

Abstract
First-order rate constants, determined by 1H NMR, are reported for deuterium exchange between solvent D2O and the α-amino carbon of glycine in the presence of increasing concentrations of carbonyl compounds (acetone, benzaldehyde and salicylaldehyde) and at different pD and buffer concentrations. These rate data were combined with 1H NMR data that define the position of the equilibrium for formation of imines/iminium ions from addition of glycine to the respective carbonyl compounds, to give second-order rate constants kDO for deprotonation of α-imino carbon by DO−. The assumption that these second-order rate constants lie on linear structure-reactivity correlations between log kOL and pKa was made in estimating the following pKas for deprotonation of α-imino carbon: pKa = 22, glycine–acetone iminium ion; pKa = 27, glycine–benzaldehyde imine; pKa ≈ 23, glycine–benzaldehyde iminium ion; and, pKa = 25, glycine–salicylaldehyde iminium ion. The much lower pKa of 17 [Toth, K.; Richard, J. P. J. Am. Chem. Soc. 2007, 129, 3013–3021] for carbon deprotonation of the adduct between 5′-deoxypyridoxal (DPL) and glycine shows that the strongly electron-withdrawing pyridinium ion is unique in driving the extended delocalization of negative charge from the α-iminium to the α-pyridinium carbon. This favors carbanion protonation at the α–pyridinium carbon, and catalysis of the 1,3-aza-allylic isomerization reaction that is a step in enzyme-catalyzed transamination reactions. An analysis of the effect of incremental changes in structure on the activity of benzaldehyde in catalysis of deprotonation of glycine shows the carbonyl group electrophile, the 2-O− ring substituent and the cation pyridinium nitrogen of DPL each make a significant contribution to the catalytic activity of this cofactor analog. The extraordinary activity of DPL in catalysis of deprotonation of α–amino carbon results from the summation of these three smaller effects.
Introduction
We have worked over the last 15 years to characterize kinetic and thermodynamic barriers for deprotonation of the α-amino carbon of amino acids1,2 (Chart 1) and peptides3 in water and to understand the mechanism by which enzymes lower these barriers in catalysis of deprotonation of amino acids.2,4 Natural selection of small molecule catalysts has seen the evolution of pyridoxal 5′-phosphate as a cofactor for an enormous range of enzymes 5–9 that catalyze deprotonation of α-amino acid carbon as the first step of more complex reactions. These enzymatic reactions include, racemization 10–12 and decarboxylation 13–18 of the amino acid, transamination to form an α–keto acid and pyridoxamine 5′-phosphate,19–22 replacement of good nucleofuges at the β-amino position through elimination of the leaving group from a pyridoxal-stabilized carbanion to form an activated alkene, which undergoes addition of a second nucleophile,23,24 and aldol type addition of the pyridoxal stabilized carbanion to formaldehyde 25 or acetaldehyde.26
Chart 1.

Our earliest studies on catalysis of deprotonation of amino acids focused on determining the effect of alkylation of the amino nitrogen and the carboxylate oxygen on the carbon acidity of glycine (Chart 1).1,2 We next examined electrophilic catalysis of deprotonation of glycine methyl ester by the simple ketone acetone (1, Chart 1)27,28 and of glycine by the pyruvoyl analog phenylglyoxylate (PG)28 through the corresponding α-iminium ion reaction intermediates (e.g., 1-IMH). Finally, we have examined catalysis of deprotonation of glycine by 5′-deoxypyridoxal (DPL, Chart 2) over a broad range of pH.29
Chart 2.

The effective catalysis of deprotonation of the α-amino carbon of glycine by acetone28 shows that the carbonyl group of PLP is the underlying source of the catalytic power of PLP. This carbonyl group catalysis is assisted by other functionality at the cofactor. Most importantly, by the pyridinium nitrogen and the 2-O− substituent at the pyridine ring. There has been speculation about the role of these substituents in catalysis of deprotonation of α-amino carbon,30 but these roles have not been rigorously evaluated. An understanding of these substituent effects will provide assistance to the development of effective small molecule catalysts of these reactions,31 and insight into the mechanism of action of enzymes that utilize PLP as a cofactor.9
The 5′-phosphate of PLP should not strongly activate the cofactor for deprotonation of α-amino acids. The most likely role for this functional group is to provide binding energy for stabilization of the Michaelis complex and the transition states for reactions in which PLP serves as a cofactor.32–34 The α-amino acid carbanion should be stabilized by delocalization of negative charge onto the pyridinium nitrogen of DPL-IMH2, compared to delocalization onto the neutral pyridine nitrogen of DPL-IMH or onto the phenyl ring of 3-IMH. However, the amino acid carbanion is already highly delocalized at 3-IMH, and this should cause an attenuation of the effect of the cationic nitrogen at the 4-position of the pyridine ring on the stability of the carbanion.35 We report here the results of experiments that allow a comparison of the activities of DPL and salicylaldehyde (3) as catalysts of deprotonation of glycine.
The 2-O− substituent at DPL and salicylaldehyde activates aldehydes for catalysis by stabilization of the iminium ion glycine adducts DPL-IMH2 and 3-IMH by an intramolecular hydrogen bond to the iminium nitrogen.36 This effect is probably attenuated, because partial proton transfer from the cationic nitrogen to oxygen will cause the charge at this nitrogen and the resulting electrostatic stabilization of the α-amino acid carbanion to decrease. We report here the results of a study of deprotonation of glycine catalyzed by 2 and by 3 that defines the contribution of this 2-O− substituent to catalysis of deprotonation of glycine by DPL.
2. Experimental Section
2.1 Materials
Glycine and salicylaldehyde were purchased from Fluka. Benzaldehyde, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), 2,2,2-trifluoroethanol (TFE), deuterium chloride (37 wt %, 99.5% D), potassium deuterioxide (40 wt %, 98 + % D), deuterium oxide (99.9% D), and acetone-d6 (99.9 atom % D) were from Aldrich. All other organic and inorganic chemicals were reagent grade and were used without further purification.
2.2. General Methods
The acidic protons of glycine, K2HPO4 and KH2PO4, were exchanged for deuterium before preparation of solutions of these compounds in D2O.2 Salicylaldehyde, HFIP and TFE were dissolved directly in D2O, which introduced < 1 atom % of protium into this solvent. Phosphate buffers were prepared by mixing stock solutions of K2DPO4 and KD2PO4 in D2O at I = 1.0 (KCl) to give the desired acid/base ratio. Pyrophosphate and carbonate buffers were prepared by dissolving a measured amount of KCl and the basic form of the buffer in D2O, and adding DCl to give the desired acid/base ratio at I = 1.0 (KCl). Buffers of HFIP and TFE were prepared by dissolving a measured amount of KCl and adding KOD to give the desired acid/base ratio at I = 1.0 (KCl).
Solution pD was determined at 25 °C using an Orion model 350 pH meter equipped with a Radiometer pHC4006-9 electrode. Values of pD were obtained by adding 0.4 to the observed reading of the pH meter.37 The concentration of deuterioxide ion at any pD was calculated using eq 1, where Kw = 10−14.87 is the ion product of deuterium oxide at 25 °C and γOL = 0.78 is the apparent activity coefficient of lyoxide ion under our experimental conditions.38
| (1) |
2.3. 1H NMR Analyses
1H NMR spectra at 500 MHz were recorded in D2O at 25 °C on a Bruker AMX500 NMR spectrometer as described previously.38,39 In all cases, the relaxation delay between pulses was at least 10-fold greater than the longest relaxation time of the protons of interest. Spectra were obtained with a sweep width of 2600 Hz, a 90° pulse angle and an acquisition time of 6 s. Baselines were corrected for drift before integration of the peaks. Chemical shifts are reported relative to (CH3)4N+ at 2.94 ppm.
2.4. Determination of Equilibrium Constants
The position of the equilibrium for addition of glycine to acetone to form the corresponding imine in D2O at 25 °C and I = 1.0 (KCl) was determined by 1H NMR spectroscopy as described in previous work.28,36 Formation of the imine was monitored in solutions that contained 0.1 M glycine and 1.0 M acetone at I = 1.0 (KCl). The pD was maintained by use of 0.10 M of the following buffers: phosphate, pD 6.1 – 8.0; pyrophosphate, pD 8.6 – 9.5; carbonate, pD 10.0 – 11.1 and TFE, pD 12.0 – 13.0.
Values of the observed equilibrium constant (Kadd)obsd (M−1) for imine formation were determined from the ratio of the integrated areas of the peaks for the methylene protons of the imine product ( ), and of the reactant glycine ( ) according to eq 2, where [1-IM]e and [Gly]e are the total equilibrium concentrations of imine and glycine, respectively. The ratio of equilibrium concentrations of imine adduct and amino acid was determined 1–2 hours after mixing the reactants except at pD > 8, where it was determined after 24 hours in order to ensure that chemical equilibrium had been reached. Acetone-catalyzed deuterium incorporation into glycine also took place during this time period at pD 13. However, 1H NMR analysis showed the same deuterium enrichment of the α–CH2 groups of glycine and of the imine within experimental error. Therefore, the ratio of the equilibrium concentrations of imine adduct and glycine was determined as the ratio of the sum of peak areas for the -CH2- and -CHD- groups.
| (2) |
2.5. Kinetic Measurements
All reactions were carried out in D2O at 25 °C and I = 1.0 (KCl). The deuterium exchange reactions of glycine in the presence of acetone or 3 were initiated by mixing solutions of glycine, the catalyst and the buffer at the same pD and ionic strength (I = 1.0 (KCl)). The final concentration of the amino acid ranged from 5 to 15 mM. The deuterium exchange reactions of glycine in the presence of 2, in alkaline solutions of D2O, were initiated by mixing solutions of glycine, 2 and KOD at the same ionic strength (I = 1.0 (KCl)) to give a final glycine concentration of 5 mM.
The exchange for deuterium of the first α-proton of glycine was followed by monitoring the disappearance of the singlet due to the α-CH2 group of the substrate and the appearance of the upfield-shifted triplet due to the α-CHD group of the monodeuterated product by 1H NMR spectroscopy at 500 MHz, as described in previous work.2,27,28 Values of R, which is a measure of the progress of the deuterium exchange reaction, were calculated using eq 3, where ACH2 and ACHD are the integrated areas of the peaks for the α-CH2 and the α-CHD groups, respectively. The reactions in the presence of acetone, 2 and 3 were followed during exchange for deuterium of 20–30%, 40–90%, and 60–90%, respectively, of the first proton of the α-CH2 group of the substrate. In all cases an equilibrium mixture of glycine and the corresponding imine was observed before the formation of a significant amount of deuterium labelled glycine. Semilogarithmic plots of reaction progress, R, against time according to eq 4 were linear with negative slopes equal to kobsd (s−1), which is the rate constant for exchange of a single proton of the α-CH2 group of the substrate. The values of the first-order rate constant for exchange of the first α-CH2 proton to give the monodeuterated product, kex (s−1), were determined as kex = 2 kobsd.
| (3) |
| (4) |
3. Results
3.1. Equilibrium Constants for Imine Formation
Figure 1 shows the change with changing pD in the observed chemical shift of the signal for the α–CH2 hydrogens of the mixture of 1-IM and 1-IM-D (Scheme 1) which forms in D2O from the reaction of 0.1 M glycine and 1.0 M acetone at 25 °C and I = 1.0 (KCl). The change in the chemical shift of the α–methylene group is due to the change in the protonation state of the imine nitrogen. The solid line in Figure 1 shows the nonlinear least squares fit of the data to eq 5, derived for Scheme 1, where (a) and are the chemical shifts for the α–CH2 group observed at limiting low and high pD, and (b) the value of (Ka)1-IM-D = 10−9.64 is determined by treating this acidity constant as a variable parameter.
Figure 1.

The dependence on pD of (δCH2)obsd (ppm) for the -CH2- hydrogen of the imine formed in D2O that contains 0.1 M glycine and 1 M acetone at 25 °C and I = 1.0 (KCl). The solid line shows the fit of the data to eq 5 derived for Scheme 1.
Scheme 1.

| (5) |
Values of the apparent equilibrium constants (Kadd)obsd (M−1) for addition of glycine to acetone to form the corresponding imine in D2O at 25 °C were determined by 1H NMR analysis, as described in the Experimental Section. Figure 2 shows the change, with changing pD, in log (Kadd)obsd for addition of glycine to acetone. The line shows the fit of the data to eq 6, derived for Scheme 1, using (Ka)GlyD = 3.5 × 10−11 M, (Ka)1-IM-D = 2.3 × 10−10 M (Table 1) and (Kadd)1-IM-D = 0.0056 M−1 determined for the formation of the iminium ion at pD < 8.
Figure 2.

Logarithmic dependence of (Kadd)obsd (M−1) on pD for conversion of glycine to the acetone imine in D2O at 25 °C and I = 1.0 (KCl). The solid line was calculated using eq 6 and the equilibrium constants from Table 1.
Table 1.
Equilibrium Constants in D2O for Addition of Glycine to Acetone.a
| Amino compound | Imine | p(Ka)Glyb | p(Ka)X-IM-Dc | Kaddd (M−1) |
|---|---|---|---|---|
| +D3 NCH2 COO− | 1-IMD | 10.44 ± 0.03 | 9.64 ± 0.03 | (5.6 ± 0.1) × 10−3 |
| D2 NCH2 COO− | 1-IM | (3.5 ± 0.5) × 10−2 |
At 25 °C and I = 1.0 (KCl).
The acidity constant of the amino acid in D2O at 25 °C and I = 1.0 (KCl) determined by 1H NMR titration.
Apparent acidity constant in D2O at 25 °C and I = 1.0 (KCl), determined by NMR titration as described in the text.
Equilibrium constant for the addition of the amino acid to acetone to form the imine adduct.
| (6) |
3.2. Deuterium Exchange Reactions
3.2.1. Catalysis by Acetone
The exchange for deuterium of the first α-proton of glycine in D2O at 25 °C and I = 1.0 (KCl) was monitored by 1H NMR spectroscopy (500 MHz) as described previously.2,28 Table S1 of the Supporting Information gives the observed first-order rate constants kex (s−1) for deuterium exchange in the presence of various concentrations of acetone and HFIP buffer at pD 9.3, 9.9 and 10.5, determined as described in the Experimental Section. Figure 3 shows the effect of increasing concentrations of HFIP buffer (50% free base) on kex (s−1) for reactions at different fixed concentrations of acetone. The intercepts of these linear correlations are the first-order rate constants ko = kw f1-IM-D (s−1, Table 2) for deuterium exchange catalyzed by the solvent at the given concentration of acetone, where f1-IM-D is the fraction of glycine present as the iminium ion adduct 1-IM-D (eq 7, derived for Scheme 1)40 and kw is the apparent first-order rate constant for the solvent reaction. Table 2 reports values of kw, calculated as kw = ko/f1-IM-D, for reactions at different concentrations of acetone.
Figure 3.

Dependence of kex (s−1) for exchange of the first α-proton of glycine in the presence of acetone on the total concentration of HFIP buffer at pD 9.98 ([RO−]/[ROH] = 1.0) in D2O at 25 °C and I = 1.0 (KCl). Key: (●) 0.10 M acetone; (■) 0.25 M; (┤) 0.50 M.
Table 2.
Rate Constants for Exchange of the First α-Proton of Glycine in the Presence of Acetone and HFIP Buffers in D2Oa
| Base | fBc | pD | [acetone]/M | f1-IM-Dd | ko/s−1e | kw/s−1f | (kbuf)obsd/M−1s−1g | kB/M−1s−1h |
|---|---|---|---|---|---|---|---|---|
| HFIP | 0.8 | 10.48 | 0.25 | 6.8 × 10−4 | 1.5 × 10−7 | 2.2 × 10−4 | 1.3 × 10−7 | 2.4 × 10−4 |
| pKBD = 9.9b | ||||||||
| 0.5 | 9.87 | 0.10 | 4.4 × 10−4 | 2.3 × 10−8 | 5.1 × 10−5 | 6.6 × 10−8 | 3.0 × 10−4 | |
| 9.94 | 0.25 | 1.1 × 10−3 | 7.0 × 10−8 | 6.5 × 10−5 | 1.2 × 10−7 | 2.3 × 10−4 | ||
| 9.98 | 0.50 | 2.1 × 10−3 | 1.5 × 10−7 | 7.1 × 10−5 | 2.8 × 10−7 | 2.6 × 10−4 | ||
| 0.2 | 9.27 | 0.25 | 1.3 × 10−3 | 2.1 × 10−8 | 1.6 × 10−5 | 5.8 × 10−8 | 2.2 × 10−4 | |
At 25 °C and I = 1.0 (KCl).
Apparent pKa of the conjugate acid in D2O at 25 °C and I = 1.0 (KCl).
Fraction of the buffer present in the basic form.
Fraction of glycine present as the reactive iminium ion 1-IM-D, calculated from eq 7 using the values of the equilibrium constants reported in Table 1.
The intercept of plots of kex against [buffer]T (Figure 3).
Apparent first-order rate constants for solvent-catalyzed deprotonation of 1-IM-D, calculated from the values of ko as described in the text.
The slope of plots of kex against [buffer]T (Figure 3).
Second-order rate constants for HFIP base-catalyzed deprotonation of 1-IM-D, calculated from the values of (kbuf)obsd as described in the text.
| (7) |
A similar analysis of data for reactions in 20 and 80% free base HFIP buffers (Table S1) gave kw = 1.6 × 10−5 s−1 and kw = 2.2 × 10−4 s−1 for deprotonation of the iminium ion at pD 9.27 and 10.48, respectively. Figure 4 (▼) shows the pD-rate profile of log kw for deprotonation of 1-IM-D in D2O. The solid line of unit slope shows the least-squares fit of the data to eq 8 using (kDO)1-IM-D = 4.2 M−1s−1 for deprotonation of 1-IM-D by DO− (Scheme 1), where Kw = 10−14.87 M2 is the ion product of D2O at 25 °C and γOL = 0.78 is the apparent activity coefficient of lyoxide ion under our experimental conditions.
Figure 4.

pD-rate profiles of kw (s−1) for exchange for deuterium of the first α–CH2 hydrogen of: (▼), 1-IM-D; (┤), 2-IM; (●), 3-IM-D; (▽), N-protonated glycine;2 (□) N-protonated glycine methyl ester;2 (■), the acetone-glycine methyl ester iminium ion.27,28
| (8) |
The slopes of the linear correlations in Figure 3 are equal to (kbuf)obsd = (kB)1-IM-D fB f1-IM-D for the deuterium exchange reaction catalyzed by buffer at the given concentration of acetone, where (kB)1-IM-D (M−1s−1) is the second-order rate constant for deprotonation of 1-IM-D by the buffer base (Scheme 1) and fB is the fraction of the catalyst in the basic form. The values of kB reported in Table 2 were calculated from (kbuf)obsd using the appropriate values of fB and f1-IM-D. A value of (kB)1-IM-D = 2.4 × 10−4 M−1s−1 for deprotonation of 1-IM-D by the HFIP base (Scheme 1) was calculated as the average of the values of kB determined at different fractions of buffer base.
3.2.2. Catalysis by Benzaldehyde (2)
The exchange for deuterium of the first α-proton of glycine in the presence of 2 and various concentrations of deuterioxide ion in D2O at 25 °C and I = 1.0 (KCl) was followed by 1H NMR (500 MHz). Table S2 gives values of the first-order rate constant kex (s−1) for the deuterium exchange reaction together with values of f2-IM, the fraction of amino acid converted to the imine (Scheme 2) under the experimental reaction conditions, determined by 1H NMR analysis of the mixture of glycine and 2 in D2O, as described in previous work.36 The rate constants for solvent-catalyzed deprotonation of the imine adduct, kw = kex/f2-IM, increase linearly with the concentration of deuterioxide ion (not shown). The slope of this correlation gives (kDO)2-IM = 2.90 × 10−4 M−1s−1 as the second-order rate constant for deprotonation of 2-IM catalyzed by deuterioxide ion. Figure 4 (┤) shows the pD-rate profile of log kw for deprotonation of 2-IM in D2O. The solid line of unit slope shows the fit of the data to eq 8 ((kDO)2-IM = 2.90 × 10−4 M−1s−1). It was not possible to determine a value of the second-order rate constant for deprotonation of 2-IM-D by DO− because the deuterium exchange reaction is too slow to monitor at pD < 11, where the concentration of 2-IM-D would begin to become significant.
Scheme 2.

3.2.3. Catalysis by Salicylaldehyde (3)
The exchange for deuterium of the first α-proton of glycine was examined in D2O at 25 °C and I = 1.0 (KCl) in solutions buffered with HFIP (pD 9.3–10.5). Table S3 of the Supporting Information gives the observed first-order rate constants kex (s−1) for deuterium exchange in the presence of various concentrations of 3 and HFIP buffer at pD 9.3, 9.8 and 10.5, determined as described in the Experimental Section. Table S3 also reports values of f3-IM-D, the fraction of total glycine that is converted to the reactive iminium ion adduct (Scheme 3) at each concentration of 3 at the given pD, determined by 1H NMR analysis of the mixture of glycine and 3, as described in previous work.36 Figure 5 shows the linear dependence of kex on the total concentration of HFIP buffer (pD 9.8) for reactions at two fixed concentrations of 3. The intercepts of these linear plots are the first-order rate constants ko = kw f3-IM-D (s−1, Table 3) for solvent-catalyzed exchange at the given pD, where kw is the apparent first-order rate constant for the solvent reaction and f3-IM-D is the fraction of glycine present as the iminium ion 3-IM-D (Table S3). Table 3 reports values of kw, calculated as kw = ko/f3-IM-D, for reactions at two different concentrations of 3. The same treatment of the data for reactions at pD 10.5 and 9.3 (Table 3) gives kw = 1.1 × 10−5 s−1 and kw = 7.8 × 10−7 s−1, respectively. The slope of a linear plot (not shown) of kw against [DO−] gives the second-order rate constant (kDO)3-IM-D = 0.19 M−1s−1 for deprotonation of 3-IMD by deuterioxide ion. Figure 4 (●) shows the pD-rate profile of log kw for deprotonation of 3-IMD in D2O. The solid line of unit slope through the data was calculated from the value of kDO using eq 8 and (kDO)3-IM-D = 0.19 M−1s−1.
Scheme 3.

Figure 5.

Dependence of kex (s−1) for exchange for deuterium of the first α-proton of glycine in the presence of salicyaldehyde on the total concentration of HFIP buffer at pD 9.8 in D2O at 25 °C and I = 1.0 (KCl). Key: (●) 10 mM 3; (○) 52 mM 3.
Table 3.
Rate Constants for Exchange of the First α-Proton of Glycine in the Presence of 3 and HFIP Buffers in D2Oa
| Base | fBc | pD | [aldehyde]/M | f3-IM-Dd | ko/s−1e | kw/s−1f | (kbuf)obsd/M−1s−1g | kB/M−1s−1h |
|---|---|---|---|---|---|---|---|---|
| HFIP | 0.8 | 10.53 | 6.0 × 10−3 | 0.15 | 1.7 × 10−6 | 1.1 × 10−5 | 3.1 × 10−6 | 2.6 × 10−5 |
| 10.52 | 2.0 × 10−2 | 0.39 | 4.4 × 10−6 | 1.1 × 10−5 | 8.8 × 10−6 | 2.8 × 10−5 | ||
| pKBD = 9.9b | ||||||||
| 0.5 | 9.80 | 1.0 × 10−2 | 0.32 | 6.9 × 10−7 | 2.2 × 10−6 | 4.2 × 10−6 | 2.6 × 10−5 | |
| 9.86 | 5.2 × 10−2 | 0.73 | 1.8 × 10−6 | 2.5 × 10−6 | 1.2 × 10−5 | 3.3 × 10−5 | ||
| 0.2 | 9.35 | 2.0 × 10−2 | 0.45 | 3.5 × 10−7 | 7.8 × 10−7 | 2.9 × 10−6 | 3.2 × 10−5 | |
At 25 °C and I = 1.0 (KCl).
Apparent pKa of the conjugate acid in D2O at 25 °C and I = 1.0 (KCl).
Fraction of the buffer present in the basic form.
Fraction of glycine present as the reactive iminium ion 3-IM-D at the given concentration of 3, determined by 1H NMR analysis of an equilibrium mixture of glycine and the imine.
The intercept of plots of kex against [buffer]T (Figure 5).
Apparent first-order rate constants for solvent-catalyzed deprotonation of 3-IM-D, calculated from the values of ko as described in the text.
The slope of plots of kex against [buffer]T (Figure 5).
Second-order rate constants for HFIP base-catalyzed deprotonation of 3-IM-D, calculated from the values of (kbuf)obsd as described in the text.
The slopes of the linear correlations in Figure 5 are apparent second-order rate constants, (kbuf)obsd = (kB)3-IM-D fB f3-IM-D for the buffer-catalyzed deuterium exchange reaction at the given concentration of 3, where (kB)3-IM-D (M−1s−1) is the second-order rate constant for deprotonation of 3-IM-D by the buffer base and fB is the fraction of the catalyst in the basic form. Table 3 reports values of kB that were calculated from (kbuf)obsd using the appropriate values of fB and f3-IM-D. A value of (kB)3-IM-D = 2.9 × 10−5 M−1s−1 for deprotonation of 3-IM-D by the HFIP base was calculated as the average of the values of kB determined at different fractions of buffer base.
4. DISCUSSION
Acetone (1), salicylaldehyde (3) and benzaldehyde (2) are effective catalysts of the deprotonation of the α–amino carbon of glycine in D2O. This reaction leads to exchange of the α–hydrogen for deuterium from solvent, which is monitored by 1H NMR. The observation that the deuterium exchange reactions are first-order in the concentration of the electrophilic catalyst and deuterioxide ion shows that they proceed by formation of imine or iminium ion adducts to glycine, which undergo deprotonation by deuterioxide ion to form enolates (Schemes 1, 2 and 3). Table 4 reports equilibrium constants Kadd for formation of adducts between glycine and 1, 2 or 3; and, second-order rate constants kDO for deprotonation of these adducts. Second-order rate constants kHO = 2.9 M−1s−1, 0.13 M−1s−1 and 2.0 × 10−4 M−1 s−1 (Table 5) for deprotonation of 1-IMH, 3-IMH and 2-IM respectively, by hydroxide ion in H2O were calculated from the experimental values of kDO and an estimated secondary solvent deuterium isotope effect of kDO/kHO = 1.46.41 Table 5 summarizes the values of kHO for deprotonation of glycine and derivatives of glycine reported here and in earlier work.2,28,29
Table 4.
Equilibrium Constants in D2O for Addition of Glycine to Carbonyl Compounds, and Second-Order Rate Constants kDO for Carbon Deprotonation of the Imine or Iminium Ion Adduct.a
| Aldehyde | Carbon Acid | Kaddb (M−1) | (Kadd)+c (M−1) | kDOd (M−1 s−1) |
|---|---|---|---|---|
 |
 |
3.5 × 10−2 | 5.6 × 10−3 | 4.2 |
 |
 |
44e | 0.0033e | 2.90 × 10−4 |
 |
 |
1.76e | 72e | 0.19 |
At 25 °C and I = 1.0 (KCl).
Equilibrium constant for addition of the basic amino form of glycine to the carbonyl compound.
Equilibrium constant for addition of glycine zwitterion to the carbonyl compound.
Second-order rate constant for carbon deprotonation of the iminium ion adduct by deuterioxide ion in D2O.
Data from ref 36.
Table 5.
Rate and Equilibrium Constants for Carbon Deprotonation of Glycine and Glycine Derivatives in Water at 25 °C and I = 1.0 (KCl)
| Carbon Acid | kHOa (M−1s−1) | pKab | Carbon Acid | kHOa (M−1s−1) | pKab |
|---|---|---|---|---|---|
 |
4.5 × 10−5 c | 28.9c | 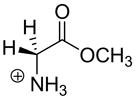 |
4.1c | 21.0c |
 |
3.3 × 10−4 c | 27.3c | 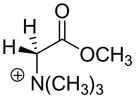 |
390c | 18.0c |
 |
2.9 | 21.9 | 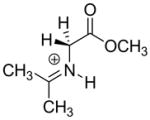 |
9.0 × 103 d | 14d |
 |
2.0 × 10−4 | 27 |  |
ca. 1 | ca. 23 |
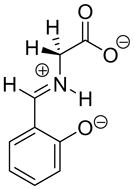 |
0.13 | 25 |  |
1.1 × 104 d | 14d |
 |
not determined | ca. 23 e |  |
7.5 × 102 f | 17f |
Second-order rate constant for deprotonation of the carbon acid by hydroxide ion calculated from data reported in this work, unless noted otherwise.
pKa for ionization of the carbon acid in water calculated from data reported in this work, unless noted otherwise.
Data from ref 2.
Data from ref 28.
Estimated as described in the text.
Data from ref 29.
4.1. Carbon Acid pKa’s
Figure 6 (■) shows the linear relationship, with slope 0.44, between the statistically corrected values of log kHO for deprotonation of cationic ketones and esters, and the pKa of the carbon acid.2 The carbon acid pKas for 1-IMH and 3-IMH reported in Table 5 were estimated by assuming that the values of log kOH for these carbon acids lie on the linear correlation for other cationic ketones and esters. Figure 6 (●) also shows the linear correlation between the statistically corrected values of log kHO for deprotonation of neutral monocarbonyl carbon acids and the carbon acid pKa.38,39,42–44 A pKa of 27 (Table 5) for the ionization of 2-IM was calculated with the assumption that the value of log kHO for this carbon acid lies on this linear correlation.
Figure 6.

Rate-equilibrium correlations of rate constants, kHO (M−1 s−1), for deprotonation of α-carbonyl carbon acids by hydroxide ion with the pKa of the carbon acid. The values of kHO and pKa were statistically corrected for the number of acidic protons p at the carbon acid. Correlation of log(kHO/p) for deprotonation of neutral aldehydes, ketones, esters, and acetamide by hydroxide ion, constructed using data from earlier work (●). 38,39,42–44 Excluding the point for acetate anion (pKa = 33.5), the data are correlated by log(kHO/p) = 6.496 − 0.401(pKa + log p). Correlation of log(kHO/p) for deprotonation of cationic ketones and esters (■).2 The data are correlated by log(kHO/p) = 10.044 − 0.444(pKa + log p). Data for deprotonation of the acetone-glycine methyl ester iminium ion (□).27 Data for deprotonation of 1-IM-H and 3-IM-H (▵). Data for deprotonation of 2-IM (○).
The large equilibrium constant of Kadd = 44 M−1 for formation of the Schiff base 2-IM from 2 and glycine facilitates detection of the 2-catalyzed deuterium exchange reaction of glycine at high pD (Figure 4). It was not possible to detect the 2-catalyzed deuterium exchange reaction of glycine at low pD, where 2-IMD is the reacting carbon acid, because (Kadd)+ = 0.0033 M−1 for formation of 2-IMD is very small (Table 4) and the low solubility of benzaldehyde in water prevents experiments with concentrations of aldehyde larger than 0.02 M.
We have estimated kHO for carbon-deprotonation of 2-IMH by making the assumption that N-protonation of glycine and of 2-IM will cause similar changes in the thermodynamic and kinetic acidity of the α–amino and α–imino carbon. The α-NH3+ substituent causes a 4.6 unit decrease in carbon acid pKa of acetate anion from 33.5 to 28.9 for glycine zwitterion 2,42 and of ethyl acetate from 25.6 to 21.0 for glycine methyl ester.2,38 By comparison the α-NH2 substituent is expected to have a small effect on carbon acidity. For example, there is only a 0.5 unit difference between carbon acid pKas of 26.5 for acetone and 26.0 for 2-dimethylaminoacetone in DMSO.45 The weak acidifying effect of an α-dimethylamino group of acetone shows that the inductive effect of this electron-withdrawing group on pKa is roughly canceled by the opposing enolate destabilization from repulsive interaction between the nonbonding electron lone pair at nitrogen and the delocalized electron pair at the enolate anion.46 A similar cancellation of inductive and electronic substituent effects has been proposed to explain the small effects of α-hydroxy and α-alkoxy substituents on the kinetic acidity of α-carbonyl carbon.46–49
Combining the 4.6 unit effect of the α-NH3+ substituent on carbon acidity with the ca. 0.5 unit effect of the α-NH2 substituent gives a ca. 4 unit effect for protonation of the α-amino nitrogen on the carbon acid pKa of glycine zwitterion and glycine methyl ester (Scheme 4). We assume that protonation of the α-imino nitrogen of 2-IM will cause a similar 4 unit decrease in the pKa of the carbon acid, from 27 to 23. The value of kHO = 1 M−1 s−1 for carbon-acid deprotonation of 2-IMH reported in Table 5 was calculated from the estimated carbon acid pKa of 23 using the linear correlation log(kHO/p) = 10.044 − 0.444(pKa + log p) established in earlier work (Figure 6).2
Scheme 4.

A comparison of kinetic data for deprotonation of DPL-IMH2 and 3-IMH by HO− shows that substitution of the ring 4–CH by –NH+ causes a 6,000-fold increase in kHO for deprotonation of this carbon by hydroxide ion. The linear correlation between log kHO and carbon acid pKa shown in Figure 6 shows that this change in kHO corresponds to a ca. 8-unit decrease in the pKa of the α-imino carbon. We expect that a large fraction of this 8-unit effect is due to the unit increase in positive charge at the aromatic ring of DPL-IMH2. For example, ca. 30% (2.1 pKa units) of the similar 7.8-unit overall effect of the protonated pyridine nitrogen on the acidity constant for the ionization of the carbon acid benzyl phenyl ketone is observed for the charge-conservative nitrogen substitution, and the remaining 70% of this substituent effect (5.7 pKa units) is expressed when nitrogen is protonated (Chart 3).50,51 We estimate a pKa of ca. 23 (Table 5) for ionization of DPL-IMH by making the assumption that 70% of the total 8-unit effect of the 4-CH for 4-NH+ substitution on carbon acidity is lost upon deprotonation of nitrogen.
Chart 3.

4.2. Substituent Effects on the Carbon Acidity of Glycine
4.2.1 Effect of the α-Iminium Nitrogen
The addition of acetone to glycine methyl ester to form the iminium ion results in a seven unit decrease in the carbon acid pKa from 21 to 14 (Table 5),27 while the addition of acetone to the glycine zwitterion results in a similar seven unit decrease in the carbon acid pKa from 29 to 22 (Table 5). The overall effect of iminium ion formation on carbon acid pKa is due to two contributing effects:
The enhancement of intramolecular electrostatic stabilization of the enolate anion by interaction with the cationic nitrogen when the hydrogen-bonded amino protons are replaced by nonpolar organic functionality.2 This results in a 3 unit larger acidifying effect of the α-NMe3+ group at betaine methyl ester (pKCH = 18.0) than of the α-NH3+ group at glycine methyl ester (pKCH = 21.0).2
The stabilization of the enolate by direct delocalization of negative charge onto the α-imino carbon (Scheme 2). A similar delocalization of charge is a contributing factor in the ca. 3 unit lower pKa of 15.2 for the C-2 proton of 3-cyclohexenone52 compared with the pKa of 18.1 for cyclohexanone.43
The following two experimental observations suggest that the effect of delocalization of negative charge onto the α-imino carbon on the pKa of these acetone iminium ions is small.
Oxygen methylation of glycine and betaine zwitterion, which relieves destabilizing electrostatic interactions between neighboring negatively charged oxygen and carbon atoms, causes 8 and 9 unit decreases, respectively, in carbon acid pKa (Table 5). The observation that oxygen methylation of 1-IMH causes a similar 8 unit decrease in carbon acid pKa is consistent with similar destabilizing electrostatic interactions between neighboring negatively charged oxygen and carbon atoms at 1-IMH, so that there is no detectable relief of this interaction from delocalization of negative charge onto the α-imino carbon of 1-IMH.
The higher estimated pKa of 23 for 2-IMH compared with 22 for 1-IMH shows that there is no significant stabilization of the glycine enolate of 1-IMH by the phenyl for methyl substitution. If there is little stabilization of 2-IMH by delocalization of negative charge from the α-imino carbon to the phenyl ring substituent, then the negative charge density at this carbon is almost certainly small.
4.2.2 Effect of Aromatic Substituents at the α-Iminium Nitrogen
Chart 4 summarizes the results of our studies of substituent effect of formation of iminium ions to simple aldehydes R-CHO on the pKa for deprotonation of the α–amino carbon. The carbon acid pKa of the iminium ion adduct of glycine to salicylaldehyde (25) is much higher than the pKa of the iminium ion adduct of glycine to DPL (17). Apparently, a very strongly electron-withdrawing group such as the pyridinium ion is required to drive the extended delocalization of negative charge onto the α-iminium carbon and cause a further large increase in carbon acidity (Chart 5A). The low carbon acid pKa of ca. 18 for the pyridoxamine analog 4-(aminomethyl)pyridine dication53 provides supporting evidence that the α–pyridinium substituent provides powerful stabilization of negatively charged carbon.
Chart 4.

Chart 5.

Table 5 shows that the addition of the strongly resonance electron withdrawing –CO2− substituent to the α-imino carbon of 2-IMH also drives the extended delocalization of negative charge onto the α-iminium carbon and causes a large decrease in carbon acid pKa from ca. 23 to 1428 for the PG-glycine iminium ion (Chart 5B). We conclude that the second resonance electron-withdrawing group present in the pyruvoyl prosthetic group (-CO2−) and PLP (pyridinium cation) provides a large kinetic push to electrophilic catalysis of deprotonation of glycine to catalysis of enzymatic reactions that proceed through α-amino carbanion intermediates.
4.3. Relevance to Enzymatic Catalysis
It has been suggested for PLP enzymes that protonation of the pyridine ring of the resonance delocalized α-imino carbanion causes a shift in negative charge density from the α-imino to the α–pyridyl carbon that favors protonation of the α–pyridyl carbon and the 1,3-aza-allylic isomerization reaction that is a step in enzyme-catalyzed transamination reactions.9 This proposal is consistent with the observation that the PLP cofactor bound to alanine racemase is stabilized by a hydrogen bond to the weakly acidic cation side chain of Arg-219,54 while the cofactor bound to D-amino acid transaminase 55,56 and alanine glyoxylate aminotransferase 57 is almost certainly protonated by more strongly acidic carboxylic acid side-chains of Glu-177 and Asp-180, respectively. The 6 unit difference in the carbon acid pKas for DPL-IMH and DPL-IMH2 shows that protonation of the pyridine ring causes a substantial increase in the stability of negative charge at the distant α–amino carbon. This provides direct evidence for an increased delocalization of negative charge across the extended π-system that will favor the enyzme catalyzed 1,3-aza-allylic isomerization reaction. In summary, PLP-dependent racemases sacrifice part of the cofactor’s intrinsic catalytic power by using the cofactor in the neutral pyridine form. Apparently, the mechanistic imperative to direct the reaction towards proton transfer at the α–amino carbon, and avoid the wasteful dead-end transamination reaction overrides the competing imperative to utilize the cofactor in its most active form. This shows that the latent catalytic power of PLP-dependent racemases is so great that a portion may be sacrificed to ensure the proper reaction specificity.
4.4. Anatomy of the Rate Acceleration of PLP-Catalyzed Deprotonation of Glycine
Chart 6 compares the first-order rate constants ko (s−1) for solvent-catalyzed deprotonation of glycine at neutral pL in the absence and presence of 0.01 M carbonyl electrophiles.58 These data allow a dissection of the contribution of component pieces of the PLP analog DPL to the enormous observed rate acceleration for deprotonation of glycine.
Chart 6.

The rate constant ko = 2 × 10−11 s−1 for the reaction in the presence of 0.01 M acetone provides a metric for the intrinsic reactivity of the electrophilic carbonyl group as a catalyst of deprotonation of glycine.
The rate constant of ko = 1 × 10−11 s−1 for the reaction in the presence of 0.01 M benzaldehyde shows that the phenyl for alkyl substitution does not greatly enhance the catalytic reactivity of the carbonyl group.
A comparison of ko = 1 × 10−11 and ko = 5.4 × 10−9 s−1 for catalysis by 0.01 M benzaldehyde and salicylaldehyde, respectively, shows that the ortho-phenoxy substituent stabilizes the transition state for deprotonation of glycine by 3.8 kcal/mol (Chart 6). This is due to the stabilizing intramolecular hydrogen bond between the ring oxygen anion and the iminum ion.36 The strong intramolecular hydrogen bond causes the carbon acid pKa to increase from 23 for 2-IMH to 25 for 3-IMH. This decrease in carbon acidity attenuates the transition state stabilization from hydrogen bonding, by causing a decrease in the reactivity of 3-IMH toward deprotonation by hydroxide ion.
The substitution of the strongly electron-withdrawing pyridinium cation for the phenyl ring at 3-IMH causes ko to increase from 5.4 × 10−9 to ca 4.6 × 10−6 s−1. Neglecting the small effect of the o-methyl group on the reactivity of DPL-IMH2, there is an additional 4 kcal/mol stabilization of the transition state for deprotonation of glycine (Chart 6). This reflects the strong electron demand of the pyridinium cation, which drives the stabilization delocalization of charge across the extended π-system.
In conclusion, these results provide an impressive example of how the summation of several relatively small effects in the assembly of the complex molecules PLP and DPL has produced a type of catalyst of truly extraordinary power.33 It is an open question of whether modern chemists will be able to match the brute-force success of the trial and error of evolution, and succeed in designing either small or large molecule catalysts with the power of those observed in nature.
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health (Grant GM 39754 to J. P. R.), and the Ministerio de Educación y Ciencia and the European Regional Development Fund (ERDF) (Grant CTQ2008-03462 to A. R. and J. C.) for generous support of this work.
Footnotes
Supporting Information Available: Table S1 of first-order rate constants, kex, for exchange for deuterium of the first α-proton of glycine in the presence of acetone and HFIP buffers; Table S2 of first-order rate constants, kex, for exchange for deuterium of the first α-proton of glycine in the presence of benzaldehyde; Table S3 of first-order rate constants, kex, for exchange for deuterium of the first α-proton of glycine in the presence of salicylaldehyde and HFIP buffers. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rios A, Richard JP. J Am Chem Soc. 1997;119:8375–8376. [Google Scholar]
- 2.Rios A, Amyes TL, Richard JP. J Am Chem Soc. 2000;122:9373–9385. [Google Scholar]
- 3.Rios A, Richard JP, Amyes TL. J Am Chem Soc. 2002;124:8251–8259. doi: 10.1021/ja026267a. [DOI] [PubMed] [Google Scholar]
- 4.Richard JP, Amyes TL. Bioorg Chem. 2004;32:354–366. doi: 10.1016/j.bioorg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 5.John RA. Biochim Biophys Acta. 1995;1248:81–96. doi: 10.1016/0167-4838(95)00025-p. [DOI] [PubMed] [Google Scholar]
- 6.Jansonius JN. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 7.Christen P, Mehta PK. Chem Rec. 2001;1:436–447. doi: 10.1002/tcr.10005. [DOI] [PubMed] [Google Scholar]
- 8.Eliot AC, Kirsch JF. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 9.Toney MD. Arch Biochem Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 10.Sun S, Toney MD. Biochemistry. 1999;38:4058–65. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe A, Yoshimura T, Mikami B, Hayashi H, Kagamiyama H, Esak N. J Biol Chem. 2002;277:19166–19172. doi: 10.1074/jbc.M201615200. [DOI] [PubMed] [Google Scholar]
- 12.Major DT, Gao J. J Am Chem Soc. 2006;128:16345–16357. doi: 10.1021/ja066334r. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X, Toney MD. Biochemistry. 1999;38:311–320. doi: 10.1021/bi981455s. [DOI] [PubMed] [Google Scholar]
- 14.Zhou X, Jin X, Medhekar R, Chen X, Dieckmann T, Toney MD. Biochemistry. 2001;40:1367–1377. doi: 10.1021/bi001237a. [DOI] [PubMed] [Google Scholar]
- 15.Jackson LK, Brooks HB, Osterman AL, Goldsmith EJ, Phillips MA. Biochemistry. 2000;39:11247–11257. doi: 10.1021/bi001209s. [DOI] [PubMed] [Google Scholar]
- 16.Jackson LK, Brooks HB, Myers DP, Phillips MA. Biochemistry. 2003;42:2933–2940. doi: 10.1021/bi026795z. [DOI] [PubMed] [Google Scholar]
- 17.Fogle EJ, Liu W, Woon S-T, Keller JW, Toney MD. Biochemistry. 2005;44:16392–16404. doi: 10.1021/bi051475b. [DOI] [PubMed] [Google Scholar]
- 18.Lin Y-L, Gao J. Biochemistry. 2010;49:84–94. doi: 10.1021/bi901790e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN, Gehring H, Christen P. J Mol Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi H, Kagamiyama H. Biochemistry. 1997;36:13558–13569. doi: 10.1021/bi971638z. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi H, Mizuguchi H, Miyahara I, Islam MM, Ikushiro H, Nakajima Y, Hirotsu K, Kagamiyama H. Biochim Biophys Acta. 2003;1647:103–109. doi: 10.1016/s1570-9639(03)00068-2. [DOI] [PubMed] [Google Scholar]
- 22.Liu W, Peterson PE, Langston JA, Jin X, Zhou X, Fisher AJ, Toney MD. Biochemistry. 2005;44:2982–2992. doi: 10.1021/bi048657a. [DOI] [PubMed] [Google Scholar]
- 23.Miles EW. Chem Rec. 2001;1:140–151. doi: 10.1002/tcr.4. [DOI] [PubMed] [Google Scholar]
- 24.Dunn MF, Niks D, Ngo H, Barends TRM, Schlichting I. Trends Biochem Sci. 2008;33:254–264. doi: 10.1016/j.tibs.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Schirch V, Szebenyi DME. Curr Opin Chem Biol. 2005;9:482–487. doi: 10.1016/j.cbpa.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 26.Paiardini A, Contestabile R, D’Aguanno S, Pascarella S, Bossa F. Biochim Biophys Acta. 2003;1647:214–219. doi: 10.1016/s1570-9639(03)00050-5. [DOI] [PubMed] [Google Scholar]
- 27.Rios A, Crugeiras J, Amyes TL, Richard JP. J Am Chem Soc. 2001;123:7949–7950. doi: 10.1021/ja016250c. [DOI] [PubMed] [Google Scholar]
- 28.Crugeiras J, Rios A, Riveiros E, Amyes TL, Richard JP. J Am Chem Soc. 2008;130:2041–2050. doi: 10.1021/ja078006c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toth K, Richard JP. J Am Chem Soc. 2007;129:3013–3021. doi: 10.1021/ja0679228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jencks WP. Catalysis in Chemistry and Enzymology. 2. Dover; New York: 1987. [Google Scholar]
- 31.Felten AE, Zhu G, Aron ZD. Org Lett. 2010;12:1916–1919. doi: 10.1021/ol100319b. [DOI] [PubMed] [Google Scholar]
- 32.Denesyuk AI, Denessiouk KA, Korpela T, Johnson MS. J Mol Biol. 2002;316:155–172. doi: 10.1006/jmbi.2001.5310. [DOI] [PubMed] [Google Scholar]
- 33.Richard JP, Amyes TL, Crugeiras J, Rios A. Curr Opin Chem Biol. 2009;13:1–9. doi: 10.1016/j.cbpa.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrow JR, Amyes TL, Richard JP. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bordwell FG, Fried HE. J Org Chem. 1981;46:4327–4331. [Google Scholar]
- 36.Crugeiras J, Rios A, Riveiros E, Richard JP. J Am Chem Soc. 2009;131:15815–15824. doi: 10.1021/ja906230n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glasoe PK, Long FA. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 38.Amyes TL, Richard JP. J Am Chem Soc. 1996;118:3129–3141. [Google Scholar]
- 39.Amyes TL, Richard JP. J Am Chem Soc. 1992;114:10297–10302. [Google Scholar]
- 40.Derived by assuming that the concentration of imine/iminium ion is negligible because only 1 % of glycine is converted to the imine/iminium ion adduct in the presence of 0.5 M acetone at pD 10.5.
- 41.The secondary solvent deuterium isotope effect for deprotonation of 1-IM-L, 3-IM-L and 2-IM by lyoxide ion is assumed to be the same as that for deprotonation of acetone: Pocker Y. Chem Ind. 1959:1383–1384.
- 42.Richard JP, Williams G, O’Donoghue AC, Amyes TL. J Am Chem Soc. 2002;124:2957–2968. doi: 10.1021/ja0125321. [DOI] [PubMed] [Google Scholar]
- 43.Keeffe JR, Kresge AJ. In: The Chemistry of Enols. Rappoport Z, editor. John Wiley and Sons; Chichester: 1990. pp. 399–480. [Google Scholar]
- 44.Chiang Y, Griesbeck AG, Heckroth H, Hellrung B, Kresge AJ, Meng Q, O’Donoghue AC, Richard JP, Wirz J. J Am Chem Soc. 2001;123:8979–8984. doi: 10.1021/ja0107529. [DOI] [PubMed] [Google Scholar]
- 45.Bordwell FG, Gallagher T, Zhang X. J Am Chem Soc. 1991;113:3495–3497. [Google Scholar]
- 46.Hine J. Structural Effects on Equilibria in Organic Chemistry. Wiley; New York: 1975. [Google Scholar]
- 47.Hine J, Hampton KG, Menon BC. J Am Chem Soc. 1967;89:2664–2668. [Google Scholar]
- 48.Hine J, Mahone LG, Liotta CL. J Am Chem Soc. 1967;89:5911–5920. [Google Scholar]
- 49.Crugeiras J, Richard JP. J Am Chem Soc. 2004;126:5164–5173. doi: 10.1021/ja049489c. [DOI] [PubMed] [Google Scholar]
- 50.Carey ARE, Eustace S, More O’Ferrall RA, Murray BA. J Chem Soc, Perkin Trans. 1993;2:2285–2296. [Google Scholar]
- 51.Carey ARE, More O’Ferrall RA, Murray BA. J Chem Soc, Perkin Trans. 1993;2:2297–2302. [Google Scholar]
- 52.Dzingeleski GD, Blotny G, Pollack RM. J Org Chem. 1990;55:1019–1023. [Google Scholar]
- 53.Crugeiras J, Rios A, Amyes TL, Richard JP. Org Biomol Chem. 2005;3:2145–2149. doi: 10.1039/b504399a. [DOI] [PubMed] [Google Scholar]
- 54.Shaw JP, Petsko GA, Ringe D. Biochemistry. 1997;36:1329–1342. doi: 10.1021/bi961856c. [DOI] [PubMed] [Google Scholar]
- 55.Sugio S, Petsko GA, Manning JM, Soda K, Ringe D. Biochemistry. 1995;34:9661–9669. doi: 10.1021/bi00030a002. [DOI] [PubMed] [Google Scholar]
- 56.Van Ophem PW, Peisach D, Erickson SD, Soda K, Ringe D, Manning JM. Biochemistry. 1999;38:1323–1331. doi: 10.1021/bi982414z. [DOI] [PubMed] [Google Scholar]
- 57.Han Q, Robinson H, Gao YG, Vogelaar N, Wilson SR, Rizzi M, Li J. J Biol Chem. 2006;281:37175–37182. doi: 10.1074/jbc.M607032200. [DOI] [PubMed] [Google Scholar]
- 58.The first-order rate constants ko (s−1) given in Chart 6 for solvent-catalyzed deprotonation of glycine at neutral pL in the presence of salicyladehyde and DPL were calculated as ko = (kLO)X-IM [LO−]fX-IM, where (kLO)X-IM is the second-order rate constant for deprotonation by lyoxide ion of the corresponding iminium ion (3-IMH or DPL-IMH2) and fX-IM is the fraction of amino acid that is converted to the iminium ion at the given pL in the presence of 0.01 M salicylaldehyde monoanion and 0.01 M DPL zwitterion, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.