

Abstract
Major controversy exists as to whether increased C-reactive protein (CRP) contributes to individual components of the metabolic syndrome or is just a secondary response to inflammatory disease processes. We measured blood pressure and metabolic phenotypes in spontaneously hypertensive rats (SHR) in which we transgenically expressed human CRP in liver under control of the apoE promoter. In SHR transgenic rats, serum levels of human CRP approximated the endogenous levels of CRP normally found in the rat. Systolic and diastolic blood pressures measured by telemetry were 10–15 mmHg greater in transgenic SHR expressing human CRP than in SHR controls (P<0.01). During oral glucose tolerance testing, transgenic SHR exhibited hyperinsulinemia compared to controls (insulin area under the curve 36±7 versus 8±2 nmol/L/2h, respectively, P<0.05). Transgenic SHR also exhibited resistance to insulin stimulated glycogenesis in skeletal muscle (174±18 versus 278±32 nmol glucose/g/2h, P<0.05), hypertriglyceridemia (0.84±0.05 versus 0.64±0.03 mmol/L, P<0.05), reduced serum adiponectin (2.4±0.3 versus 4.3±0.6 mmol/L, P<0.05), and microalbuminuria (200±35 versus 26±5 mg albumin/g creatinine, respectively, P<0.001). Transgenic SHR had evidence of inflammation and oxidative tissue damage with increased serum levels of interleukin 6 (IL6) (36.4±5.2 versus 18±1.7 pg/ml, P<0.005) and increased hepatic and renal TBARS (1.2±0.09 versus 0.8±0.07 and 1.5±0.1 versus 1.1±0.05 nM/mg protein, respectively, P<0.01), suggesting that oxidative stress may be mediating adverse effects of increased human CRP. These findings are consistent with the hypothesis that increased CRP is more than just a marker of inflammation and can directly promote multiple features of the metabolic syndrome.
Keywords: C-reactive protein, metabolic syndrome, oxidative stress, transgenic, spontaneously hypertensive rat
Introduction
There is mounting interest in the role of inflammation in the pathogenesis of obesity, metabolic disturbances, diabetes, and cardiovascular disease. Among the various biomarkers of inflammation that are associated with increased risk for these disorders, C-reactive protein (CRP) has attracted the most attention1,2. However, major controversy exists as to whether increased CRP contributes to disease pathogenesis or is just a secondary response to inflammatory disease processes3. Although most of the experimental focus has been on the relationship between CRP and risk for coronary heart disease, increased CRP levels have also been reported to correlate with increased risk for diabetes and multiple features of the metabolic syndrome including obesity, insulin resistance, dyslipidemia, and increased blood pressure4,5. Yet a huge knowledge gap exists as to whether CRP is just a marker or an active mediator of any of these inflammatory metabolic and cardiovascular disorders.
Although the results of recent “mendelian randomization” studies have failed to support a role for common CRP gene polymorphisms in the pathogenesis of coronary heart disease, they have not addressed whether genetic or non-genetic variation in CRP levels can influence risk for other forms of cardiovascular disease or features of the metabolic syndrome and diabetes6,7. Because metabolic and hemodynamic disturbances affect millions of people and significantly increase the risk for a variety of cardiovascular disorders and for diabetes, there is intense interest in understanding the significance of increased CRP in pathogenesis of features of the metabolic syndrome and related conditions. However, research progress on CRP has been hampered by the lack of effective animal models for studying the role of human CRP in pathogenesis of the metabolic syndrome or its components and for preclinical testing of novel CRP inhibitors. In the current studies, we sought to investigate whether increased levels of human CRP per se can promote increases in blood pressure and disturbances in glucose and lipid metabolism characteristic of the metabolic syndrome. To accomplish this goal, we transgenically expressed human CRP in the spontaneously hypertensive rat (SHR), a widely-studied animal model of hypertension that is genetically predisposed to development of multiple features of the metabolic syndrome.
Methods
Animals
We transgenically expressed human CRP in a highly inbred strain of SHR (SHR/OlaIpcv) that has been brother x sister mated for well over 130 generations. Transgenic SHR were derived by microinjections of zygotes with a previously described construct containing the cDNA for human CRP under control of the apoE promoter8 with the objective of driving expression of the CRP transgene in liver where CRP is normally produced. Because hypertension begins to develop at a relatively young age in SHR whereas metabolic disturbances can take longer to become apparent, we performed blood pressure studies in 3 month old transgenic SHR (N=9) and age-matched nontransgenic controls (N=8) and metabolic studies in 13 month old transgenic and control animals (N=8 per group). In all experiments, we studied male CRP transgenic SHR together with male, age-matched, non-transgenic SHR controls. The rats were housed in an air-conditioned animal facility and allowed free access to standard diet and water. All experiments were performed in agreement with the Animal Protection Law of the Czech Republic (311/1997) and were approved by the Ethics Committee of the Institute of Physiology, Academy of Sciences of the Czech Republic, Prague.
Expression of the transgene for human CRP and the endogenous gene for rat CRP determined by real time PCR
Total RNA was extracted using Trizol reagent (Invitrogen), and cDNA was prepared and analyzed by real-time PCR testing using QuantiTect SYBR Green reagents (Qiagen, Inc.) on an Opticon continuous fluorescence detector (MJ Research). Gene expression levels were normalized relative to the expression of peptidylprolyl isomerase A (Ppia) (cyclophilin) gene, which served as the internal control, with results being determined in triplicate. For the detection of the human CRP transgene we used these primers: hCRP151F 5′-CTT TTG GCC AGA CAG ACA TG-3′ and hCRP280R 5′-GTG TAG AAG TGG AGG CAC A-3′. For the detection of the endogenous gene encoding rat CRP, we used these primers: rCrp117F 5′-GCT TTT GGT CAT GAA GAC ATG-3′ and rCrp255R 5′-TCA CAT CAG CGT GGG CAT AG-3′.
Parameters of glucose and lipid metabolism and blood pressure measurements
The methods for oral glucose tolerance testing, assessment of skeletal muscle insulin sensitivity, all biochemical measurements in serum and tissue, and telemetric blood pressure measurements are described in the online Data Supplement (please see http://hyper.ahajournals.org).
Urine collection, microalbuminuria, cGMP and parameters of oxidative stress
Rats were placed into metabolic cages for 6 h to obtain urine samples for analysis of urinary excretion of albumin and cGMP. The level of albumin in urine was analyzed by HPLC method with UV-VIS detection according to Contois et al9. Urine albumin was adjusted for creatinine concentration (mg/g creatinine). Creatinine was measured by Jaffe rate assay (Pliva-Lachema, Brno, Czech Republic). The activities of antioxidant enzymes and lipoperoxidation products were determined as previously described10. Detailed descriptions of the methods are available in the online Data Supplement (please see http://hyper.ahajournals.org).
Statistical analysis
The data are expressed as means ± SEM. Individual groups were compared by unpaired Student t-test. The 24 h mean values of systolic blood pressure were analyzed by repeated measures ANOVA with grouping effect of strain and repeated measurements in time. Statistical significance was defined as P<0.05.
Results
Derivation of transgenic ratsand expression of transgenic and endogenous CRP proteins
Because the human CRP transgene was directly introduced on the highly inbred genetic background of the SHR, the transgenic SHR and non-transgenic SHR controls are genetically identical except for the presence of the human CRP transgene. We confirmed successful germline transmission and functional expression of the transgene by genotype analysis and by measuring mRNA and protein levels of human CRP in the offspring. The real time-PCR measurements of CRP mRNA expression using primers that distinguish the sequences of the genes encoding human CRP and endogenous rat CRP are shown for the transgenic rats in Figure 1A. In transgenic SHR (Figure 1A), the human CRP transgene and the endogenous rat CRP gene were both abundantly expressed in liver with relatively little or no transgene expression in other tissues as anticipated given use of the apoE promoter. In the SHR controls, the hepatic expression level of the endogenous rat CRP gene was not significantly different from that in the transgenic SHR and there was no detectable expression of the human CRP transgene in the controls as expected (data not shown).
Figure 1.

Gene expression pattern and serum protein levels for human CRP and for endogenous rat CRP. A. Real time PCR results in the transgenic SHR comparing expression levels of the human CRP transgene and the endogenous rat CRP gene across different tissues (in the transgenic animals), normalized relative to the expression of the peptidylprolyl isomerase A (Ppia) gene which served as an internal control. Both the human CRP transgene and the endogenous rat CRP gene were predominantly expressed in liver of the transgenic animals. In the SHR controls (data not shown), the endogenous rat CRP gene was also predominantly expressed in liver as expected and its expression level was not significantly different from that in transgenic SHR. B. Serum levels of human CRP and endogenous rat CRP in transgenic SHR compared to nontransgenic SHR controls. Serum from transgene negative SHR controls showed little or no cross-reactivity in the human CRP assay.
Figure 1B shows serum levels of human CRP and rat CRP detected in the transgenic SHR strain and in transgene negative SHR controls using immunoassays with high specificity for rat or human CRP. The serum levels for human CRP in the transgenic SHR were similar to serum levels of endogenous rat CRP in the SHR controls (Figure 1B). The overall concentration of CRP in the transgenic rats (contributed by both human CRP and endogenous rat CRP) was increased by approximately 50% compared to the normal serum concentration of endogenous rat CRP in the SHR controls. These findings confirm that the transgene is being functionally expressed and produces serum levels of human CRP that are close to the endogenous levels of CRP normally found in the SHR and in other strains of rats as well11.
Transgenic expression of human CRP promotes multiple features of the metabolic syndrome
Having established functional expression of the human CRP transgene, we next phenotyped the transgenic SHR and non-transgenic SHR controls for key clinical features of the metabolic syndrome. Figure 2 shows that transgenic expression of human CRP significantly increased blood pressure in the SHR. The daily 24 hour average systolic and diastolic blood pressures measured byradiotelemetry in conscious, unrestrained transgenic SHR expressing human CRP were significantly greater than in transgene negative SHR controls (Figure 2). The average mean arterial pressure was signficantly greater in the CRP transgenic SHR than in the transgene negative controls, 156±3 mmHg versus 142±4 mmHg, respectively, P<0.0001, with blood pressures being elevated in both the daytime and nightime hours (data not shown). Supplemental dietary sodium chloride induced similar increases in blood pressure in the transgenic SHR and in the transgene negative controls. In addition, we have found that the levels of the major nitric oxide second messenger cyclic GMP (cGMP) were significantly reduced in urine of the CRP transgenic rats when compared to controls (cGMP/creatinine ratio 0.177±0.021 nmol/mmol versus 0.235±0.015 nmol/mmol, respectively, P<0.05) suggesting that transgenic CRP might be influencing blood pressure regulation through nitric oxide related pathways.
Figure 2.

Systolic and diastolic blood pressures. The daily 24 hour average systolic and diastolic blood pressures measured by radiotelemetry in conscious, unrestrained transgenic SHR expressing human CRP were significantly greater than in transgene negative SHR controls (P<0.01).
In the transgenic SHR, fasting glucose levels before oral glucose loading were similar to those in the transgene negative controls (Figure 3A). During the oral glucose tolerance test, there was a tendency for increased serum glucose levels in the transgenic SHR but the results did not achieve statistical significance. However, serum insulin levels were significantly increased in the transgenic SHR compared to controls during the glucose tolerance test (Figure 3B). These findings are consistent with an impairment in insulin action and are further supported by the observation of skeletal muscle insulin resistance in the SHR transgenic rats versus transgene negative SHR controls (Figure 4); transgenic SHR were almost completely resistant to the effects of insulin on nonoxidative glucose metabolism (glucose incorporation into skeletal muscle glycogen). The basal level of glycogenesis in SHR-CRP transgenic rats was also reduced compared to that in SHR controls but the difference did not achieve statistical significance (P=.07) (Figure 4). Transgenic expression of human CRP was associated with increased serum levels of triglycerides and liver triglyceride concentrations while muscle triglyceride concentrations were not significantly different (Table 1). HDL cholesterol levels were similar between the two groups (Table 1). The above findings show that transgenic expression of human CRP in the SHR can promote multiple features of the metabolic syndrome including insulin resistance, hypertriglyceridemia, and increased blood pressure. In addition, we have found that transgenic expression of human CRP induces decreases in serum adiponectin levels compared to those in non-transgenic SHR controls (Table 1). These findings establish that the SHR strain provides a favorable genetic background for investigating the adverse effects of human CRP on both hemodynamic and biochemical components of the metabolic syndrome.
Figure 3.

Oral glucose tolerance test (OGTT). A. Serum glucose levels during the glucose tolerance test; B. Serum insulin levels during the OGTT; * and ** denote P<0.01 and P<0.001, respectively.
Figure 4.

Basal and insulin stimulated incorporation of glucose into soleus muscle glycogen in CRP transgenic rats (solid bars) and in their transgene negative SHR controls (open bars). In the transgene negative SHR controls, insulin stimulated the incorporation of glucose into skeletal muscle glycogen. In contrast, the CRP transgenic SHR were almost completely resistant to stimulatory effects of insulin on skeletal muscle glycogenesis. The basal level of glycogenesis in SHR-CRP transgenic rats was also somewhat reduced compared to SHR controls but the difference did not achieve statistical significance (P=0.07). * denotes P<0.01.
Table 1.
Metabolic and inflammatory parameters in nonfasted rats
| Trait | Control SHR | CRP transgenic SHR |
|---|---|---|
| Body weight | 412±13 | 410±4 |
| Serum adiponectin (ng/ml) | 4.32±0.59 | 2.37±0.31* |
| Serum triglycerides (mmol/L) | 0.64±0.03 | 0.84±0.05* |
| Liver triglycerides (μmol/g) | 4.30±0.57 | 7.62±0.90* |
| Muscle triglycerides (μmol/g) | 1.00±0.73 | 1.45±0.38 |
| Serum NEFA (mmol/L) | 0.44±0.07 | 0.36±0.02 |
| Serum HDL cholesterol (mmol/L) | 1.35±0.05 | 1.36±0.06 |
| Serum interleukin 6 (pg/ml) | 18±1.8 | 36.4±5.2* |
| Serum TNFα (pg/ml) | 0.94±0.25 | 0.82±0.19 |
| Serum ALT (U/L) | 104±3 | 120±5* |
| Serum AST (U/L) | 403±20 | 376±22 |
| Serum creatinine (μmol/L) | 43.6±2.5 | 45.9±2.2 |
denotes P<0.05
Transgenic expression of human CRP promotes inflammation and oxidative tissue damage in the SHR model
It has recently been proposed that CRP might be influencing risk for hypertension, diabetes, and cardiovascular disease by increasing oxidative stress in tissues involved in the pathogenesis of these disorders12,13. It has long been suspected that increased oxidative stress in liver might influence glucose metabolism and risk for diabetes and that increased oxidative stress in kidney might play a role in the pathogenesis of hypertension. Therefore, we measured activities of key anti-oxidant enzymes in liver and kidney and assessed tissue levels of TBARS and conjugated dienes as indices of tissue oxidative damage in transgenic SHR. Table 2 shows that expression of transgenic CRP was associated with impaired tissue activity of glutathione peroxidase, reduced glutathione levels, and increased hepatic and renal oxidative tissue damage. As shown in Table 1, serum levels of ALT were increased in the SHR CRP transgenic rat consistent with the findings of oxidative liver damage. Expression of transgenic CRP was also associated with increased levels of interleukin 6 suggesting the presence of an inflammatory state
Table 2.
Parameters of oxidative stress
| Parameters | Control SHR Liver | CRP transgenic SHR |
|---|---|---|
| Liver | ||
| SOD U/mg protein | 0.363±0.038 | 0.297±0.014‡ |
| GSH-Px, μM GSH/min/mg protein | 636±18 | 467±22* |
| GR, μM NADPH/min/mg protein | 115±14 | 142±14 |
| CAT μM H2O2/min/mg protein | 798±36 | 899±21‡ |
| GSH μM/mg protein | 22.0±1.6 | 16.9±1.1‡ |
| TBARS nM/mg protein | 0.829±0.045 | 1.328±0.086* |
| CD nM/mg protein | 19.5±1.1 | 24.2±2‡ |
| Renal cortex | ||
| SOD U/mg protein | 0.386±0.019 | 0.304±0.022‡ |
| GSH-Px, μM GSH/min/mg protein | 512±18 | 292±19* |
| GR, μM NADPH/min/mg protein | 236±23 | 268±19 |
| CAT μM H2O2/min/mg protein | 671±19 | 595±25‡ |
| GSH μM/mg protein | 12.7±0.6 | 9.0±0.8* |
| TBARS nM/mg protein | 1.111±0.055 | 1.456±0.102† |
| CD nM/mg protein | 20.8±1.1 | 26.8±1.6† |
p<0.001,
p<0.01,
p<0.05; SOD – superoxide dismutase; GSH-Px – seleno-dependent glutathione peroxidase; GR – glutathione reductase; CAT – catalase; GSH – glutathione; TBARS – thiobarbiturate acid reactive substances; CD – conjugated dienes
In addition to finding direct evidence of renal oxidative damage in the CRP transgenic SHR (as reflected by increased TBARS and conjugated dienes in kidney tissue shown in Table 2), we found marked increases in microalbuminuria in the CRP transgenic rats (200±35 versus 26±5 mg/g albumin/creatinine ratio, respectively, P<0.001) which was accompanied by histopathological changes in the kidneys (Figure 5). These observations raise the possibility that increased levels of CRP might be promoting renal damage not onlythrough effects on blood pressure but also through direct actions of CRP on the kidney. However, it is also possible that renal damage in the CRP transgenic rat could have been mediated by increases in blood pressure coupled with disturbances in glucose and lipid metabolism rather than by a direct effect of CRP on the kidney.
Figure 5.
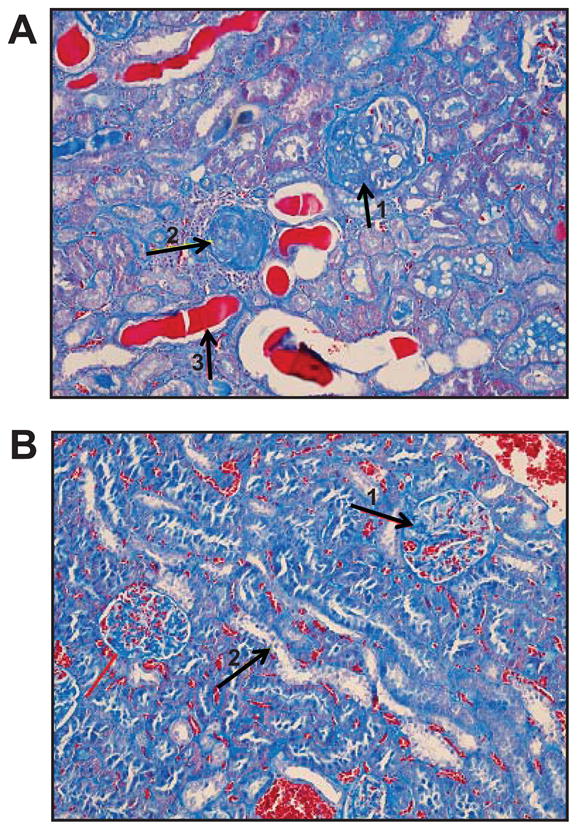
Histology of kidney using staining with trichrome blue (20×). A. SHR-CRP transgenic rat exhibited partial and total sclerosis of glomeruli (arrows 1 and 2), dystrophy of segments of tubular epithelium, proteinaceous casts in the lumen of some tubules (arrow 3), and evidence of fibrosis and inflammatory cellular infiltrates in the interstitium. B. SHR nontransgenic controls showed mild mesangial expansion of the glomeruli (arrow 1) and vacuolar dystrophy of epithelium in some tubules (arrow 2).
Discussion
Many epidemiologic studies have shown associations between increased CRP levels and increased risk for cardiovascular disease, diabetes, and multiple components of the metabolic syndrome including obesity, insulin resistance, dyslipidemia, and increased blood pressure1,4,5. However, epidemiologic studies cannot resolve issues of pathogenesis and considerable controversy exists as to whether increased CRP functions as a disease marker, a disease mediator, or both. To directly investigate the effects of increased levels of human CRP on disease pathogenesis, it is necessary to determine the impact of actively perturbing CRP levels in the whole organism.
Infusions of highly purified CRP can be used to manipulate CRP levels in humans and in animals, however, such studies are typically performed on a relatively short term basis and are inconvenient for studying the pathogenic effects of chronic exposure to increased levels of CRP. Delivery of human CRP to animals by virally mediated gene transfer can facilitate chronic studies but can be complicated by immune reactions to the administration of heterologous forms of CRP. In contrast, transgenic techniques can be used to manipulate CRP levels over an extended period of time in vivo without many of the limitations associated with CRP injections or delivery of CRP by viral vectors.
Given the potential benefits of transgenic approaches for investigating the biologic effects of increased levels of CRP, many investigators have used transgenic mice expressing human CRP to study its role in atherosclerosis and cardiovascular function14–19. However, relatively little work has been done in these models with respect to pathogenesis of features of the metabolic syndrome and risk factors for diabetes. Owing to the fact that CRP is not an acute phase reactant in mice and is synthesized in very low amounts in mice, some investigators have also raised questions about the utility of mouse models for studying the pathogenic effects of human CRP20–24. Recently, several groups have advocated use of the rat as an alternative model for studying the biologic actions of human CRP24–26. Because human CRP can activate rat complement and can be pro-inflammatory in rats, we investigated the blood pressure and metabolic effects of transgenically expressed human CRP in the SHR model that is genetically predisposed to developing multiple features of the metabolic syndrome.
In the current studies, we have found that in the SHR model, transgenic expression of human CRP in liver promotes insulin resistance, hypertriglyceridemia, as well as increased blood pressure. The serum level of human CRP in the transgenic rats was similar to the serum level of endogenous rat CRP in the transgene negative controls. Accordingly, in the transgenic SHR, the overall CRP level (the level of rat and human CRP combined) was increased by less than two fold compared to the normal CRP level observed in the SHR controls. This relative increase in total CRP level in the transgenic SHR is similar in magnitude to the relative increase in CRP observed in humans with metabolic syndrome. Thus, in the SHR model, serum levels of human CRP that are close to the endogenous levels of CRP normally found in rats can be associated with effects on the main hemodynamic and biochemical features of the metabolic syndrome. However, it should also be noted that the absolute increase in CRP in our rat model was very large compared to the absolute increase of CRP observed in humans with the metabolic syndrome. Therefore, in the SHR transgenic rats, as with any animal model, caution should be taken when extrapolating the results to humans.
Insulin resistance is considered to be a central component of the metabolic syndrome and in the current studies, we have found that transgenic expression of human CRP can promote disturbances in insulin and glucose metabolism including hyperinsulinemia and impaired insulin stimulated glucose incorporation into skeletal muscle glycogen. These findings are consistent with recent in vitro studies in which exposure of L6 myocytes to human CRP was found to decrease both insulin stimulated glucose uptake and glucose incorporation into glycogen27. The in vitro studies by D’Alessandris et al. indicate that human CRP affects insulin stimulated phosphorylation of IRS-1 and suggest that human CRP causes disturbances in glucose metabolism by impairing insulin signaling pathways that regulate cell glucose transport27.
In the current studies, transgenic expression of human CRP was also associated with reduced urinary excretion of cGMP suggesting that increased levels of CRP may be attenuating NO production. It has been reported that CRP might reduce the activity of endothelial nitric oxide synthase (eNOS) by affecting the stability of eNOS mRNA either directly or through the action of IL6 or TNFα28. In addition, it has been shown that CRP can activate NADPH oxidase and cause increases in reactive oxygen species that may decrease NO production by inducing eNOS uncoupling26,29. Consistent with the current findings, Guan et al.30 recently reported that virus mediated expression of human CRP in Wistar rats also induces increases in blood pressure (measured by direct puncture of carotid artery under anesthesia) and decreases in urinary cGMP. In addition, Vongpatanasin et al.31 have reported that transgenic expression of rabbit CRP in mice augments pressor responses to angiotensin II and promotes hypertension by inducing a decline in bioavailable NO and secondary downregulation of angiotensin II type 2 receptors in the vasculature. In contrast, alterations in angiotensin II type 1 receptor activity did not appear to play a role in the effects of CRP on blood pressure31. Taken together, these observations support the possibility that increases in human CRP might be aggravating hypertension and features of the metabolic syndrome in part by reducing NO bioavailability. In future studies, it will be of interest to confirm whether the effects of human CRP on blood pressure in transgenic SHR rat are more likely mediated by downregulation of NO activity and AT2 receptors than by activation of AT1 receptors.
Increased oxidative stress represents a well known mechanism that may mediate some of the adverse effects of increased CRP levels on risk for hypertension, diabetes, and cardiovascular disease12,13,26. It has long been suspected that increased oxidative stress in liver and skeletal muscle might play an important role in risk for diabetes and that increased oxidative stress in the kidney may be an important determinant of hypertension. In the current studies, we found that transgenic expression of human CRP causes increased oxidative tissue damage in both liver and kidney. These observations suggest that increased CRP levels maybe influencing risk for the metabolic syndrome by inducing oxidative damage in a variety of tissues. As previously shown by Hein et al., activation of NADPH oxidase is one of the principal mechanisms whereby human CRP stimulates production of reactive oxygen species26. The current studies suggest that impaired activity of glutathione peroxidase and reductions in glutathione levels could also be contributing to the effects of human CRP on oxidative stress and tissue damage.
It should be emphasized that the current studies on the metabolic effects of human CRP were performed in the SHR model which is a strain known to be genetically susceptible to developing multiple features of the metabolic syndrome. It is possible that other hypertensive models or strains might differ from the SHR with respect to their susceptibility to the adverse metabolic effects of human CRP. Having established that human CRP can aggravate features of the metabolic syndrome in the most widely used animal model of essential hypertension, future studies can now be planned to determine whether other strains or models exist that are more or less susceptible than the SHR to the adverse metabolic effects of human CRP. If so, this could open the door to linkage mapping of genetic factors that influence metabolic responses to increased levels of human CRP.
Perspectives
The current findings are consistent with the proposal that human CRP is more than just a secondary marker of inflammation and that increased levels of human CRP might directly contribute to the pathogenesis of features of the metabolic syndrome and risk for diabetes. The humanized CRP transgenic SHR represents a new model for investigating mechanisms whereby increased CRP levels may promote multiple components of the metabolic syndrome and could be further used to search for genetic factors that might influence susceptibility to the adverse metabolic effects of human CRP. This transgenic SHR model should also be of interest for testing the therapeutic effects of novel CRP inhibitors and a variety of other drugs such as anti-oxidants, anti-inflammatory agents, etc. that might block or attenuate the adverse effects of human CRP on tissues involved in the pathogenesis of the metabolic syndrome, diabetes, and related cardiovascular disorders.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the Internal Grant Agency of the Ministry of Health of the Czech Republic (NS9757-3); the Ministry of Education of the Czech Republic (ME08006, 1M0520, MSM6046070901); the Grant Agency of the Czech Republic (301/10/0290 and P303/10/0505); Grant Agency of the Academy of Sciences of the Czech Republic (IAA500110805); by the European Community’s Seventh Framework Program (FP7/2007-2013) under grant agreement no. HEALTH-F4-2010-241504 (EURATRANS); and the National Heart, Lung and Blood Institute of the US National Institutes of Health. M.P. is an international research scholar of the Howard Hughes Medical Institute.
Footnotes
Disclosures
None
References
- 1.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ JUPITER Trial Study Group. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 2.Lavie CJ, Milani RV, Verma A, O’Keefe JH. C-reactive protein and cardiovascular diseases is it ready for primetime? Am J Med Sci. 2009;338:486–492. doi: 10.1097/MAJ.0b013e3181c61b66. [DOI] [PubMed] [Google Scholar]
- 3.Scirica BM, Morrow DA. Is C-reactive protein an innocent bystander or proatherogenic culprit? The verdict is still out. Circulation. 2006;113:2128–2134. doi: 10.1161/CIRCULATIONAHA.105.611350. [DOI] [PubMed] [Google Scholar]
- 4.Ndumele CE, Pradhan AD, Ridker PM. Interrelationships between inflammation, C-reactive protein, and insulin resistance. J Cardiometab Syndr. 2006;1:190–196. doi: 10.1111/j.1559-4564.2006.05538.x. [DOI] [PubMed] [Google Scholar]
- 5.Haffner SM. The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am J Cardiol. 2006;97:3A–11A. doi: 10.1016/j.amjcard.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, Erdmann J, Braund P, Engert JC, Bennett D, Coin L, Ashby D, Tzoulaki I, Brown IJ, Mt-Isa S, McCarthy MI, Peltonen L, Freimer NB, Farrall M, Ruokonen A, Hamsten A, Lim N, Froguel P, Waterworth DM, Vollenweider P, Waeber G, Jarvelin MR, Mooser V, Scott J, Hall AS, Schunkert H, Anand SS, Collins R, Samani NJ, Watkins H, Kooner JS. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordestgaard BG, Zacho J. Lipids, atherosclerosis and CVD risk: is CRP an innocent bystander? Nutr Metab Cardiovasc Dis. 2009;19:521–524. doi: 10.1016/j.numecd.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Koike T, Kitajima S, Yu Y, Nishijima K, Zhang J, Ozaki Y, Morimoto M, Watanabe T, Bhakdi S, Asada Y, Chen YE, Fan J. Human C-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. 2009;120:2088–2094. doi: 10.1161/CIRCULATIONAHA.109.872796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contois JH, Hartigan C, Rao LV, Snyder LM, Thompson MJ. Analytical validation of an HPLC assay for urinary albumin. Clin Chim Acta. 2006;367:150–155. doi: 10.1016/j.cca.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Malínská H, Oliyarnyk O, Hubová M, Zídek V, Landa V, Simáková M, Mlejnek P, Kazdová L, Kurtz TW, Pravenec M. Increased liver oxidative stress and altered PUFA metabolism precede development of non-alcoholic steatohepatitis in SREBP-1a transgenic spontaneously hypertensive rats with genetic predisposition to hepatic steatosis. Mol Cell Biochem. 2010;335:119–125. doi: 10.1007/s11010-009-0248-5. [DOI] [PubMed] [Google Scholar]
- 11.de Beer FC, Baltz ML, Munn EA, Feinstein A, Taylor J, Bruton C, Clamp JR, Pepys MB. Isolation and characterization of C-reactive protein and serum amyloid P component in the rat. Immunology. 1982;45:55–70. [PMC free article] [PubMed] [Google Scholar]
- 12.Devaraj S, Dasu MR, Singh U, Rao LV, Jialal I. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis. 2009;203:67–74. doi: 10.1016/j.atherosclerosis.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeller JM, Sullivan BL. C-reactive protein selectively enhances the intracellular generation of reactive oxygen products by IgG-stimulated monocytes and neutrophils. J Leukoc Biol. 1992;52:449–455. doi: 10.1002/jlb.52.4.449. [DOI] [PubMed] [Google Scholar]
- 14.Zhang R, Zhang YY, Huang XR, Wu Y, Chung AC, Wu EX, Szalai AJ, Wong BC, Lau CP, Lan HY. C-reactive protein promotes cardiac fibrosis and inflammation in angiotensin II-induced hypertensive cardiac disease. Hypertension. 2010;55:953–960. doi: 10.1161/HYPERTENSIONAHA.109.140608. [DOI] [PubMed] [Google Scholar]
- 15.Xing D, Hage FG, Chen YF, McCrory MA, Feng W, Skibinski GA, Majid-Hassan E, Oparil S, Szalai AJ. Exaggerated neointima formation in human C-reactive protein transgenic mice is IgG Fc receptor type I (Fc gamma RI)-dependent. Am J Pathol. 2008;172:22–30. doi: 10.2353/ajpath.2008.070154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirschfield GM, Gallimore JR, Kahan MC, Hutchinson WL, Sabin CA, Benson GM, Dhillon AP, Tennent GA, Pepys MB. Transgenic human C-reactive protein is not proatherogenic in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2005;102:8309–8314. doi: 10.1073/pnas.0503202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovacs A, Tornvall P, Nilsson R, Tegnér J, Hamsten A, Björkegren J. Human C-reactive protein slows atherosclerosis development in a mouse model with human-like hypercholesterolemia. Proc Natl Acad Sci U S A. 2007;104:13768–13773. doi: 10.1073/pnas.0706027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teoh H, Quan A, Lovren F, Wang G, Tirgari S, Szmitko PE, Szalai AJ, Ward ME, Verma S. Impaired endothelial function in C-reactive protein overexpressing mice. Atherosclerosis. 2008;201:318–325. doi: 10.1016/j.atherosclerosis.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 19.Danenberg HD, Grad E, Swaminathan RV, Chen Z, Seifert P, Szalai AJ, Lotan C, Simon DI, Edelman ER. Neointimal formation is reduced after arterial injury in human crp transgenic mice. Atherosclerosis. 2008;201:85–91. doi: 10.1016/j.atherosclerosis.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tennent GA, Hutchinson WL, Kahan MC, Hirschfield GM, Gallimore JR, Lewin J, Sabin CA, Dhillon AP, Pepys MB. Transgenic human CRP is not pro-atherogenic, pro-atherothrombotic or pro-inflammatory in apoE−/− mice. Atherosclerosis. 2008;196:248–255. doi: 10.1016/j.atherosclerosis.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Torzewski J. C-reactive protein and atherogenesis: new insights from established animal models. Am J Pathol. 2005;167:923–925. doi: 10.1016/S0002-9440(10)61182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reifenberg K, Lehr HA, Baskal D, Wiese E, Schaefer SC, Black S, Samols D, Torzewski M, Lackner KJ, Husmann M, Blettner M, Bhakdi S. Role of C-reactive protein in atherogenesis: can the apolipoprotein E knockout mouse provide the answer? Arterioscler Thromb Vasc Biol. 2005;25:1641–1646. doi: 10.1161/01.ATV.0000171983.95612.90. [DOI] [PubMed] [Google Scholar]
- 23.Verma S, Devaraj S, Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation. 2006;113:2135–2150. [PubMed] [Google Scholar]
- 24.Devaraj S, Singh U, Jialal I. The evolving role of C-reactive protein in atherothrombosis. Clin Chem. 2009;55:229–238. doi: 10.1373/clinchem.2008.108886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, Hawkins PN, Myers RM, Smith MD, Polara A, Cobb AJ, Ley SV, Aquilina JA, Robinson CV, Sharif I, Gray GA, Sabin CA, Jenvey MC, Kolstoe SE, Thompson D, Wood SP. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. 2006;440:1217–1221. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 26.Hein TW, Singh U, Vasquez-Vivar J, Devaraj S, Kuo L, Jialal I. Human C-reactive protein induces endothelial dysfunction and uncoupling of eNOS in vivo. Atherosclerosis. 2009;206:61–68. doi: 10.1016/j.atherosclerosis.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alessandris C, Lauro R, Presta I, Sesti G. C-reactive protein induces phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 in L6 myocytes, thereby impairing the insulin signalling pathway that promotes glucose transport. Diabetologia. 2007;50:840–849. doi: 10.1007/s00125-006-0522-y. [DOI] [PubMed] [Google Scholar]
- 28.Verma S, Wang CH, Li SH, Dumont AS, Fedak PW, Badiwala MV, Dhillon B, Weisel RD, Li RK, Mickle DA, Stewart DJ. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106:913–919. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- 29.Jialal I, Verma S, Devaraj S. Inhibition of endothelial nitric oxide synthase by C-reactive protein: Clinical relevance. Clin Chem. 2009;55:206–208. doi: 10.1373/clinchem.2008.119206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guan H, Wang P, Hui R, Edin ML, Darryl C, Zeldin DC, Wang DW. Adeno-associated virus-mediated human C-reactive protein gene delivery causes endothelial dysfunction and hypertension in rats. Clin Chem. 2009;55:274–284. doi: 10.1373/clinchem.2008.115857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vongpatanasin W, Thomas GD, Schwartz R, Cassis LA, Osborne-Lawrence S, Hahner L, Gibson LL, Black S, Samols D, Shaul PW. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation. 2007;115:1020–1028. doi: 10.1161/CIRCULATIONAHA.106.664854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.