

Abstract
Packaged molecular machines are available in large amounts using dual expression vectors that guide the preparation of Qβ virus-like particles encapsulating multiple copies of functional enzymes. Packaging is promoted by RNA aptamer sequences that bridge between the coat protein and a peptide tag fused to the desired cargo. Peptidase E and luciferase were thus encapsulated and shown to be catalytically active inside the particle. The encapsulated enzymes are less sensitive to inactivation by heating and surface adsorption than the corresponding free enzymes. This system represents a modular way to marry catalytic activity with robust scaffolding for the development of multifunctional materials.
Keywords: virus-like particles, Qβ bacteriophage, enzyme catalysis, enzyme protection, RNA-protein interactions, encapsulation
The sequestration of functional units from the environment is a hallmark of biological organization. In addition to encapsulation within lipid membrane-bound organelles, proteinaceous cages serve this purpose for many prokaryotes.[1] From a chemical perspective, the outstanding advantages of such packages are their capabilities for high selectivity and activity, both achieved by encapsulating only those catalysts required for the desired task in confined space, and the potential for the container to control its position in a complex environment. Artificial encapsulation or immobilization on solid supports has been shown to confer stability as well as facilitate purification and reuse.[2] While chemists have sequestered enzymes in or on a wide variety of non-biological compartments, Nature remains the undisputed master of the art.
Protein nanoparticles represent a uniquely useful bridge between chemistry, materials science, and biology because they combine robust self-assembly properties with genetically-enabled atomic control of chemical reactivity. The synthetic biomimetic packaging of functional proteins has been accomplished with several different types of protein nanoparticles. Two general strategies have been employed: genetic fusion of the cargo to a component that directs localization to the particle interior,[3] and non-specific packaging by in vitro assembly.[4] However, yields of the encapsulated protein products have been low, and, while examples of increased stability towards a variety of treatments have been noted,[3b, 3e, 4b] no quantitative kinetic comparisons of enzymes in free vs. protein-encapsulated forms have been described. We report here the use of a virus-like particle for this purpose, providing a general and robust method for the encapsulation of highly active enzymes.
Bacteriophage Qβ form icosahedral virus-like particles (VLPs) from 180 copies of a 14.3 kD coat protein (CP).[5] These VLPs are highly stable under a variety of conditions and have been used to display functional small molecules,[6] immungenic ligands,[7] and peptides and proteins on their exterior surface.[8] The infectious phage particle packages its single-stranded RNA genome by virtue of a high-affinity interaction between a hairpin structure and interior-facing residues of the CP.[9] This interaction is preserved when the CP is expressed recombinantly to form VLPs[10] and we used this to direct the packaging of cargo materials (Figure 1). A related approach has been reported by Franzen and coworkers to entrain gold nanoparticles inside red clover necrotic mosaic virus.[11]
Figure 1.

Schematic representation of the technique used to package protein inside Qβ VLPs. Dual-plasmid transformation of E. coli with compatible T7 expression vectors is the only input into the system. IPTG induction results in the expression of capsid protein (CP), Rev-tagged cargo enzyme, and bifunctional mRNA. The Rev-tag binds to the α-Rev aptamer (apt) and Qβ genome packaging hairpin (hp) binds to the interior of the CP monomers, thus tethering the enzyme to the interior of the VLP with the Coat protein RNA sequence (cp) acting as the linker.
To facilitate RNA-directed encapsidation, two binding domains were introduced to the CP mRNA, carried on a ColE1-group plasmid. An RNA aptamer developed by in vitro selection to bind an arginine-rich peptide (Rev) derived from HIV-1[12] was inserted just upstream of the ribosome binding site. The sequence of the Qβ packaging hairpin was positioned immediately downstream of the stop codon. The cargo enzyme was N-terminally tagged with the Rev peptide and inserted into a compatible CloDF13-group plasmid. Transformation with both plasmids and expression in BL21(DE3) E. coli yielded VLPs encapsidating the Rev-tagged protein. Such species are designated Qβ@(protein)n, where n = the average number of proteins packaged per particle, determined by electrophoretic analysis as in Figure 2a and Supporting Figure S1a. We report here the packaging of the 25-kD N-terminal aspartate dipeptidase peptidase E (PepE)[13], 62-kD firefly luciferase (Luc), and a thermostable mutant of Luc (tsLuc)[14] inside VLPs.
Figure 2.

Characterization of Qβ@(RevPepE)18. (A) Electrophoretic analysis: lane M = protein ladder marker, 1 = E.coli cell lysate 4 h after induction, 2 = purified particles showing CP and Rev-pepE bands. (B) Transmission electron micrograph; images are indistinguishable from those of WT Qβ VLPs. (C) Size-exclusion FPLC (Superose 6) showing the intact nature of the particles.
The enzyme-filled VLPs were indistinguishable from standard VLPs by techniques that report on the exterior dimensions of the particles (transmission electron microscopy, size-exclusion chromatography, and dynamic light scattering; Fig. 2b,c Supporting Figures S2b,c and Table S3). However, the particles exhibited different densities by analytical ultracentrifugation: non-packed Qβ VLPs, 76S; Qβ@(RevLuc)4, 79S; and Qβ@(RevPepE)18, 86S (Fig. S2). These values agree with variations expected in overall molecular weights calculated from estimates of the RNA and protein content of each particle (Supporting Information).
The average number of encapsidated cargo proteins was controlled by changing expression conditions or by removing interaction elements from the plasmids (Table S2). In this way, PepE incorporation could be reproducibly varied between 2 and 18 per particle. Fewer copies of Luc proteins were packaged, with less variation in the number: 4–8 copies per particle were found for most conditions, whereas the number of packaged tsLuc molecules was varied between 2 and 11 per VLP. In addition to its larger size, Luc is less stable than PepE and its gene was not optimized for expression in E. coli (Supporting Fig. S1a, lane 1), all factors that could contribute to the lower numbers of packaged enzyme in this case. Yields of purified particles ranged from approximately 50–75 mg per liter of culture for the typical particles encapsidating PepE, and 75–140 mg per liter for the Luc or tsLuc particles (Table S2).
To test the functional capabilities of the packaged enzymes, the activities of encapsidated Rev-PepE and free PepE were compared using the fluorogenic substrate Asp-AMC[15] (Fig. 3). The kinetic parameters, obtained by standard Michaelis-Menten analysis, were found to be quite similar for the two forms of the enzyme, with kcat/Km for free PepE exceeding that of Qβ@(RevPepE)9 by a factor of only three (1.8±0.2 ×10−2 vs. 6.3±0.9 ×10−3). The observed Km values are comparable to those reported for cleavage of Asp-Leu (0.3 mM)[13]. For this analysis, all copies of encapsidated RevPepE in Qβ@(RevPepE)9 were assumed to be independently and equivalently active, and the substrate and product were assumed to diffuse freely in and out of the capsid. The close correspondence between the reactions of free and encapsidated enzyme appear to support these assumptions.
Figure 3.
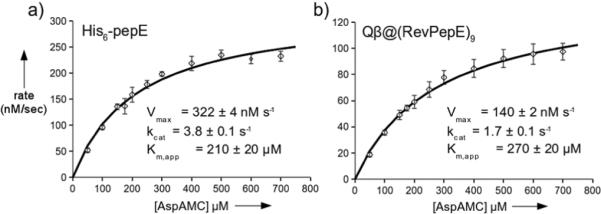
Kinetics of PepE-catalyzed hydrolysis of fluorogenic Asp-AMC. Squares show the average of three independent initial rate measurements (<4 min) with standard deviation as the error bars. Solid curves show the best fit using the Michaelis-Menten equation, giving the parameters shown.
Peptidase E was also significantly stabilized by encapsidation. Free PepE retained only half of its initial activity after incubation for 30 min at 45 °C and 20% of its activity at 50 °C (Fig. 4A). In contrast, Qβ@(RevPepE)9 showed no loss of activity at temperatures up to 50 °C for 30 min. Extended incubation at these temperatures showed the packaged enzyme was about 60 times more resistant than the free enzyme to thermal deactivation (Supporting Fig. S3). Heating did not disrupt the particle structure (Supporting Fig. S4), suggesting that at least partial denaturation of the packaged protein can occur inside the capsid shell. Packaged RevPepE was also protected from protease digestion, maintaining more than 80% activity under conditions in which the activity of the free enzyme was entirely degraded by proteinase K (Fig. 4B).
Figure 4.

Protection from thermal and protease inactivation of peptidase E by encapsidation. (A) Relative initial (<10 min) rates of substrate hydrolysis after incubation of the enzyme for 30 min at the indicated temperature followed by cooling to room temperature before assay. The rate exhibited by enzyme incubated at 4 °C was set at 100%. (B) Relative initial rates of substrate hydrolysis after incubation at specified time with proteinase K. Data is represented as a percentage of a buffer control at each time point. Points are averages of independent measurements in triplicate and error bars are the standard deviation.
The activity of Qβ@(RevLuc) was similarly compared to free recombinant firefly luciferase. In this case, packaging of the enzyme did not substantially change kcat, but Km in both luciferin and ATP substrates was significantly higher for the packaged enzyme (Table 1, Supporting Fig. S5). Luciferase is quite unstable toward thermal denaturation in both free and immobilized forms,[16] the free tsLuc variant having a half-life at 37 °C of only 16 minutes.[14] No improvement in thermal sensitivity was observed for Qβ@(Rev-tsLuc)9, but both packaged enzymes were protected from inactivation (presumably from adsorption) to unblocked polystyrene plates, to which the free enzyme was highly susceptible (Supporting Fig. S6).
Table 1.
Kinetic constants for free and packaged luciferase enzymes.
| Km,app (μM), luciferin | Km,app (μM), ATP | kcat (s−1) | |
|---|---|---|---|
| free luciferase | 7.9 ± 0.1 | 60 ± 10 | 38 ± 1.9 |
| Qβ@(RevLuc)4 | 140 ± 7 | 460 ± 30 | 22 ± 0.4 |
| Qβ@(RevtsLuc)2 | 77 ± 3 | 360 ± 20 | 35 ± 8 |
| Qβ@(RevtsLuc)9 | 171 ± 8 | 550 ± 30 | 20 ± 2 |
*Calculated from specific activity of luciferase (4.89 × 1010 light units/mg) and conversion to moles of pyrophosphate released.
Discussion
The increase in apparent Km for packaged Luc and tsLuc, but not PepE, may reflect a variety of factors. The encapsidated enzymes are apparently able to easily access small-molecule substrates and release products, presumably through the 20 large pores that exist at the quasi-six fold axes of the icosahedral capsid.[17] However, since it requires a ternary complex for catalysis (enzyme, luciferin, and ATP), luciferase may have a greater sensitivity to lower diffusion rates of the VLP-enzyme capsule. (The diffusion coefficient of icosahedral nanoparticles of this type and size is approximately 2 × 10−7 cm2 s−1,[18] 10-fold lower than that of the free enzymes.) Alternatively, the packaged enzyme may have less dynamic flexibility in a manner that affects its kinetic performance. Consistent with the latter hypothesis, we found superior kinetic parameters for particles containing two copies of RevtsLuc compared to particles containing nine copies (Table 1). Similar observations and issues have been described for luciferase immobilized to a variety of heterogeneous supports.[19]
These results represent the first examples of polynucleotide-mediated packaging of functional enzymes inside a protein shell, and the first kinetic comparisons between free and protein-encapsulated catalysts. While some differences were noted in kinetic parameters, the free and encapsidated enzymes exhibited very similar activities at saturation on a per-enzyme basis, showing that the enzyme-filled capsids can be highly potent catalytic engines.
The RNA-mediated packaging method combines the binding functions of two linked RNA aptamers, the first a natural hairpin sequence that engages in a strong association with the inside of the VLP, and the second an artificial aptamer selected by in vitro methods to bind to an oligopeptide tag fused to the desired cargo. The fact that the second of these aptamers works is especially significant, since it shows that the active conformation of the aptamer is accessible even when the sequence is coded into a larger piece of expressed and packaged messenger RNA.
This method of packaging enzymes inside protective protein shells has several attributes that distinguish it from existing technologies. First, since the entire packaging scheme is present within the host bacteria, the complete structure is assembled by the end of the expression. There is no need to purify separate elements and bring them together in vitro as in other systems.[3e, 4] These time-consuming steps are often low-yielding, requiring large amounts of starting material. Secondly, purification is largely independent of the packaged material, allowing the same efficient procedures to be used for a large range of packaged proteins. Thirdly, in contrast to most other co-expression systems,[3a–e] we use a scaffold that was evolved in E. coli, and expression in the native host provides high yields of pure VLP in a short amount of time. Finally, other systems have packaged functional enzymes[3a, 3b, 3d, 4b] and showed activity, but none have supplied kinetic analyses compared to the free enzyme. Such testing is critical for further development of therapeutically relevant targets.
The active nature of the encapsulated enzymes, and the ability of the capsid shell to stabilize them against thermal degradation, protease attack, and hydrophobic adsorption, shows that this method may be generally applicable to the production of fragile or difficult-to-purify enzymes. All production and assembly steps occur within the bacterial cell, with indirect control of amount of packaged cargo possible by simply changing the expression media or the nature of the components of the packaging system. VLPs are produced in high yields and are purified by a convenient standard procedure, independent of the protein packaged inside. This system therefore represent a unique method for the harnessing of enzymatic activity in a process-friendly fashion.
Supplementary Material
Acknowledgments
We are grateful to Dr. So-Hye Cho for assistance in acquiring TEM images, and to Dr. Joshua Price for analytical ultracentrifugation. The pRevTRE-Luc vector was a kind gift from Ms. Ashley Pratt. This project was supported by the National Institutes of Health (CA112075 and RR021886), the Skaggs Institute for Chemical Biology, and the W.M. Keck Foundation.
Footnotes
Dedicated to Prof. John E. Johnson in honor of his 65th birthday.
References
- [1].Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO. Science. 2005;309:936. doi: 10.1126/science.1113397. [DOI] [PubMed] [Google Scholar]
- [2].a) Tischer W, Wedekind F. Top. Curr. Chem. 1999;2000:95. [Google Scholar]; b) Hanefeld U, Gardossi L, Magner E. Chem. Soc. Rev. 2009;38:453. doi: 10.1039/b711564b. [DOI] [PubMed] [Google Scholar]
- [3].a) Beterams G, Bottcher B, Nassal M. FEBS Letters. 2000;481:169. doi: 10.1016/s0014-5793(00)01927-x. [DOI] [PubMed] [Google Scholar]; b) Kickhoefer VA, Garcia Y, Mikyas Y, Johansson E, Zhou JC, Raval-Fernandes S, Minoofar P, Zink JI, Dunn B, Stewart PL, Rome LH. Proc. Natl. Acad. Sci. USA. 2005;102:4348. doi: 10.1073/pnas.0500929102. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Seebeck FP, Woycechowsky KJ, Zhuang W, Rabe JP, Hilvert D. J. Am. Chem. Soc. 2006;128:4516. doi: 10.1021/ja058363s. [DOI] [PubMed] [Google Scholar]; d) Inoue T, Kawano MA, Takahashi RU, Tsukamoto H, Enomoto T, Imai T, Kataoka K, Handa H. Biotechnol. 2008;134:181. doi: 10.1016/j.jbiotec.2007.12.006. [DOI] [PubMed] [Google Scholar]; e) Goldsmith LE, Pupols M, Kickhoefer VA, Rome LH, Monbouquette HG. ACS Nano. 2009;3:3175. doi: 10.1021/nn900555d. [DOI] [PubMed] [Google Scholar]; f) Minten IJ, Hendriks LJA, Nolte RJM, Cornelissen JJLM. J. Am. Che. Soc. 2009;131:17771. doi: 10.1021/ja907843s. [DOI] [PubMed] [Google Scholar]
- [4].a) Lee KW, Tan WS. J. Virol. Methods. 2008;151:172. doi: 10.1016/j.jviromet.2008.05.025. [DOI] [PubMed] [Google Scholar]; b) Comellas-Aragones M, Engelkamp H, Claessen VI, Sommerdijk NAJM, Rowan AE, Christianen PCM, Maan JC, Verduin BJM, Cornelissen JJLM, Nolte RJM. Nat. Nanotech. 2007;2:635. doi: 10.1038/nnano.2007.299. [DOI] [PubMed] [Google Scholar]
- [5].a) Kozlovska TM, Cielens I, Dreilinņa D, Dislers A, Baumanis V, Ose V, Pumpens P. Gene. 1993;137:133. doi: 10.1016/0378-1119(93)90261-z. [DOI] [PubMed] [Google Scholar]; b) Golmohammadi R, Fridborg K, Bundule M, Valegård K, Liljas L. Structure. 1996;4:543. doi: 10.1016/s0969-2126(96)00060-3. [DOI] [PubMed] [Google Scholar]
- [6].Strable E, Finn MG. Curr.Top.Microbiol.Immunol. 2009;327:1. doi: 10.1007/978-3-540-69379-6_1. [DOI] [PubMed] [Google Scholar]
- [7].a) Kaltgrad E, Sen Gupta S, Punna S, Huang C-Y, Chang A, Wong C-H, Finn MG, Blixt O. ChemBioChem. 2007;8:1455. doi: 10.1002/cbic.200700225. [DOI] [PubMed] [Google Scholar]; b) Cornuz J, Zwahlen S, Jungi WF, Osterwalder J, Klingler K, van Melle G, Bangala Y, Guessous I, Muller P, Willers J, Maurer P, Bachmann MF, Cerny T. PLoS ONE. 2008;3:e2547. doi: 10.1371/journal.pone.0002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Vasiljeva I, Kozlovska T, Cielens I, Strelnikova A, Kazaks A, Ose V, Pumpens P. FEBS Lett. 1998;431:7. doi: 10.1016/s0014-5793(98)00716-9. [DOI] [PubMed] [Google Scholar]; b) Banerjee D, Liu AP, Voss NR, Schmid SL, Finn MG. ChemBioChem. 2010 doi: 10.1002/cbic.201000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Witherell GW, Uhlenbeck OC. Biochemistry. 1989;28:71. doi: 10.1021/bi00427a011. [DOI] [PubMed] [Google Scholar]
- [10].Weber H. Biochim. Bioophys. Acta. 1976;418:175. doi: 10.1016/0005-2787(76)90067-8. [DOI] [PubMed] [Google Scholar]
- [11].Loo L, Guenther RH, Lommel SA, Franzen S. J. Am. Chem. Soc. 2007;129:11111. doi: 10.1021/ja071896b. [DOI] [PubMed] [Google Scholar]
- [12].Xu W, Ellington AD. Proc. Natl. Acad. Sci. USA. 1996;93:7475. doi: 10.1073/pnas.93.15.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lassy RA, Miller CG. J. Bacter. 2000;182:2536. doi: 10.1128/jb.182.9.2536-2543.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].White PJ, Squirrell DJ, Arnaud P, Lowe CR, Murray JA. Biochem. J. 1996;319(Pt 2):343. doi: 10.1042/bj3190343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Larsen RA, Knox TM, Miller CG. J. Bacteriol. 2001;183:3089. doi: 10.1128/JB.183.10.3089-3097.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mononen IT, Kaartinen VM, Williams JC. Anal. Biochem. 1993;208:372. doi: 10.1006/abio.1993.1063. [DOI] [PubMed] [Google Scholar]
- [16].Wang CY, Hitz S, Andrade JD, Stewart RJ. Anal. Biochem. 1997;246:133. doi: 10.1006/abio.1997.2006. [DOI] [PubMed] [Google Scholar]
- [17].For an example in which the protein shell provides some barrier to the interactions of small molecule substrates with a packaged catalyst, see: Ueno T, Suzuki M, Goto T, Matsumoto T, Nagayama K, Watanabe Y. Angew. Chem. Int. Ed. 2004;43:2527–2530. doi: 10.1002/anie.200353436..
- [18].a) Neurath H, Cooper GR. J. Biol. Chem. 1940;135:455. [Google Scholar]; b) Bayer ME, Deblois RW. J. Virol. 1974;14:975. doi: 10.1128/jvi.14.4.975-980.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Lee Y, Jablonski I, DeLuca M. Anal. Biochem. 1977;80:496. doi: 10.1016/0003-2697(77)90672-8. [DOI] [PubMed] [Google Scholar]; b) Blum LJ, Coulet PR. Biotechnol. Bioeng. 1986;28:1154. doi: 10.1002/bit.260280804. [DOI] [PubMed] [Google Scholar]; c) Eu J, Andrade J. Luminescence. 2001;16:57. doi: 10.1002/bio.612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.