

Abstract
Background
Juvenile polyps are distinct hamartomatous malformations of the gastrointestinal tract that may occur in the heritable juvenile polyposis syndrome (JPS) or sporadically. Histologically, juvenile polyps are characterised by a marked increase of the stromal cell compartment but, an epithelial phenotype has also been reported. JPS has an increased risk of colorectal cancer but sporadic juvenile polyps do not. In 50–60% of JPS patients a germline mutation of the TGF-β/BMP pathway genes SMAD4 or BMPR1A is found. This study compares the histological phenotype of juvenile polyps with a SMAD4 or BMPR1A germline mutation and sporadic juvenile polyps.
Methods
H&E slides of 65 JPS polyps and 25 sporadic juvenile polyps were reviewed for histological features and dysplasia. Systematic random crypt and stroma counts were obtained by count stereology and a crypt-stroma ratio was determined. All polyps were subsequently categorised as type A (crypt-stroma ratio <1.00) or type B (crypt-stroma ratio ≥1.00), the latter referring to the epithelial phenotype. Cell cycle activity was assessed using immunohistochemistry of the proliferation marker Ki67, and mutation analysis was conducted for KRAS and APC to determine the involvement of the adenoma-carcinoma sequence.
Results
Juvenile polyps with a SMAD4 germline mutation were predominantly type B, whereas, type A was more common among juvenile polyps with a BMPR1A germline mutation, but this distinction could not be ascribed to differences in cell cycle activity. Dysplasia was equally common in JPS polyps with either a SMAD4 or BMPR1A germline mutation, where the involvement of the adenoma-carcinoma sequence does not seem to play a distinct role.
Conclusion
juvenile polyps in the setting of JPS exhibit distinct phenotypes correlating with the underlying genetic defect.
Keywords: Juvenile Polyposis, juvenile polyp, phenotype, genotype, colorectal cancer, SMAD4, BMPR1A
INTRODUCTION
Juvenile polyposis syndrome (JPS) is a rare autosomal dominant disorder characterised by the presence of multiple juvenile polyps in the gastrointestinal tract. Patients present during the first or second decade of life and have a markedly increased risk of colorectal cancer at later age.5,7,9. Clinical diagnosis is made when any one of the following criteria is met: (1) more than 3–5 juvenile polyps in the colorectum; (2) juvenile polyps throughout the intestinal tract; or (3) any number of juvenile polyps in combination with a positive family history of JPS.9,19,25. Sporadic juvenile polyps are a more common finding, occurring in up to 2% of the paediatric population, and are considered benign solitary lesions of the colorectum 10,19.
Juvenile polyps most often have a spherical appearance with extensive surface erosion; a marked increase of the stromal cell compartment; inflammatory and reactive changes of the epithelium; and distorted and dilated crypts. Several reports have noticed a more lobulated and epithelial phenotype in a subset of juvenile polyps 19.
In 50–60% of JPS patients a germline defect in SMAD4 or BMPR1A of the TGF-β/BMP signalling pathway, is found 16,17,28. Inactivation of these genes in mice leads to a JPS-like phenotype. Smad4 mutant mice develop gastrointestinal polyps characterised by elongated and dilated tubular structures lined by hyperplastic or serrated epithelium and a moderate expansion of the stromal cell compartment. Although mostly hyperplastic, the epithelium may show some atypia with occasional foci of dysplasia 15,27. In Bmpr1a mutant mice, polyps had cystically dilated and distorted glands filled with mucin and inflammatory cells surrounded by fibrous stroma 14. Moreover, inhibition of BMP signalling in mice (by transgenic expression of noggin, a BMP inhibitor, under control of a villin promoter) leads to branching and budding of the intestinal epithelium, crypt dilatation and reactive inflammatory changes. At later stages these mice develop foci of dysplastic epithelium and adenomatous change13.
These mouse models all display features reminiscent of JPS but, differences may exist in the phenotype depending on the genetic background. In this study, we compared the histological phenotype of human juvenile polyps with a SMAD4 or BMPR1A germline mutation and sporadic juvenile polyps. In addition, the possible role of the conventional adenoma-carcinoma sequence in the neoplastic progression in JPS was evaluated.
METHODS
Patients and tissue
Archival material from patients with one or more juvenile polyps was collected from The Johns Hopkins Polyposis Registry and clinic (Baltimore, MD, USA) and two academic hospitals in the Netherlands (Academic Medical Centre, Amsterdam, and University Medical Centre, Utrecht). The study was carried out according to the guidelines and with approval of the ethical committee of these institutions. Clinical and family history data were examined and polyps were carefully reviewed by an experienced GI pathologist (GJAO) to confirm the diagnosis of JPS or sporadic juvenile polyps. All JPS patients were analysed for germline defects of SMAD4, BMPR1A, PTEN, ENG and TGFBRII through direct sequencing and multiplex ligation-dependent probe amplification (MLPA).6,28 Thirty-nine patients (90 polyps) were included in this study, including 8 patients (21 polyps) with a SMAD4 germline defect, 6 patients (44 polyps) with a BMPR1A germline defect and 25 sporadic juvenile polyps.
Histological characterization
H&E stained slides of all juvenile polyps were systematically scored for individual histological features possibly associated with juvenile polyps (i.e. crypt distortion and dilatation, crypt density, stromal expansion, surface erosion, inflammatory and reactive change of the epithelium, vascular proliferation, Paneth cell metaplasia and thickening of the basal membrane, eosinophilia). Evaluation revealed 2 general phenotypes, namely that of a classic juvenile polyp comprising a prominent stromal compartment, dilated glands and surface erosion, as well as polyps with a predominantly epithelial phenotype (Figure 1a and 1b). Several features best describing the encountered phenotypes were then grouped together creating 2 categories as is shown in Table 1. Subsequently, all polyps were classified according to these 2 categories. In case polyps displayed heterogenic or intermediate features, overall crypt density served as decisive, most discriminatory feature. In addition, all polyps were graded for dysplasia according to the standard criteria 11.
Figure 1.
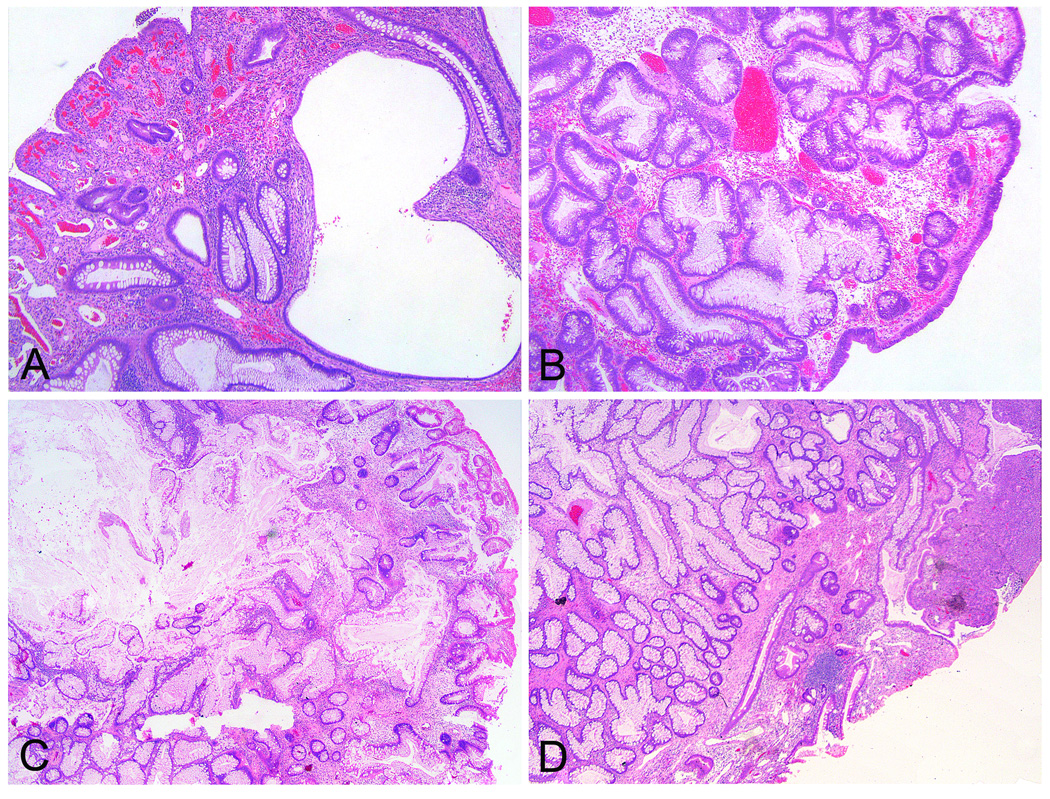
Histological appearance of the classic juvenile polyp and the epithelial phenotype. A shows histological image of a classic juvenile polyp with prominent stromal compartment, an eroded surface, inflammation and reactive changes of the epithelium, and distortion and dilation of the glands. B is illustrative of the epithelial phenotype devoid of an expanded stromal compartment but with an intact surface and with abundant tall columnar mucus secreting epithelium. C shows an intermediate phenotype initially graded as a classic type polyp, but was rendered type B by stereologic means. Conversely, panel D shows a juvenile polyp that was scored histologically of the epithelial type, yet stereology revealed a type A phenotype.
Table 1.
Features of the classic juvenile polyp versus the epithelial variant.
| Classic juvenile polyp | Epithelial juvenile polyp |
|---|---|
| Spherical | Lobulated |
| Eroded and granular surface | Villous-like surface |
| Stromal compartment expanded | Stromal compartment not expanded |
| Low crypt density | High crypt density |
| Flattened reactive epithelium | Columnar hypermucinous epithelium |
Count stereology
To better appreciate our histological findings a quantitative evaluation of crypt density was performed. A crypt-stroma ratio was determined by means of count stereology on H&E stained slides of all juvenile polyps. Using the Q-prodit software fields of vision were systematically distributed throughout the entire polyp at a 10X magnification. An overlying four points Weibel-grid was used to score epithelial or stromal counts. A discriminator of 1.00, describing equal counts of stroma and epithelium, was chosen arbitrarily to determine the predominant feature. A ratio <1.00 indicating a low crypt density was designated type A and a ratio ≥1.00 was designated type B. All polyps were categorised according to these criteria and results were compared to the histological classification.
Immunohistochemistry and scoring
Tissue was formalin-fixed and paraffinized in accordance with standard procedures. Immunohistochemistry was performed using a monoclonal antibody for Ki67 (DAKO MIB-1, Cat.no. M7240, 1:200). Briefly, 4 µm sections were deparaffinised and blocked for endogenous peroxidase activity by immersion in 0.3% H2O2 in methanol for 20 min. Antigen retrieval was performed in Tris/EDTA buffer (10 mM/1 mM; pH 9.0) for 10 min at 120°C. Nonspecific binding sites were blocked in PBS with 10% normal goat serum for 10 min, followed by antibody incubation for 1h at room temperature. Antibody binding was visualized using the Powervision+poly-HRP detection system (ImmunoVision Technologies, Co, Daly City, CA, USA) with 3,3-diamino-benzidine (DAB, Sigma D5637) as chromogen. Slides were counterstained with hematoxylin.
Ki67 is a nuclear proliferation marker expressed during all phases of actively growing cells but not in quiescent cells. Normal colon mucosa shows a distinctive nuclear Ki67 staining pattern with positive cells limited to the bottom third of the crypt, i.e. the proliferative compartment. Although in juvenile polyps the glands are often distorted, proliferative activity may remain confined to a restricted crypt compartment underneath the adjacent differentiated – non-proliferative – epithelium and thus retaining a compartmentalized phenotype. Loss of compartmentalization was defined as an overall increase in proliferative cells and the dissemination thereof throughout the epithelium resulting in loss of a clear distinction between the proliferative zone and the overlying differentiated epithelium. Slides were scored in a dichotomous manner describing retention or loss of compartmentalisation, the latter indicating an expansion of cell cycle activity.
Statistical analysis
Statistical analysis was performed using the SPSS 15.0 software package. The chi-square test was utilized to determine whether correlation between phenotypes and Ki67 expression were statistically significant at a p-value <0,05.
Laser microdissection and DNA isolation
Dysplastic epithelium was manually isolated from 8 µm sections counterstained with haematoxylin. DNA was obtained using TK buffer [400 µg/ml of proteinase K and 0.5% Tween 20, 50 mmol/l Tris (pH 9), 1 mmol/l NaCl, 2 mmol/L EDTA]. After overnight incubation in 50 µl TK buffer at 56°C, proteinase K was inactivated through incubation at 95°C for 10 minutes 4.
APC and K-ras mutation analysis
APC and K-ras mutation analysis was conducted through PCR amplification using Platinum®Taq DNA Polymerase (Invitrogen Corporation, Carsbad, California, USA) of DNA samples. For APC, 4 specific primer sets covering the mutation cluster region (MCR) in exon 15 were used 24. (1For-GAAATAGGATG TAATCAGACG, 1Rev-CGCTCCTGAAGAAA ATTCAAC, 2For-ACTGCAGGGTTCTAGT TTATC, 2Rev-GAGCTGGCAATCGAACGA CT, 3For-TACTTCTGTCAGTTCACTTGA TA, 3Rev-ATTTTTAGGTACTTCTCGCTTG, 4For-AAACACCTCCACCACCTCC, 4Rev-GCATTATTCTTAATTCCACATC). Amplification was performed at a Tm of 55°C for primer sets 1, 2 and 3, and at 58°C for primer set 4. Two primer sets were used for K-ras mutation analysis for exon 1 and 2, where mutational hotspots codon 12, 13 and 61 are located 8 (Exon1 For-CTGGTGGAGTATTT GATAGT, Exon1 Rev-ATG GTCCTGCACCAGTAATA, Exon2For-GTGCACTGTAATAA TCCAGAC, Exon2 Rev-CCACCTATAATGGTGAATATCT). Sequencing was conducted using the ABI Prism® 3130 genetic analyzer.
RESULTS
Histological characterization
Two categories of individual features were created in order to best describe the 2 phenotypes encountered upon initial evaluation of the slides, namely the classical and the epithelial type juvenile polyp (Table 1) (Figure 1a and 1b). All polyps were classified according to these 2 categories. Intermediate features notwithstanding, a dichotomous decision with regard to juvenile polyp phenotype was always rendered with crypt density serving as decisive feature. The phenotype described as classic juvenile polyp was found in 39 of 65 (60%) JPS polyps and the epithelial variant in 26 of 65 (40%) polyps. The epithelial variant was more common in polyps with a SMAD4 germline mutation compared to polyps with a BMPR1A germline mutation (p<0.001). (Table 2) All of the 25 sporadic juvenile polyps were of the classic phenotype.
Table 2.
Results of the classification based on histology and crypt-stroma ratio.
| Germline mutation |
Histological classification | Crypt-stroma classification | ||
|---|---|---|---|---|
| Classic | Epithelial | A | B | |
| SMAD4 | 6 (29%) | 15 (71%) | 8 (38%) | 13 (62%) |
| BMPR1A | 33 (75%) | 11 (25%) | 30 (68%) | 14 (32%) |
| Total | 39 | 26 | 38 | 27 |
Crypt-stroma ratio
Crypt density was considered the most discriminatory in differentiating between the classic juvenile polyp and the epithelial type juvenile polyp based upon initial evaluation of the earlier mentioned features. To better appreciate our histological findings a quantitative evaluation of crypt density was performed. Systematic random crypt and stroma counts were obtained by stereologic methodology to determine a crypt-stroma ratio. (Figure 2a) The crypt-stroma ratio was significantly higher in juvenile polyps with a SMAD4 germline mutation compared to those with a BMPR1A germline mutation (p=0.001) (Figure 2b). Juvenile polyps with a BMPR1A germline mutation had a higher ratio than sporadic juvenile polyps (p<0.001).
Figure 2.

Crypt-stroma ratio. Crypt and stroma counts as obtained by systematic random count stereology (A) and the crypt-stroma ratio displayed by genetic background (B). Juvenile polyps with a SMAD4 germline defect had a significantly higher crypt-stroma ratio compared to juvenile polyps with a BMPR1A germline defect (p=0.001).
A crypt-stroma ratio <1.00 was designated type A referring to the classic juvenile polyp category, and a ratio ≥1.00 was called type B specifying to the epithelial variant. According to these criteria, 38 out of 65 JPS polyps were of type A (58%) and 27 of type B (42%). Classification according to crypt-stroma ratio confirmed the observations made on histological evaluation that the epithelial variant (type B polyp) is more frequently found in patients with a SMAD4 germline mutation compared to individuals with a BMPR1A germline mutation (p<0.05) (Table 2). In 8 JPS polyps, of which 3 had a SMAD4 germline mutation and 5 had a BMPR1A germline mutation, histological classification did not concur with stereological findings (Figure 1c and 1d). Regarding the sporadic juvenile polyps all but one were classified type A.
Dysplasia
All polyps were graded for dysplasia. The frequency in which different grades of dysplasia were seen was similar for polyps with either a SMAD4 or BMPR1A germline mutation (Table 3). However, evaluation by polyp type revealed a distinct pattern of dysplasia in polyps with a SMAD4 or BMPR1A background. Focal dysplasia in a SMAD4 setting was found only in type B polyps, but in a BMPR1A setting focal dysplasia was seen in both type B and type A polyps. (Table 4) All sporadic polyps were negative for dysplasia.
Table 3.
Dysplasia in juvenile polyps.
| Dysplasia | Germline mutation | |
|---|---|---|
| SMAD4 | BMPR1A | |
| Negative | 8 (38%) | 17 (39%) |
| Indefinite | 4 (19%) | 8 (18%) |
| Low grade | 7 (33%) | 15 (34%) |
| High grade | 2 (9%) | 4 (9%) |
| Total | 21 | 44 |
Table 4.
Dysplasia in juvenile polyps organised by phenotype and germline defect.
| Dysplasia | Germline mutation and polyp type | |||
|---|---|---|---|---|
| SMAD4 | BMPR1A | |||
| A | B | A | B | |
| Negative | 7 (88%) | 1 (12%) | 15 (88%) | 2 (12%) |
| Indefinite | 1 (25%) | 3 (75%) | 5 (63%) | 3 (37%) |
| Low grade | - | 7 (100%) | 8 (53%) | 7 (47%) |
| High grade | - | 2 (100%) | 2 (50%) | 2 (50%) |
| Total | 8 | 13 | 30 | 14 |
Immunohistochemistry
To investigate whether variations in crypt density in juvenile polyps could be attributed to differences in proliferative activity, immunostaining of the Ki67 proliferation marker was performed. Focal loss of compartmentalisation of Ki67 indicating expanded cell cycle activity, was observed in 12 of 21 (57%) juvenile polyps with a SMAD4 germline mutation and in 25 of 44 (57%) juvenile polyps with a BMPR1A germline mutation. Evaluation of the immunostaining per polyp type per germline defect showed a correlation between a B phenotype and focal loss of Ki67 compartmentalisation especially in juvenile polyps with a SMAD4 germline mutation (p=0.006), but not in those with a BMPR1A mutation (p=0.131) (Table 5). However, when stratified by presence or absence of dysplasia no correlation between de-compartmentalisation of Ki67 and the B phenotype was seen in juvenile polyps with either a SMAD4 (p=1.000) or BMPR1A (p=0.668) germline mutation. Focal loss of compartmentalisation was found in 2 sporadic juvenile polyps.
Table 5.
Results Ki67 immunohistochemistry on juvenile polyps organised by phenotype and germline mutation.
| Compartmentalisation of Ki67 |
Germline mutation and polyp type | |||
|---|---|---|---|---|
| SMAD4 | BMPR1A | |||
| A | B | A | B | |
| Normal | 7 (88%) | 2 (15%) | 15 (50%) | 4 (29%) |
| Loss | 1 (12%) | 11 (85%) | 15 (50%) | 10 (71%) |
| Total | 8 | 13 | 30 | 14 |
APC and KRAS mutation analysis
To explore the role of the conventional adenoma-carcinoma sequence in the development of neoplastic change in JPS patients, APC and K-ras mutation analysis was performed. Only those polyps graded for dysplasia were analysed. Of the 16 tissues available, 2 polyps showed a K-ras point-mutation in exon 1 (GGT ➔ GAT). The K-ras mutations were found in areas of low grade dysplasia in one type A polyp from a patient with a germline BMPR1A mutation and in one type B polyp from a patient with a SMAD4 germline mutation. None of the polyps showed a mutation in the MCR of the APC gene (data not shown). In addition 10 nonsyndromic juvenile polyps were found negative for K-ras mutation.
DISCUSSION
JPS is caused by a germline defect in SMAD4 or BMPR1A. 16,17 Transgenic mice develop distinct JPS-like phenotypes depending on which of the JPS causing genes is targeted. Smad4 mutant mice show hyperplastic or serrated epithelium and minor stromal overgrowth whereas Bmpr1a mutant mice or mice with inhibited BMP signalling through transgenic expression of noggin show polyps with reactive changes of the epithelium, crypt dilatation and a prominent stromal compartment.13–15,27 We investigated and compared the histological phenotype of human juvenile polyps from patients with a SMAD4 or BMPR1A germline mutation.
Consistent with earlier reports, histological evaluation revealed a subset of JPS polyps featuring an epithelial phenotype (40%), deviating from classic juvenile polyps characterised by a prominent stromal compartment (60%).10,19 The epithelial phenotype was more prevalent in cases with a SMAD4 germline mutation, whereas, juvenile polyps with a BMPR1A germline mutation predominantly had the classic juvenile polyp phenotype (p<0.001).
Interestingly, quantitative evaluation of the crypt-stroma ratio confirmed our initial histological findings. Juvenile polyps with a SMAD4 germline mutation had a significantly higher crypt-stroma ratio compared to those with a BMPR1A germline mutation indicating a higher crypt density in the former and confirming the epithelial phenotype. These results underscore the relevance of crypt-density as discriminatory feature between the classic and epithelial type juvenile polyp.
Nevertheless, 8 JPS polyps showed a discrepancy between histological and stereological classification and may thus be considered to display intermediate features (Figure 1c and 1d). Re-evaluation of these polyps revealed that massive crypt dilatation may result in a juvenile polyp initially labelled as classic phenotype to be considered a B type polyp by stereologic means. On the other hand, surface erosion and subsequent inflammation in juvenile polyps with a proliferative core may cause a polyp to be classified histologically as an epithelial phenotype yet stereologically be scored as type A.
Similar frequencies of indefinite, low grade and high grade dysplasia were found in juvenile polyps from patients with either a SMAD4 or BMPR1A germline defect, contradicting earlier reports of a more dysplasia prone intestinal phenotype in polyps with a SMAD4 germline mutation.12,26 Interestingly, 50% of all foci of low or high grade dysplasia in juvenile polyps with a BMPR1A germline defect were found in type A polyps, whereas, none of the type A polyps with a SMAD4 germline defect contained dysplasia.
To investigate whether the neoplastic change in the juvenile polyps could be attributed to mutations in the conventional adenoma-carcinoma sequence, the dysplastic areas were investigated for APC and K-ras mutations. Our results revealed only 2 polyps with K-ras mutations, consequently, prior reports of APC mutations in dysplastic polyps 31 could not be confirmed. These data suggest that the conventional adenoma-carcinoma sequence may not play a distinct role in JPS tumour formation, as has been concluded by other investigators.32 Moreover, the different phenotypes could not be attributed to either an APC or K-ras mutation. Evaluation of the Ki67 proliferation marker demonstrated that focal loss of compartmentalisation of Ki67 i.e. expanded cell cycle activity, could not be linked to an A or B phenotype when stratified by dysplasia. This finding is consistent with the concept that loss of compartmentalisation of the proliferative zone is a general feature of dysplasia regardless of the underlying genetic defect.
Few prior studies have been dedicated to the evaluation of a relation in genetic make-up and histological phenotype in juvenile polyps. Handra-Luca et al analyzed a series of juvenile polyps for percentage and morphology of epithelial and stromal components, blood vessels, level of inflammation, hyperplasia and dysplasia.12 They characterized several distinctive features of polyps with a SMAD4 mutation: association with various grades of dysplasia, upper digestive tract location and malformative vessels in the stroma. BMPR1A polyps were exclusively of the lower digestive tract and were not associated with dysplasia or malformative vessels. As mentioned, our results showed no distinction with regard to presence and/or grade of dysplasia between juvenile polyps in the setting of a SMAD4 or BMPR1A germline mutation. Although presence of vascular malformation was evaluated in the initial screening it was not a discriminatory feature in our series of juvenile polyps (data not shown). No polyps of the upper digestive tract were present in our cohort.
Also, Aretz et al describe various histological phenotypes ranging from juvenile polyps to hyperplastic polyps and pseudopolyps albeit with adenomatous components or even adenomas in juvenile polyposis patients with an established germline mutation in one of the associated genes but no correlation between genotype and histological phenotype is provided.1 Nevertheless, our results confirm the wide array of histological phenotype variations encountered in the setting of juvenile polyposis.
SMAD4 and BMPR1A are both key components of the TGF-β/BMP signalling pathway maintaining homeostasis of the intestinal lining through processes of cellular proliferation (TGF-β) and differentiation and apoptosis (BMP). Signal transduction takes place through phosphorylation of the type 1 transmembrane receptor kinase (i.e. BMPR1A) by the type 2 receptor. The activated type 1 receptor phosphorylates the pathway restricted SMAD2 and 3 (TGF-β) or SMAD1,5 and 8 (BMP) which, in complex with the common mediator SMAD4, is translocated to the nucleus where target gene transcription is regulated.23
Individuals with germline defects in SMAD4 or BMPR1A and consequent disrupted TGF-β/BMP signalling develop multiple hamartomatous malformations in the gastrointestinal tract. These hamartomas are often characterized by an abnormal stromal component suggesting a prominent role for the stroma in polyp formation. The polyp epithelium initially shows normal maturation, although inflammation is common and may cause reactive changes. Subsequent dysplastic progression of the epithelium has been proposed to be the result of the altered microenvironment.21
Recent studies provide evidence that conditional inactivation of Bmpr2 in the intestinal mesenchyme leads to mice developing hamartoma-like polyps, whereas, conditional deletion of Bmpr1a in the epithelium showed elongation of the villi, but no de-novo crypt or polyp formation.2,3 Consistent loss of heterozygosity (LOH) of the BMPR1A locus has thus far not been detected in the epithelium or stroma of JPS polyps from patients with a BMPR1A germline mutation 16; although one study reports somatic loss of the 10q22 region exclusively in the lamina propria and not in the epithelium suggesting inactivation of BMPR1A might be a stromal event.18
Selective loss of Smad4-dependent signalling in T cells leads to a JPS-like phenotype reminiscent of what we described as a type A polyp with cystic spaces lined by columnar epithelium surrounded by abundant stroma.20 Smad4 heterozygous mice on the other hand develop polyps with an epithelial phenotype (type B) and show LOH specifically in the epithelium of larger polyps.27,32 Likewise, LOH of the SMAD4 locus occurs in the epithelium of juvenile polyps from patients with a SMAD4 germline mutation.22,29 The exact role of SMAD4 and timing of SMAD4 inactivation in polyp initiation and progression remains poorly understood but it seems that these polyps develop mainly through an epithelial defect.22,30 In addition to phenotype classification, SMAD4 immunohistochemistry may provide a specific marker for the detection of a SMAD4 germline mutation.22
Although the number of polyps in this study is limited, we propose that juvenile polyps with a SMAD4 germline defect have a higher crypt density regardless of the dysplastic status. On the contrary, juvenile polyps with a BMPR1A defect are more often classic juvenile polyps with a prominent stromal compartment. Crypt density in these polyps is initially low but may increase due to neoplastic change of the epithelium. Investigation of Ki67 immunohistochemistry reveals that the difference in crypt density in juvenile polyps with a SMAD4 or BMPR1A germline mutation is not a result of altered proliferative activity.
We conclude that juvenile polyps in the setting of juvenile polyposis syndrome may exhibit distinct phenotypes. Juvenile polyps with a SMAD4 germline mutation more likely express an epithelial phenotype with a relatively high crypt density, whereas, juvenile polyps with a BMPR1A mutation are usually the classic juvenile polyp phenotype with a prominent stromal compartment. Importantly, we find similar rates for all grades of dysplasia in juvenile polyps with either a SMAD4 or BMPR1A background.
Acknowledgments
Funding: Supported by The Netherlands Digestive Disease Foundation (MLDS WS 04-06), The John G. Rangos, Sr. Charitable Foundation, The Clayton Fund, and NIH grant P50 CA 93-16. The study sponsors were not involved in study design, collection, analysis, and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication.
Abbreviations
- JPS
juvenile polyposis syndrome
- LOH
loss of heterozygosity
- MCR
mutation cluster region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: No competing interests to declare.
REFERENCES
- 1.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44:702–709. doi: 10.1136/jmg.2007.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auclair BA, Benoit YD, Rivard N, et al. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology. 2007;133:887–896. doi: 10.1053/j.gastro.2007.06.066. [DOI] [PubMed] [Google Scholar]
- 3.Beppu H, Mwizerwa ON, Beppu Y, et al. Stromal inactivation of BMPRII leads to colorectal epithelial overgrowth and polyp formation. Oncogene. 2008;27:1063–1070. doi: 10.1038/sj.onc.1210720. [DOI] [PubMed] [Google Scholar]
- 4.Brosens LA, Iacobuzio-Donahue CA, Keller JJ, et al. Increased cyclooxygenase-2 expression in duodenal compared with colonic tissues in familial adenomatous polyposis and relationship to the -765G -> C COX-2 polymorphism. Clin Cancer Res. 2005;11:4090–4096. doi: 10.1158/1078-0432.CCR-04-2379. [DOI] [PubMed] [Google Scholar]
- 5.Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56:965–967. doi: 10.1136/gut.2006.116913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brosens LA, van Hattem WA, Kools MC, et al. No TGFBRII germline mutations in juvenile polyposis patients without SMAD4 or BMPR1A mutation. Gut. 2009;58:154–156. doi: 10.1136/gut.2008.161232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desai DC, Neale KF, Talbot IC, et al. Juvenile polyposis. Br J Surg. 1995;82:14–17. doi: 10.1002/bjs.1800820106. [DOI] [PubMed] [Google Scholar]
- 8.Fearon ER. Molecular genetic studies of the adenoma-carcinoma sequence. Adv Intern Med. 1994;39:123–147. [PubMed] [Google Scholar]
- 9.Giardiello FM, Hamilton SR, Kern SE, et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Arch Dis Child. 1991;66:971–975. doi: 10.1136/adc.66.8.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grotsky HW, Rickert RR, Smith WD, et al. Familial juvenile polyposis coli. A clinical and pathologic study of a large kindred. Gastroenterology. 1982;82:494–501. [PubMed] [Google Scholar]
- 11.Hamilton SR, Vogelstein B, Kudo S, et al. Carcinoma of the colon and rectum. In: Hamilton SR, Aaltonen LA, editors. Pathology and genetics of tumours of the digestive system. Lyon: IARC Press; 2000. pp. 101–119. [Google Scholar]
- 12.Handra-Luca A, Condroyer C, de Moncuit C, et al. Vessels' morphology in SMAD4 and BMPR1A-related juvenile polyposis. Am J Med Genet A. 2005;138:113–117. doi: 10.1002/ajmg.a.30897. [DOI] [PubMed] [Google Scholar]
- 13.Haramis AP, Begthel H, van den Born M, et al. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science. 2004;303:1684–1686. doi: 10.1126/science.1093587. [DOI] [PubMed] [Google Scholar]
- 14.He XC, Zhang J, Tong WG, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- 15.Hohenstein P, Molenaar L, Elsinga J, et al. Serrated adenomas and mixed polyposis caused by a splice acceptor deletion in the mouse Smad4 gene. Genes Chromosomes Cancer. 2003;36:273–282. doi: 10.1002/gcc.10169. [DOI] [PubMed] [Google Scholar]
- 16.Howe JR, Bair JL, Sayed MG, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 17.Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 18.Jacoby RF, Schlack S, Cole CE, et al. A juvenile polyposis tumor suppressor locus at 10q22 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 1997;112:1398–1403. doi: 10.1016/s0016-5085(97)70156-2. [DOI] [PubMed] [Google Scholar]
- 19.Jass JR, Williams CB, Bussey HJ, et al. Juvenile polyposis--a precancerous condition. Histopathology. 1988;13:619–630. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 20.Kim BG, Li C, Qiao W, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441:1015–1019. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- 21.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 22.Langeveld D, van Hattem WA, de Leng WW, et al. SMAD4 Immunohistochemistry Reflects Genetic Status in Juvenile Polyposis Syndrome. Clin Cancer Res. 2010;16:4126–4134. doi: 10.1158/1078-0432.CCR-10-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 24.Miyoshi Y, Nagase H, Ando H, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 25.Sachatello CR, Hahn IS, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: report of a case and review of the literature of a recently recognized syndrome. Surgery. 1974;75:107–114. [PubMed] [Google Scholar]
- 26.Sayed MG, Ahmed AF, Ringold JR, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol. 2002;9:901–906. doi: 10.1007/BF02557528. [DOI] [PubMed] [Google Scholar]
- 27.Takaku K, Miyoshi H, Matsunaga A, et al. Gastric and duodenal polyps in Smad4 (Dpc4) knockout mice. Cancer Res. 1999;59:6113–6117. [PubMed] [Google Scholar]
- 28.van Hattem WA, Brosens LA, de Leng WW, et al. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut. 2008;57:623–627. doi: 10.1136/gut.2007.142927. [DOI] [PubMed] [Google Scholar]
- 29.Woodford-Richens K, Williamson J, Bevan S, et al. Allelic loss at SMAD4 in polyps from juvenile polyposis patients and use of fluorescence in situ hybridization to demonstrate clonal origin of the epithelium. Cancer Res. 2000;60:2477–2482. [PubMed] [Google Scholar]
- 30.Woodford-Richens KL, Rowan AJ, Poulsom R, et al. Comprehensive analysis of SMAD4 mutations and protein expression in juvenile polyposis: evidence for a distinct genetic pathway and polyp morphology in SMAD4 mutation carriers. Am J Pathol. 2001;159:1293–1300. doi: 10.1016/S0002-9440(10)62516-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu TT, Rezai B, Rashid A, et al. Genetic alterations and epithelial dysplasia in juvenile polyposis syndrome and sporadic juvenile polyps. Am J Pathol. 1997;150:939–947. [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Brodie SG, Yang X, et al. Haploid loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and cancer in mice. Oncogene. 2000;19:1868–1874. doi: 10.1038/sj.onc.1203504. [DOI] [PubMed] [Google Scholar]