 64 × 1). {Sacq} is formulated as the product of a crosstalk matrix ([C], 64 × 64) and the vector of the original distribution of the source signals in the sample plane ({Ss}, 64 × 1), {Sacq} = [C] ×{Ss}. The crosstalk matrix ([C]) is constructed based on PSFem which is a function of imaging depth.
Although a significant portion of scattered emission photons are still distributed outside the correct pixels of the MAPMT-based MMM, these photons can be effectively restored to the correct MAPMT pixels by applying a post-acquisition image processing. The intensity ratios of those repeated images in Fig. 7(d) provide critical information for deconvolution. The convolution matrix [C] can be experimentally quantified on sample to sample basis by measuring PSFem and integrating it over the detection area of individual MAPMT pixels. Alternative, it can be measured by illuminating the sample with a single excitation focus and detecting the emission signal distributed at different pixels of the MAPMT. Once [C] is estimated, the deconvolution process is relatively simple and is quite immune to noise, because [C] is constructed by the spatial integration of PSFem. In the real measurements, the information of PSFem variations is not always available, because the level of emission photon scattering varies depending on the specimen. The signal of each image pixel is the combined effect from 8 × 8 excitation foci (one real signal and the rest of crosstalk signals). Sometimes, it can be difficult to estimate [C] from the acquired image. However, in a simple case where the acquired image has a sparse structure such as the image of a few microspheres in Fig. 7(a), it is possible to find one set of 8 × 8 image pixels in which only one real signal dominates. The estimated crosstalk matrix ([C]est) could be constructed by measuring the intensity ratios among those pixels in this case. The signal vector of pixels in the sample plane ({Ss}est) was calculated by the product of the inverse transform of [C]est and the acquired pixel vector ({Sacq}), .
The restored image is presented in Fig. 7(b). We measured the signal decay of the restored image with the imaging depth and found that the decay coefficient (c) was significantly reduced to 1.58 with the deconvolution algorithm applied (Fig. 6), due to correct reassignment of the scattered emission photons. The ghost images are almost completely removed in the restored image. The restoration algorithm can be further refined with more sophisticated algorithms such as using maximum likelihood estimation to minimize image structural overlap between neighboring pixels. This process may also better handle the effect of tissue heterogeneity on the crosstalk matrix [C]. Nonetheless, the simple deconvolution approach has been shown to effectively improve the performance of MAPMT-based MMM and allows this system to perform very close to SMM in tissue imaging in terms of imaging depth.
3.4 MAPMT-based MMM imaging of ex vivo human skin based on autofluorescence
The sensitivity of the MAPMT-based MMM is demonstrated by imaging an ex vivo human skin in 3D. Imaging skin tissues based on autofluorescence is a challenging task due to the low quantum yields and extinction coefficients of endogenous fluorophores compared to typical exogenous fluorophores. Furthermore, the layered skin structure with different refractive indices induces large spherical aberration. Formalin fixed cadaver skin specimens were obtained from the Penn State Milton S. Hershey Medical Center (Hershey, PA). For imaging, they were mounted and coverslipped on microscope slides using adhesive silicone isolators (JTR20-A2-1.0, Grace Bio-Labs, Bend, OR). Typical multiphoton skin imaging in a SMM system with negligible photodamage was performed with a typical pixel integration time of 67 μs and 15 mW excitation laser power [6, 36, 37]. In the MMM system, we used 7 mW on average per excitation focus at the specimen. The excitation wavelength was set at 800 nm and the objective used was the 20× water-immersion lens with 0.95 NA. In the MMM configuration we imaged at a frame rate of 2.5 fps for 320 × 320 pixel images. This imaging speed was 10 times faster than the one taken with SMM for equivalent SNR. We imaged the epidermis down to the basal cell layer using this MAPMT-based MMM and representative images from the stratum corneum, stratum granular, and the basal layer are presented in Fig. 8. Clear visualization of cellular structures in the skin specimen demonstrates that the sensitivity of MAPMT-based MMM is equivalent to that of the SMM but with a significantly enhanced imaging speed. The signal level of the image is not uniform: the intensity is higher in the center of the image than in the corners. This is caused by the Gaussian spatial power distribution of the expanded laser beam illuminating the microlens array. This drawback can be easily resolved by using other beam-let formation approaches in the future [22, 38].
Fig. 8.

Human skin images acquired with MAPMT-based MMM. Epidermal layer of the skin is imaged. From left to right, imaging depth goes deeper from surface. Stratum corneum layer (a), stratum granular (b), and basal layer (c) are shown. Cells are visible based on autofluorescence. Image size: 360 μm × 360 μm, input power: 7 mW per focus, objective: 20×, imaging speed: 2.5 frames/s with 320 × 320 pixels.
3.5 Comparing CCD-based MMM and MAPMT-based MMM in neuronal imaging
We further compared the performance of the two MMM systems by imaging ex vivo brain tissue sections with neurons expressing green fluorescent protein (GFP). A Thy1-GFP transgenic mouse [39] was deeply anesthetized with 2.5% Avertin (0.025 ml/g i.p.) and transcardially perfused with PBS, followed by 4% paraformaldehyde. Its brain was dissected and placed overnight in cold 4% paraformaldehyde. 1 mm thick coronal sections were sectioned with a vibrotome, then mounted and coverslipped on microscope slides using adhesive silicone isolators (JTR20-A2-1.0, Grace Bio-Labs, Bend, OR). The specimens were imaged in 3D with both the CCD-based MMM and the MAPMT-based MMM. The objective used was the 20× water-immersion with NA 0.95. Excitation laser power was 300 mW at 890 nm wavelength (approximately 4.7 mW per each excitation focus). The frame rate was 0.3 frames per second covering a 320 × 320 pixels image. The slow frame rate was used to image down to 120 μm deep from surface limited by the low excitation power available at 890 nm. The image stacks were acquired with a depth increment of 1.5 microns. Representative images are shown in Fig. 9. The images in the first column are from the CCD-based MMM at the surface, 30 μm, and 75 μm deep (a-c). The images in the second and third column are from the MAPMT-based MMM: raw images (d-f) and deconvoluted images (g-i). Neurons are distributed approximately equally throughout the entire imaging depth. At the surface, the thin dendritic structures and cell bodies of neurons are visible in all the images. However, the image from the CCD-based MMM has slightly lower contrast than the MAPMT-based MMM image. It may be because the emission photons, which travel forward direction into the specimen, were back scattered toward the CCD camera. These back scattered photons were then collected in the incorrect pixels of the CCD degrading the image SNR even at the surface. This is consistent with the observation that the signal initially increases with increasing imaging depth, shown in Fig. 3. At 30 μm deep, the dendrite structures are less visible in the images taken with the CCD-based MMM system than in the images taken with the MAPMT-based MMM. This is because CCD-based MMM is more sensitive to emission photon scattering and its SNR degrades faster than that of the MAPM-based system. This is consistent with the results obtained in tissue phantoms shown in Fig. 6. At 75 μm depth, the image from the CCD-based MMM shows that thin dendritic structures are completely invisible. The image from the MAPMT-based MMM (h) shows that ghost images of bright cell bodies appear in the neighboring pixels indicating a significant amount of emission photons are scattered into the neighboring pixels at this depth. The ghost images are removed after the deconvolution process is applied (i). We also note that the intensity of neurons is increased in the deconvoluted image (i) as the scattered photons are restored to the correct pixels.
Fig. 9.

Images of GFP expressing neurons in the ex vivo mouse brain acquired with CCD-based MMM (a-c) and MAPMT-based MMM (d-f) at different depth locations (surface, 30 um, and 75 um deep). For images with MAPMT-based MMM, a deconvolution algorithm was applied to remove the effect of emission photon scattering (g-i). The objectiveused is a 20× water immersion with NA 0.95. The input laser power is 300 mW at 890 nm wavelength. The frame rate is 0.3 frames per second with a frame size of 320 × 320 pixels.
3.6 Potential future improvements in MAPMT-based MMM
The images in Fig. 9 show that the MAPMT-based MMM can provide superior images as compared to the CCD based MMM for tissue imaging. However, additional improvements of this system are possible. First, since the MAPMT is positioned in the image plane, the location of each excitation focus corresponds to the center position of the matching pixel of the MAPMT. The effective detector area is scaled quadratically with the separation distance of the foci. Therefore, with wider foci separation, each pixel of the MAPMT will have better collection efficiency for scattered emission photons. In the current configuration, the excitation foci are separated by 45 μm so that the effective detector area for each channel of the MAPMT is 45 μm × 45 μm. The size of the imaging field of view with 8 × 8 foci is 360 μm × 360 μm. As the excitation foci are separated more, the system becomes less sensitive to emission photon scattering effect. The maximum separation of excitation foci is limited by either the field of view of the objective or the apertures of other optical elements in the collection light path. The 20× water immersion objective used in our experiments has a field of view of 1100 μm in diameter. This will allow us to separate the foci to approximately 100 μm from each other. It should be noted that further separation of excitation foci while maintaining the same image resolution will result in larger field of view but lower frame rate.
Second, having a lower signal collection efficiency is a disadvantage of a MAPMT-based MMM design compared with a CCD-based MMM design. The signal collection efficiency is lower, because the emission light is de-scanned and emission photons pass through more optical elements, including the scanner mirror, until they are collected at the MAPMT. Further, the de-scanned geometry has a longer optical path which contributes to the loss of some scattered photons due to the finite aperture of the optics. We find that the signal collection efficiency is approximately 70 % of the collection efficiency of directly detection using the CCD camera without de-scanning. In the future, a non-descanned system may be designed based on using a lenslet array in the detection path that can further improve signal collection efficiency.
Third, the MAPMT used in this experiment is composed of bi-alkali cathodes with very low efficiency in the red. However newer MAPMTs with multi-alkali cathodes have a more uniform spectral response having a quantum efficiency of 15-20% throughout the visible spectrum. Further, these MAPMTs have very low noise. 100 dark counts per second are typical and cooling can reduce this further by several orders of magnitude. For example, with a readout rate of 30 KHz, the typical dark count per pixel is less than 3×10−2. Therefore, when there is very low signal or when the system is operating at a very high frame rate, the MAPMT system can perform better than a CCD-based system even for non-scattering applications. The production of MAPMTs with higher sensitivity cathode materials such as GaAsP is being considered by the manufacturer and this can bring the quantum efficiency up to almost 40 % at blue-green wavelength range, within a factor of two of the sensitivity of back-thinned CCD-based systems with quantum efficiency between 70-90%.
Fourth, the photon sensitivity of each MAPMT pixel can vary up to 50%. This effect can be further compounded by the Gaussian intensity spatial distribution of the excitation beam. However, the problem of unequal excitation power has been solved previously using a multiple reflecting beam splitter to generate equal intensity beam-lets [22, 38]. The MAPMT-based MMM system can be further improved by utilizing this type of beam splitter with additional flat field correction algorithm to remove the inherent, but constant, non-uniformity of the MAPMT sensitivity.
Fifth, there is also electronic crosstalk among neighboring pixels of the MAPMT. The typical crosstalk is about 2 – 4% when the photons are collected at the center of each pixel. The level of crosstalk is not known when photons are collected off-center. However, this crosstalk can be removed by the same deconvolution procedure as discussed previously.
Finally, MMM tissue imaging requires high laser power. Assuming that an input power of 10 mW is needed for each excitation focus on the sample surface, the generation of 64 excitation foci requires 640 mW of power at the sample. When imaging turbid tissue specimens, more input power is required at deeper sections to compensate for the signal loss due to excitation photon scattering. In the case of a tissue specimen with a mean free path length of 160 μm at the excitation wavelength, an input power of 21.8 mW and 47.7 mW are required to image at 100 and 200 μm deep respectively with a 1.2 NA objective. This estimation is based on the assumption that there is minimal loss in the excitation power delivery through the optical beam path such as the objective. Unfortunately, while the Olympus XLUMPLFL20XW objective has many positive qualities, its transmission in the infrared is poor, resulting in a total loss by a factor of 3. Therefore, the actual required power before the objective is 37.7 and 82.5 mW for 100 and 200 μm deep imaging. Therefore, over 2.4 to 5.3 W power is needed for 64 foci imaging. This is at or above the power available from typical ultrafast lasers today. The current speed limitation in MMM tissue imaging, quantified as the maximum volume imaged per unit time at diffraction limited resolution, is therefore the limited power of the available light source and the transmittance of objectives in the infrared.
4. Conclusion
Conventional MMM systems may not be the ideal high-speed methodology for imaging multiply scattering tissue specimens. These systems are highly sensitive to the emission photon scattering, because the effective area of the detector pixels is smaller than the spatial distribution of scattered emission photons in the image plane. We found that in the imaging depth from surface down to , the image resolution is not affected, because resolution is mostly determined by the unscattered emission photons. On the other hand, the image contrast is seriously degraded. Only the unscattered emission photons are collected as signal and the scattered emission photons become background noise due to their broad spatial distribution. The fast loss of the image contrast in MMM was demonstrated both in numerical simulation and in the imaging of tissue phantoms and biological specimens.
As an improvement, a new high-speed MMM is developed based on a MAPMT. High-speed imaging is achieved by scanning the specimen with an 8 × 8 array of excitation foci and by acquiring signals from those foci simultaneously with a MAPMT which has 8 × 8 pixels. The system has very low noise by collecting signal in single photon counting mode. MAPMT-based MMM system is optimized for high-speed imaging of turbid tissues, because it is significantly less sensitive to the effect of emission photon scattering as compared with a CCD-based system. The high sensitivity of the system was demonstrated in the imaging of ex vivo human skin based on weak endogenous fluorophores. We further compared the performance of MAPMT- and CCD-based MMMs in the imaging of tissue phantoms and GFP expressing neuronal structures in mouse brain slices. We demonstrated that MAPMT-based MMM achieve significantly higher imaging depth.
Acknowledgments
This research is supported by NIH R33CA091354, R21/R33CA84740, R03EY014891 and NSF BES-0093840.
Footnotes
OCIS codes: (190.4180) Multiphoton process; (180.2520) Fluorescence microscopy; (180.5810) Scanning microscopy; (180.6900) Three-dimensional microscopy.
References and links
- 1.Denk W, Strickler JH, Webb WW. 2-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 2.Grutzendler J, Kasthuri N, Gan W-B. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- 3.Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- 4.Lee WC, Huang H, Feng G, Sanes JR, Brown EN, So PTC, Nedivi E. Dynamic Remodeling of Dendritic Arbors in GABAergic Interneurons of Adult Visual Cortex. PLoS Biol. 2005;4:e29. doi: 10.1371/journal.pbio.0040029. http://biology.plosjournals.org/perlserv?request=get-document&doi=10.1371/journal.pbio.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Padera TP, Stoll BR, So PTC, Jain RK. Conventional and High-Speed Intravital Multiphoton Laser Scanning Microscopy of Microvasculature, Lymphatics, and Leukocyte.Endothelial Interactions. Molecular Imaging. 2002;1:9–15. doi: 10.1162/15353500200200004. [DOI] [PubMed] [Google Scholar]
- 6.Konig K, Riemann I. High-resolution multiphoton tomography of human skin with subcellular spatial resolution and picosecond time resolution. J. Biomed. Opt. 2003;8:432–439. doi: 10.1117/1.1577349. [DOI] [PubMed] [Google Scholar]
- 7.Koehler MJ, Konig K, Elsner P, Buckle R, Kaatz M. In vivo assessment of human skin aging by multiphoton laser scanning tomography. Opt. Lett. 2006;31:2879–2881. doi: 10.1364/ol.31.002879. [DOI] [PubMed] [Google Scholar]
- 8.Roorda RD, Hohl TM, Toledo-Crow R, Miesenbock G. Video-rate nonlinear microscopy of neuronal membrane dynamics with genetically encoded probes. J. Neurophysiol. 2004;92:609–621. doi: 10.1152/jn.00087.2004. [DOI] [PubMed] [Google Scholar]
- 9.Ragan T, Kim KH, Bahlmann K, So PTC. Two-photon tissue cytometry. Methods Cell Biol. 2004;75:23–39. doi: 10.1016/s0091-679x(04)75002-5. [DOI] [PubMed] [Google Scholar]
- 10.Yu B, Kim KH, So PTC, Blankschtein D, Langer R. Topographic Heterogeneity in Transdermal Transport revealed by High-Speed Two-Photon Microscopy: determination of representative skin sample sizes. J. Invest. Dermatol. 2002;118:1085–1088. doi: 10.1046/j.1523-1747.2002.01796.x. [DOI] [PubMed] [Google Scholar]
- 11.Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, Luster AD, Scadden DT, Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim KH, Buehler C, So PTC. High-speed two-photon scanning microscope. Appl. Opt. 1999;38:6004–6009. doi: 10.1364/ao.38.006004. [DOI] [PubMed] [Google Scholar]
- 13.Fan GY, Fujisaki H, Miyawaki A, Tsay R-K, Tsien RY, Ellisman MH. Video-rate scanning two-photon excitation fluorescence microscopy and ratio imaging with Cameleons. Biophy. J. 1999;78:2412–2420. doi: 10.1016/S0006-3495(99)77396-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cianci GC, Wu J, Berland KM. Saturation modified point spread functions in two-photon microscopy. Microsc Res Tech. 2004;64:135–141. doi: 10.1002/jemt.20071. [DOI] [PubMed] [Google Scholar]
- 15.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003;21:1369–1377. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 16.Iyer V, Losavio BE, Saggau P. Compensation of spatial and temporal dispersion for acousto-optic multiphoton laser-scanning microscopy. J. Biomed. Opt. 2003;8:460–471. doi: 10.1117/1.1580827. [DOI] [PubMed] [Google Scholar]
- 17.Reddy GD, Saggau P. Fast three-dimensional laser scanning scheme using acousto-optic deflectors. J. Biomed. Opt. 2005;10:064038. doi: 10.1117/1.2141504. [DOI] [PubMed] [Google Scholar]
- 18.Zeng S, Lv X, Zhan C, Chen WR, Xiong W, Jacques SL, Luo Q. Simultaneous compensation for spatial and temporal dispersion of acousto-optical deflectors for two-dimensional scanning with a single prism. Opt. Lett. 2006;31:1091–1093. doi: 10.1364/ol.31.001091. [DOI] [PubMed] [Google Scholar]
- 19.Goebel W, Kampa BM, Helmchen F. Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nat. Methods. 2007;4:73–79. doi: 10.1038/nmeth989. [DOI] [PubMed] [Google Scholar]
- 20.Bewersdorf J, Pick R, Hell SW. Multifocal multiphoton microscopy. Opt. Lett. 1998;23:655–657. doi: 10.1364/ol.23.000655. [DOI] [PubMed] [Google Scholar]
- 21.Buist AH, Muller M, Squier J, Brakenhoff GJ. Real time two-photon absorption microscopy using multi point excitation. J. Micros.-Oxf. 1998;192:217–226. [Google Scholar]
- 22.Nielsen T, Fricke M, Hellweg D, Andresen P. High efficiency beam splitter for multifocal multiphoton microscopy. J. Microsc. 2000;201:368–376. doi: 10.1046/j.1365-2818.2001.00852.x. [DOI] [PubMed] [Google Scholar]
- 23.Kurtz R, Fricke M, Kalb J, Tinnefeld P, Sauer M. Application of multiline two-photon microscopy to functional in vivo imaging. J. Neurosci. Methods. 2006;151:276–286. doi: 10.1016/j.jneumeth.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Jureller JE, Kim HY, Scherer NF. Stochastic scanning multiphoton multifocal microscopy. Opt. Express. 2006;14:3406–3414. doi: 10.1364/oe.14.003406. [DOI] [PubMed] [Google Scholar]
- 25.Gu M. Principles of three-dimensional imaging in confocal microscopes. World Scientific Pub Co Inc; 1996. [Google Scholar]
- 26.Egner A, Hell SW. Time multiplexing and parallelization in multifocal multiphoton microscopy. J. Opt. Soc. Am. A. 2000;17:1192–1201. doi: 10.1364/josaa.17.001192. [DOI] [PubMed] [Google Scholar]
- 27.Gan X, Gu M. Spatial distribution of single-photon and two-photon fluorescence light in scattering media: Monte Carlo simulation. Appl. Opt. 2000;39:1575–1579. doi: 10.1364/ao.39.001575. [DOI] [PubMed] [Google Scholar]
- 28.Blanca CM, Saloma C. Monte Carlo analysis of two-photon fluorescence imaging through a scattering medium. Appl. Opt. 1998;37:8092–8102. doi: 10.1364/ao.37.008092. [DOI] [PubMed] [Google Scholar]
- 29.Beaurepaire E, Mertz J. Epifluorescence collection in two-photon microscopy. Appl. Opt. 2002;41:5376–5382. doi: 10.1364/ao.41.005376. [DOI] [PubMed] [Google Scholar]
- 30.Ying J, Liu F, Alfano RR. Spatial distribution of two-photon-excited fluorescence in scattering media. Appl. Opt. 1999;38:224–229. doi: 10.1364/ao.38.000224. [DOI] [PubMed] [Google Scholar]
- 31.Dunn AK, Wallace VP, Coleno M, Berns MW, Tromberg BJ. Influence of optical properties on two-photon fluorescence imaging in turbid samples. Appl. Opt. 2000;39:1194–1201. doi: 10.1364/ao.39.001194. [DOI] [PubMed] [Google Scholar]
- 32.Dong CY, Koenig K, So PTC. Characterizing point spread functions of two-photon fluorescence microscopy in turbid medium. J. Biomed. Opt. 2003;8:450–459. doi: 10.1117/1.1578644. [DOI] [PubMed] [Google Scholar]
- 33.Oheim M, Beaurepaire E, Chaigneau E, Mertz Jerome, Charpak S. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J. Neuro. Method. 2001;111:29–37. doi: 10.1016/s0165-0270(01)00438-1. [DOI] [PubMed] [Google Scholar]
- 34.Fantini S, Franceschini MA, Gratton E. Semi-infinite-geometry boundary problem for light migration in highly scattering media: a frequency-domain study in the diffusion approximation. J. Opt. Soc. Am. B. 1994;11:2128–2138. [Google Scholar]
- 35.Buehler C, Kim KH, Greuter U, Schlumpf N, So PTC. Single-photon counting multicolor multiphoton fluorescence microscope. J. Fluoresc. 2005;15:41–51. doi: 10.1007/s10895-005-0212-z. [DOI] [PubMed] [Google Scholar]
- 36.So PTC, Kim H, Kochevar IE. Two-photon deep tissue ex vivo imaging of mouse dermal and subcutaneous structures. Opt. Express. 1998;3:339–350. doi: 10.1364/oe.3.000339. [DOI] [PubMed] [Google Scholar]
- 37.Masters BR, So PTC, Buehler C, Barry N, Sutin JD, Mantulin WW, Gratton E. Mitigating thermal mechanical damage potential during two-photon dermal imaging. J. Biomed. Opt. 2004;9:1265–1270. doi: 10.1117/1.1806135. [DOI] [PubMed] [Google Scholar]
- 38.Fittinghoff DN, Wiseman PW, Squier JA. Widefield multiphoton and temporally decorrelated multifocal multiphoton microscopy. Opt. Express. 2000;7:273–279. doi: 10.1364/oe.7.000273. [DOI] [PubMed] [Google Scholar]
- 39.Feng G, Mellor RH, Bernstein M, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]