

Abstract
Uncoupling proteins (UCPs) are modulators of mitochondrial metabolism that have been implicated in the development of both insulin resistance and insulin insufficiency, the two major pathophysiological events associated with type 2 diabetes. UCP2 mRNA is expressed in a wide range of tissues; however UCP2 protein expression is restricted to fewer tissues, including the endocrine pancreas, spleen, stomach, brain and the lung. To date, its role in the pathophysiology of diabetes has been most strongly associated with impaired glucose-stimulated insulin secretion from the β-cell, particularly after its induction by free fatty acids. The physiological role of UCP2 remains controversial, but it may act as a downstream signal transducer of superoxide. UCP3 mRNA and protein are expressed in relatively few tissues, predominately skeletal muscle, brown adipose tissue and heart. Increased expression of UCP3 in skeletal muscle is associated with protection from diet-induced insulin resistance in mice. In patients with type 2 diabetes UCP3 protein in muscle is reduced by 50% compared to healthy controls. The primary physiological role of the novel UCPs does not appear to be protection against positive energy balance and obesity; this is based largely on findings from studies of UCP2 and UCP3 knockout mice and from observed increases in UCP3 expression with fasting. The mechanism(s) of action of UCP2 and UCP3 are poorly understood. However, findings support roles for UCP2 and UCP3 as modifiers of fatty acid metabolism and in mitigating damage from reactive oxygen species.
Keywords: Uncoupling proteins, insulin secretion, insulin resistance, reactive oxygen species, fatty acid metabolism
Type 2 Diabetes: the double jeopardy of β-cell dysfunction and insulin resistance
It is generally acknowledged that the common variants of type 2 diabetes mellitus (T2DM) require both reduced insulin action at target tissues (i.e. insulin resistance) and impaired glucose-stimulated insulin secretion resulting from biochemical changes in insulin-secreting pancreatic β-cells. This review describes the accumulating evidence that uncoupling proteins are implicated in the pathophysiology of both insulin resistance and impaired insulin secretion.
Characteristics of β-cell dysfunction in type 2 diabetes
Models of pancreatic islet β-cell dysfunction can be created by exposing isolated islets or clonal cells to combination of high glucose and lipid concentrations, which is detrimental to insulin secretion and islet survival [1] and has been termed “glucolipotoxicity”. In vivo, hyperglycemia and hyperlipidemia are common in obesity, the most important predictor of the development of T2DM [2]. Several excellent recent reviews describe in more detail the changes in islet biochemistry that result in β-cell dysfunction [3–5]. Briefly, the characteristic pattern of insulin secretion in glucolipotoxic states is elevated basal secretion and reduced responsiveness to glucose stimulation. Both glucose and fatty acids influence the expression of a multitude of β-cell genes through changes in activation of transcription factors such as the peroxisome proliferator-activated receptors (PPARs), sterol response element binding proteins (SREBPs), hepatic nuclear factors (HNFs) and others [6,7]. Prolonged activation results in adaptive and eventually maladaptive responses to the glucolipotoxic conditions. In addition, obesity is now considered to be a condition of chronic inflammation [8], which implicates cytokines and adipokines (see below) as stimulants for altered β-cell gene expression and an increase in oxidative stress that leads to insufficient insulin secretion.
Insulin resistance
The other major pathophysiological event associated with T2DM is insulin resistance. Insulin resistance results collectively from maladaptive events in peripheral tissues such as skeletal muscle and adipose tissue as well as in the liver. In the liver, the negative feedback mechanisms that are normally initiated by insulin become defective and despite frank hyperglycemia, there is glycogenolysis and gluconeogenesis. In the insulin-sensitive tissues of the periphery, insulin becomes less effective in the stimulation of glucose uptake. Adipose tissue and skeletal muscle are predominantly affected.
In adipose tissue, recent research has identified the release of a wide range of factors that are important in the development of, as well as the protection from, insulin resistance. Beyond free fatty acids and fatty acid-derived molecules, adipocytes release the adipokines, defined as proteins produced by and secreted specifically from adipocytes. The importance of adipokines in the development of insulin resistance is increasingly being recognized [9]. During the development of T2DM, adipose tissue exhibits a low-grade chronic inflammation, infiltration with macrophages and release of pro-inflammatory cytokines, such as tumor necrosis factor-〈 (TNF〈) and interleukin-6 (IL6) [10,11]. While some adipokines, such as TNF〈 and resistin contribute to the development of insulin resistance in muscle, others mitigate insulin resistance, and include leptin and adiponectin. Thus, factors released from adipose tissue play important roles in whole body glucose metabolism.
Skeletal muscle is the major site of glucose disposal, which is controlled by insulin and modulated by adipokines (reviewed in [12]). Fatty acids and fatty acid-derived metabolites in muscle impair glucose metabolism, but the underlying mechanisms are as yet debated. Over forty years ago the Randle hypothesis was forwarded as a mechanistic explanation for the effect [13]. It held essentially that fatty acids compete with glucose for metabolism, and that fatty acid-derived metabolites, such as acetyl CoA and citrate, inhibit pathways of glucose oxidation, which would subsequently inhibit insulin-stimulated glucose uptake in muscle. This original concept of direct competition between energy substrates has been challenged in the last decade. In muscle of human subjects in vivo, it has been demonstrated that fatty acids and/or metabolites of fatty acids (e.g., diacylglycerol) directly inhibit insulin-stimulated glucose transport [14,15]. Moreover, impaired mitochondrial fatty acid oxidation, leading to the accumulation of fatty acid metabolites in muscle, is proposed as a key factor in the development of insulin resistance in muscle [16,17]. Recent clinical studies have identified mitochondrial dysfunction as potentially very important. In muscle of insulin-resistant offspring of T2DM patients, studies have revealed decreases in mitochondrial activity and increases in intramuscular fat [18]. Gene microarray analyses further demonstrate reductions in the expression of genes that code for key proteins involved in oxidative phosphorylation and fatty acid metabolism in muscle of subjects with T2DM, or who have a family history of T2DM [19,20].
Uncoupling proteins: Overview
The most important facet of mitochondrial metabolism is the production of ATP, which occurs when energy derived from nutrient fuels is captured from an electrochemical gradient traversing the mitochondrial inner membrane (the protonomotive force) by ATP synthase (Fig. (1)). Metabolic uncoupling refers to a state in which nutrient fuels are oxidized but the resultant energy is not linked to ATP synthesis but rather is dissipated as heat. Thus, the degree of metabolic efficiency is determined largely by the amount of uncoupling occurring in a tissue. Uncoupling can be mediated by specific proteins found in the inner mitochondrial membrane, called uncoupling proteins, which are the focus of this paper. In addition, complexes such as the mitochondrial transition pore can also contribute to uncoupling [21] and other mitochondrial inner membrane proteins, such as the adenine nucleotide translocator, participate in fatty acid cycling, one of the postulated mechanisms of uncoupling [22].
Fig. 1.
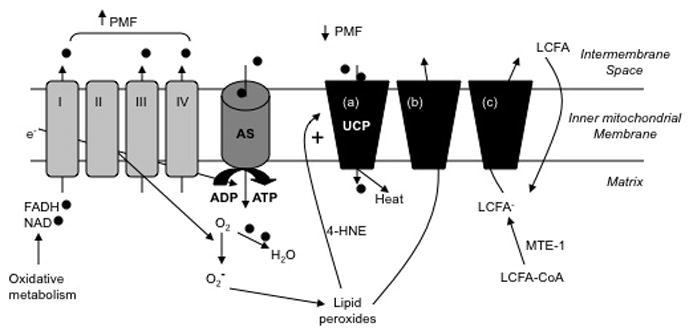
Role of uncoupling proteins in respiration and metabolism. In the course of oxidative phosphorylation, NADH and FADH2 are produced and donate electrons (e−) to the electron transport chain (complexes I-IV) and the resultant protons () are pumped into the intermembrane space to generate a large protonmotive force (DYm). Protons may reenter the matrix via ATP synthase (AS), leading to generation of ATP from ADP and reduction of DYm. The electrons join with oxygen (O2) and protons to form H2O. Alternatively, DYm may be dissipated via actions of uncoupling proteins (UCP). Three potential mechanisms for uncoupling are illustrated. In the first instance (a) UCP acts as a protonophore to allow reentry of protons and production of heat. This function may be enhanced by superoxide (O2−) or its lipid peroxidation by-product, 4-hydroxynonenal (4-HNE). Superoxide is formed when electrons “leak” from the electron transport chain and react with O2. In (b), lipid peroxides formed exit the matrix via a UCP, thus allowing for mitochondrial detoxification. Scenario (c) posits that UCP transports long chain fatty acid anion (LCFA−) either to balance diffusion of neutral fat (LCFA) across the inner mitochondrial membrane or to remove excess LCFA-CoA not metabolized through b-oxidation. The conversion of LCFA-CoA is catalyzed by mitochondrial thioesterase-1 (MTE-1).
The members of the uncoupling protein family in mammals include UCPs 1–4 and brain mitochondrial carrier protein-1 (also called UCP5) [23]. It has however been argued that UCPs 4 and 5 should not be classified as uncoupling proteins given their unknown physiological function and the low degree of homology to the archetypal UCP1 [24]. UCP1 homologues have also been found in fish [25] and birds [26]; in plants, homologues referred to as StUCP (also called PUMP) and AtUCP have been identified. [27]. Homologues of uncoupling proteins have also been documented in Drosophila and Caenorhabditis elegans [28]. The structure and function of UCP1 has been studied for over 30 years. Its biochemistry formed the basis of our hypotheses of the potential physiological roles of the other family members when they were discovered [29–33]. Yet, as will be described below, the emerging functions of UCP2 and UCP3, in particular, are markedly different than the function of UCP1 despite the high degree of molecular homology.
The first uncoupling protein discovered was UCP1, expressed in brown adipose tissue and shown to be an important thermogenic molecule in rodents and in a wide range of neonatal mammals [34–38]. UCP1 elicits uncoupling by transporting protons across the inner membrane into the matrix of the mitochondria [39–45], thus dissipating the protonmotive force as heat [29,46]. UCP1 activity in brown fat of rodents is an important determinant of whole-body energy expenditure [29,46]. Studies of UCP1 knockout mice demonstrate that UCP1 is important in regulating heat production during cold exposure in rodents [29]. The mechanism of proton transfer is still debated (Fig. (1)). In the first model, the uncoupling protein dimers form a protonophore in the membrane, thus functioning as an ion-selective channel. In the second model, cycling of protonated fatty acids across the membrane is dependent on outward movement of fatty acid anion, which interact with on amino acid moieties in the uncoupling protein transmembrane structure. As described in a recent review, these two views are not mutually exclusive and may depend upon the ambient fatty acid concentrations [27].
Uncoupling protein-2
UCP2 was discovered when UCP1 knockout mice failed to become obese as predicted [29], prompting a search for homologous proteins. UCP2 shares 56–59% identity with UCP1 at the amino acid level [30,31]. UCP2 mRNA is expressed in white adipose tissue, heart, lung, skeletal muscle and kidney of both mice and humans [31]. Like UCP1 [47], the UCP2 gene encodes a protein of ~300 amino acids, with 6 exons corresponding to transmembrane domains [48]. A postulated role for UCP2 in thermoregulation, which would mimic that of UCP1 in brown adipose tissue, was quickly ruled out. First, UCP1 knockout mice are cold-intolerant despite strong induction of UCP2 [29]. Second, UCP2 knockout mice have normal body temperature even when cold-exposed [49].
Interest in UCP2 now focuses on several tissue-specific functions: (1) response to oxidative stress in liver [50], brain [51,52] and other tissues; (2) regulation of energy availability in heart [53]; (3) regulation of fatty acid metabolism in white adipose tissue [54] and skeletal muscle [55] (4) regulation of insulin secretion in pancreatic β-cells [56].
Type 2 diabetes, insulin secretion and uncoupling protein-2
UCP2 has been cited as a candidate T2DM gene [57]. For this to be true, mutations in UCP2 may associate with T2DM susceptibility or elements of the pre-diabetes internal milieu may cause transcriptional or post-transcriptional alterations in UCP2 expression and/or function.
Genetic association of UCP2 and type 2 diabetes
Linkage studies show that the UCP2 gene is associated strongly with obesity loci in the human genome [30,58]. Of the mutations found within the coding region, the A/V55 variant is linked to energy expenditure [59–61] but not insulin resistance or diabetes [62–64]. A 45-bp insertion variant in the untranslated region of exon 8 is associated with obesity in most [61,65–67] but not all [64] populations studied. Other less common polymorphisms within the coding region have not been associated with altered metabolism or diabetes.
Interestingly, the genetic variant with the strongest association to T2DM is the −866G/A polymorphism, which is in the gene promoter region. In several populations, individuals homozygous for the A allele are more likely to be diabetic [68–70] and diabetic patients with the A allele are more likely to require insulin [71] and have dyslipidemia [72]. The proposed mechanism is that higher promoter activity of the A allele leads to increased UCP2 expression, resulting in impaired insulin secretion [71]. Nondiabetic individuals homozygous for the A allele have lower glucose-stimulated insulin secretion during oral glucose tolerance tests and in vitro than those with −866G/A or −866G/G genotypes [73]. The −866G/A polymorphism is also associated with another facet of the metabolic syndrome, hypertension [74] but generally not with obesity [75–77]. However, one other study noted an association of the G allele with T2DM [78]. Moreover, UCP2 gene interaction with PPAR© polymorphisms contributing to T2DM has been proposed [68,79]; given the polygenic origins of typical T2DM, such interactions are expected. Thus, across a range of populations from Asia to Europe to North America, there appears to be evidence that at least one genetic variant of UCP2 contributes either to the onset or severity of T2DM and perhaps other elements of the metabolic syndrome. However, changes in function or expression of UCP2 caused by altered environment rather than genetics are also thought to be important in impaired β-cell function associated with T2DM, at least in animal models. The challenge of proving such an assertion in human patients is in its earliest stages but available data suggest that induction of UCP2 is also detrimental to insulin secretion from human islets [80].
Expression studies of UCP2 in β-cell lines and rodent islets
UCP2 mRNA and/or protein has been localized in β-cell lines [81,82], rodent [83,84] and human islets [80,81]. In most reports, UCP2 expression is higher in obese and/or diabetic rodent islets, including ob/ob mice [49], partially pancreatectomized rats [85], nondiabetic fa/fa rats and glucose-infused Wistar rats [86], and mice fed high-fat diets [87,88] than in lean animals. The exception appears to be the diabetic Zucker fatty rat [89,90]. Most obese-diabetic rodents are both hyperglycemic and hyperlipidemic. Therefore, the glucolipotoxic environment may be a cause of up-regulated UCP2 expression. Evidence presented in the following two paragraphs suggests this hypothesis is true (see also Fig. (2)).
Fig. 2.
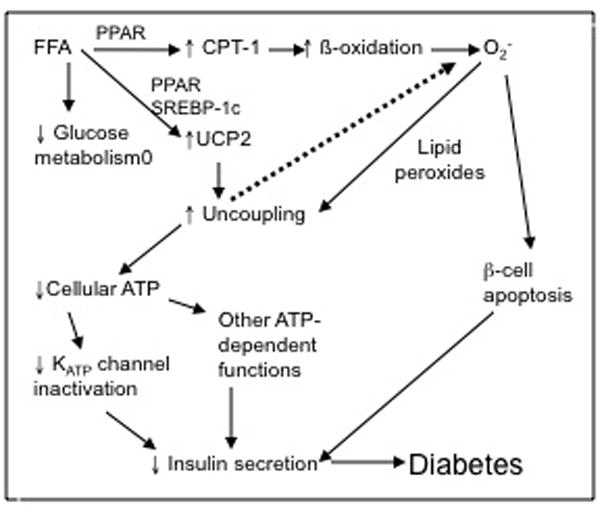
Hypothesized role of UCP2 in pathogenesis of type 2 diabetes. High circulating free fatty acids (FFA) induce UCP2 expression while suppressing glucose metabolism. A parallel increase in enzymes of b-oxidation, including carnitine palmitoyl transferase-1 (CPT-1) mediated by transcription factors such as PPAR and SREBP-1c lead to an increase in metabolic by-products including superoxide (O2−). Either O2− or lipid peroxides activate UCP2 to increase respiratory uncoupling. ATP production is reduced, leading to impaired inactivation of KATP channels and other ATP-dependent processes. Together with the pro-apoptotic effects of O2−, leading to a reduction in b-cell mass, insulin secretion is reduced and diabetes may ensue.
Elevation of free fatty acids clearly induces UCP2 transcription. INS-1 cells and rat islets have increased expression of UCP2 mRNA after exposure to either palmitic acid (saturated) or oleic acid (monounsaturated) [82,91,92]. Similar findings were obtained for mouse islets [87]. Feeding rodents high fat diets achieves a similar outcome [81,88]. Mobilization of intracellular triglyceride also induces UCP2 expression in a mouse model [93]. The effects of fatty acids on gene transcription may be mediated by PPARs, of which the natural ligands are long-chain fatty acids. Two peroxisome proliferator response elements are found in the promoter region of UCP2 gene [48]. Incubation of rat islets with a PPAR© inhibitor ablates the up-regulation of UCP2 protein by free fatty acids and normalizes the ATP/ADP ratio [92]. In addition, over-expression of PPAR© in rat islets induces UCP2 and suppresses insulin secretion [94]. Elevation of PPAR〈 with either an adenoviral vector or stimulation of endogenous activity with its ligand clofibrate induced UCP2 expression in INS-1 cells [95]. The UCP2 gene promoter also contains a response element for the transcription factor SREBP-1c [96]. Elimination of this element blocks the oleic acid-induced induction of UCP2 expression [96] while its over-expression leads to elevation of UCP2 [96–98].
Glucose is also implicated in the induction of UCP2 in islets. Although high glucose uniformly suppressed PPAR〈 expression in reports from two different groups, one study found an increase in UCP2 correlated with an increase in PPAR© [85] while another found a decrease in UCP2 [99]. A key difference is that the first study made rats hyperglycemic in vivo whereas the second exposed isolated rat islets to high glucose in culture. Therefore, in Laybutt et al., [85] insulin or other glucose-regulated hormones might have influenced the outcome. However, additional reports using both rodent [92,100] and human [80] isolated islets cultured with high glucose observed significant induction of UCP2 mRNA. The transcription factor involved in high glucose-induced UCP2 expression has not been identified. The hepatic nuclear factors HNF4〈 and HNF1〈 are shown to up-regulate UCP2 expression in β-cells [101]. Since over-riding control of HNF4〈 and HNF1〈 is provided by Foxa2 [102], an important regulator of many genes involved in insulin secretion [103], this forkhead family transcription factor might be the general controller of glucose-mediated UCP2 induction. However, overexpression of Foxa2 does not affect UCP2 transcription in β-cells [104]. Intriguingly, the transcription factor CCAAT enhancer binding protein (C/EBPβ) is up-regulated in β-cells by hyperglycemia [105] and the promoter region of UCP2 contains a consensus sequence for a C/EBPβ response element [48]. However, there is only one report showing a positive correlation between C/EBPβ activation and UCP2 expression in macrophages [106] and no information about pancreatic β-cells.
In summary, accumulating research suggests there is regulation of UCP2 gene expression by both glucose and fatty acids. Evidence that other endocrine or autocrine factors influence UCP2 expression in β-cells is still equivocal. In Zucker diabetic fatty rats, the adipokine leptin increases UCP2 mRNA [84] but in human islets, leptin suppresses UCP2 expression [80]. Cytokines such as TNF-〈 and IL-1 have also been implicated as regulators of UCP2 expression, although with variable outcomes [107–111]. In β-cells, the only report published to date suggests that down-regulation of UCP2 by IL-1 is not the cause of IL-1’s negative effects on insulin secretion [112]. Given that glucolipotoxic stimuli appear to be the most important regulators of UCP2 expression in β-cells, the next question to be addressed is whether the uncoupling activity of UCP2 is increased under such conditions.
Effects of UCP2 on β-cell function
Since the identification of the novel uncoupling proteins, the physiological role of these molecules as true uncouplers of oxidative phosphorylation has been questioned. When UCP2 is expressed in artificial systems such as yeast mitochondria [30,113], liposomes [114] or studied in isolated mitochondria [115,116] proton flux is sometimes but not always detected. Confusion in the earlier literature may have been due to less than optimal reconstitution conditions in the artificial systems. A later study by Echtay et al. [117] that optimized factors required for protein folding shows significant purine nucleotide-sensitive uncoupling by UCP2. Also, in more physiological systems, uncoupling activity of UCP2 has been demonstrated. For example, in intact thymocytes isolated from wild-type and UCP2 knockout mice, mitochondrial proton leak is greater in the wild-type cells [118]. In ®-cells, overexpression of UCP2 prevents the glucose-induced hyperpolarization of the mitochondrial membrane potential [81] and increases uncoupled respiration [119]. Whereas in UCP2 knockout mice there is a significant increase in the magnitude of the mitochondrial membrane potential when β-cells are exposed to high glucose [87]. Using permeabilized β-cells, sensitivity of endogenous UCP2 to purine nucleotides and fatty acids was demonstrated [120], similar to the properties of UCP1 [121].
Consistent with uncoupling of oxidative phosphorylation, the cellular concentration of ATP changes inversely with changes in UCP2 expression [49,81,119]. Although ATP has many actions as both an energy-storage molecule and a participant in cellular signaling, the main consequence of lower cellular ATP appears to be impaired closure of plasma membrane ATP-dependent K channels [81,122]. Interestingly, however, islets of UCP2 knockout mice also display altered oscillatory frequency and a decreased magnitude of oxygen consumption in response to glucose, compared to islets of wild-type mice [123]. Since a change in oscillatory behavior of β-cells is associated with T2DM patients, this observation could have implications for overall insulin secretion when UCP2 is up-regulated.
Evidence that UCP2 activity in β-cells is altered by glucolipotoxic conditions is emerging. β-cells cultured in monounsaturated fatty acid (MUFA) for 72 h exhibit higher levels of uncoupled respiration than control cells [120]. Moreover, MUFA-cultured cells are highly sensitive to the acute uncoupling effects of fatty acids [120]. High glucose exposure for 72 h also induces conditions in β-cells consistent with higher uncoupling activity of UCP2 [118]. For both fat- and glucose-exposed β-cells, the reactive oxygen species superoxide, which is produced as a by-product of respiration (especially during non-phosphorylating respiration), may be the endogenous activator of UCP2. In isolated kidney mitochondria, exogenously supplied superoxide strongly stimulates UCP2-mediated proton flux [124]; more recent studies have shown that the direct activators may be superoxide byproducts such as reactive alkenals, including 4-hydroxynonenal [125]. When MUFA-cultured β-cells (MIN6) are treated with a superoxide dismutase mimetic to block the actions of the reactive oxygen species, the uncoupling effect produced by acute fatty acid application is attenuated, demonstrating that endogenous superoxide activates UCP2 [100]. A similar approach was used to demonstrate that endogenous superoxide stimulates UCP2 activity in native mouse β-cells. Importantly, when superoxide activity is blocked, the high glucose-mediated impairment of glucose-stimulated insulin secretion is erased [100].
These experiments in isolated systems have provided a framework for studies in whole animals, in which the hypothesis is that induction of UCP2 expression and/or activity by glucolipotoxic conditions is detrimental to insulin secretion (Fig. (2)). The UCP2 knockout mouse is a useful model for studying the effects of high fat diet on UCP2 and β-cell function. When UCP2 knockout mice are fed a chow diet, they have slightly higher insulin secretion to glucose both in vivo and in vitro, which correlates with higher islet ATP content [49]. Studies by Joseph et al. show that UCP2 knockout mice fed a lard-based diet maintain superior glucose tolerance and in vitro glucose-stimulated insulin secretion compared with wild-type mice [88]. There appear to be several contributing factors to the ability of UCP2 knockout mice to retain a high level of β-cell function. First, the high fat induces a higher uncoupling activity as evidenced by a lower islet ATP in the wild-type mice, and a lower insulin secretion [87,88]. Second, there is 2-fold greater expansion of β-cell mass in the high fat-fed UCP2 knockout than wild-type mice, which increases the total secretory capacity of the pancreas [88]. Third, wild-type mice on high fat diet accumulate triglyceride in their islets whereas UCP2 knockout mice maintain high fatty acid oxidation rates, which prevents toxic build-up [87]. Triglyceride accumulation is associated with impaired insulin secretion and increased apoptosis of β-cells in obese rats [126]. Fourth, although chow fed UCP2 knockout mice have higher levels of superoxide in pancreatic islets than wild-type mice [87,100], fatty acids do not generate a further increase as occurred in wild-type mice [87]. Moreover, the dependence of superoxide on UCP2 to mediate its negative effects on glucose-stimulated insulin secretion [100] means that UCP2 knockout mice are able to maintain higher insulin secretion in the face of metabolic overload. To further prove this point, UCP2 knockout mice were crossed with obese-diabetic ob/ob mice. The ob/ob/UCP2 knockout mice have 7-fold higher islet superoxide production than wild-type mice, and 4-fold greater than ob/ob mice [100]. Despite this, the ob/ob/UCP2 knockout mice maintain lower blood glucose and higher plasma insulin than age-matched ob/ob mice [49].
Aside from obese-diabetic models, certain transgenic mice created to better understand the physiology of specific enzymes in the β-cell have implicated UCP2 as part of insulin-deficiency pathways. Clonal β-cells overexpressing SREBP-1c have attenuated glucose-stimulated insulin secretion together with elevated expression of UCP2 [98,127]. Transfection of small interfering RNA for UCP2 into these cells markedly improves insulin secretion [98]. Mice overexpressing hormone-sensitive lipase specifically in β-cells develop diabetes with impaired glucose-stimulated insulin secretion. Although the triglyceride content of the islets is reduced, there is strong up-regulation of UCP2 and lower islet ATP when the mice are fed a high fat diet [128]. Conversely, β-cells transfected with carnitine palmitoyl transferase-I, the rate limiting enzyme for fatty acid oxidation, also have blunted glucose-stimulated insulin secretion but no change in UCP2 expression [129]. The difference between hormone-sensitive lipase and carnitine palmitoyl transferase-I overexpression is probably related to the availability of free fatty acids to act as signaling molecules. In the case of hormone-sensitive lipase, liberation of free fatty acids from triglyceride would increase the availability of substrate for β-oxidation but it would also increase the intracellular concentration of ligands for PPAR〈 and PPAR©, thereby allowing induction of UCP2. In the case of carnitine palmitoyl transferase-I overexpression, increased oxidation of free fatty acids would decrease ligands for the PPARs. Initially there was some controversy as to whether free fatty oxidation was required for UCP2 up-regulation to occur [82,91]. Data from the carnitine palmitoyl transferase-I overexpression studies suggest that free fatty acid oxidation is not necessary [129], as supported by studies showing that bromopalmitate, which is not metabolized, can induce UCP2 expression [82].
Through the use of various diabetic rodent models, it has been relatively straightforward to establish a link between increased expression and/or activity of UCP2 and pancreatic β-cell dysfunction in T2DM. What remains less clear is the physiological role of UCP2 in healthy islets. Two general hypotheses have been developed that might apply to β-cells. The first is that alterations in UCP2 activity regulate the cellular content of coenzymes and thus the metabolic activity of cells. Increased uncoupling would permit respiration to continue without creating additional ATP (which would then slow down respiration). Such an effect would maintain higher amounts of oxidized coenzymes (eg. NAD+), thereby modulating processes like lipogenesis [27]. In the β-cell, the rate of mitochondrial metabolism, which is correlated with hyperpolarization of the mitochondrial inner membrane, strongly predicts insulin secretion [130]. Those authors suggest that molecules like UCP2, which can dissipate the mitochondrial membrane potential, are important determinants of the maximal fuel-stimulated insulin secretion. Mild uncoupling, which could occur acutely with increased fuel provision such as occurs at meal-time, might act as a metabolic brake to prevent over-secretion of insulin.
The second hypothesis is that UCP2 protects cells from the damaging effects of reactive oxygen species by limiting their production. Mild uncoupling of respiration increases the reduction of O2 to H2O without production of ATP, thereby shortening the half-life of molecular oxygen and decreasing its availability to react with stray electrons to create superoxide [27,130]. Moreover, uncoupling decreases the mitochondrial membrane potential, which, in turn, minimizes reactive oxygen species production by the electron transport chain complexes. Mice lacking UCP2 have higher superoxide production in islets and other tissues [87,100,131,132]. Studies on UCP2 in the brain suggest that absence of UCP2 increases the susceptibility of UCP2 knockout mice to damage from stroke and other excitotoxic conditions {[51,52]. Overexpression of UCP2 protects clonal β-cells from death caused by exposure to exogenous H2O2 and such exposure increases endogenous UCP2 expression [133]. Studies of UCP2 function in other tissues suggest that UCP2 is not activated directly by superoxide but rather, the peroxidation of lipids initiated by superoxide leads to an increase in UCP2-mediated proton translocation [134]. It has been proposed that lipid peroxide-mediated activation of UCP2 promotes egress of fatty acid peroxides from the matrix to the inter-membrane space of the mitochondria, where they cause less damage [135,136] (Fig. (1)). Although the mechanism(s) invoked to explain how UCP2 affords protection from reactive oxygen species differs between these 3 groups, the outcome of protection is the same. The hypothesis is attractive as a function for UCP2 in β-cells because of the relatively low expression of protective enzymes such as glutathione peroxidase, catalase and superoxide dismutase [137]; perhaps UCP2 activity can partially substitute for inadequate antioxidant activity. However, if this were true, then UCP2 knockout mice should be highly susceptible to β-cell oxidative stress. In contrast, the available evidence suggests that β-cell apoptosis is higher in UCP2 knockout than wild-type mice fed a chow diet. However, high fat diet induces more apoptosis in wild-type mice [88] and, in fact, the deleterious effects of high fat diet are absent or reduced in UCP2 knockout mice [87]. Thus, the role of UCP2 as a protector from oxidative stress in the β-cell is not yet clear.
Uncoupling protein-3
Uncoupling protein-3 (UCP3) was identified in 1997 as another member of the mitochondrial anion carrier protein family, having approximately 58% homology to the archetypal UCP1 and 71% homology to UCP2 [32,33]. UCP3 has a more restricted tissue expression pattern than does UCP2, and is highly selectively expressed in skeletal muscle, brown fat, and also in diabetic heart. It is the only UCP that is expressed at the protein level in skeletal muscle [138]. Interestingly, all evidence thus far suggests that UCP2 and UCP3 proteins do not normally coexist in any one tissue or cell type.
Type 2 diabetes mellitus, insulin resistance and UCP3
mRNA levels of UCP3 but not UCP2 are decreased in skeletal muscle of patients with T2DM compared to healthy control subjects, and there is a positive correlation between UCP3 levels in muscle and whole body insulin stimulated glucose utilization rates, consistent with the idea that UCP3 levels could be protective against the development of insulin resistance [139]. Levels of UCP3 protein are also decreased in muscle of patients with T2DM. Schrauwen and colleagues demonstrated that UCP3 protein content in vastus lateralis muscle of patients with T2DM is approximately 50% of that in healthy control subjects [140]. Characteristics of muscle mitochondrial dysfunction during T2DM include higher levels of lipid peroxidation [141] and reduced vastus lateralis mitochondrial content and oxidative capacity (NADH2:O2 oxidoreductase capacity) [142]. It is not yet known if the decreased UCP3 content of muscle is a cause or a consequence of insulin resistance. However, it is relevant that increases in plasma non-esterified fatty acids induce the expression of UCP3 in muscle, while no significant increases in UCP3 expression are observed if the infusions of triglycerides are administered during a hyperinsulinemic clamp [143]. As well, UCP3 protein content is decreased in the pre-diabetic state of impaired glucose tolerance [144]. The latter is consistent overall with the idea of a maladaptive response to increased circulating fatty acid concentrations when there are simultaneously high circulating levels of insulin and glucose, i.e., glucolipotoxicity. The possible mechanistic role(s) for UCP3 are not well understood; however, some of the possible roles in the metabolism of fatty acids, and in the protection from reactive oxygen species are described further below.
Knockouts and transgenics: What have we learned?
Like UCP2, UCP3 was originally proposed to cause uncoupling of oxidative phosphorylation through the mitochondrial proton leak so well elucidated by Martin Brand over the past 15 years (e.g., [145–148]. However, just as a function for UCP2 in physiological uncoupling is controversial, such a role for UCP3 has been questioned to an even greater extent. At the physiological level, it is clear that altered UCP3 expression patterns in skeletal muscle induced by physiological interventions are not consistent with a role for UCP3 in uncoupling thermogenesis. This has been a topic that has been covered effectively in several recent review papers (e.g., [24,149–151]. Some of the key points are outlined in the next section, beginning with an overview of the phenotypic characteristics of the UCP3 knockout and overexpressor mice. This will be followed by a description of recent in vitro studies that have shed some light on possible mechanistic aspects.
The UCP3 knockout mouse is not obese, is normophagic, has normal overall energy expenditure, and is not more predisposed to diet-induced obesity than wild-type controls [152,153]. In muscle mitochondria, higher state 4 (non-phosphorylating) membrane potentials are reported in muscle mitochondria of knockouts than controls, consistent with decreased proton conductance, but state 4 respiration is found to be normal [152,154]. In independently produced UCP3 knockout mice, Vidal Puig et al [153] observed higher respiratory control ratios (state 3 respiration: state 4 respiration) in UCP3 knockout mitochondria, while this was not observed in other lines of knockout mice [152,154,155]. Differences in mitochondrial incubation conditions and in mouse genetic backgrounds may explain at least some of the discrepancies. Results from UCP3 knockout mice are consistent with a possible role for UCP3 in protection from reactive oxygen species and in fatty acid metabolism. In muscle mitochondria of knockout mice, reactive oxygen species production is increased [153], consistent with a possible role for UCP3, like UCP2, in protection from oxidative stress. In 4–6 month old UCP3 knockout mice fed a high fat diet, circulating levels of free fatty acids are higher than in wild-type mice [153]. Moreover, indirect calorimetry indicated that fasted UCP3 knockout mice have lower whole body fat oxidation than wild-type mice [154]. Vidal Puig et al [153] measured oleate and glucose oxidation in soleus and extensor digitorum longus muscles, and while oxidation of both was consistently decreased in knockout mice, measurement error appeared high, and no statistical significance was reported. These results from UCP3 knockout mice are thus consistent with the earlier results of Argyropoulos et al. [156] where impaired fatty acid oxidation was linked to a UCP3 gene polymorphism in humans (the latter findings are reviewed more extensively in [150]).
The transgenic overexpression of UCP3 in muscle in mice has been studied by a number of groups. The first reports describes mice expressing UCP3 mRNA at levels 66-fold normal, and phenotypic characteristics include reduced adiposity despite hyperphagia, lower fasting plasma glucose and insulin levels and an increased glucose clearance rate [157]. The physiological relevance of this model is thus doubtful based on the supraphysiologic levels of mRNA and protein (66-fold and 20-fold, respectively) [155]. Moreover, it was concluded that the increased proton leak in mitochondria of overexpressor mice was not caused by a native function of UCP3 because the proton leak is not proportional to the increased amounts of UCP3 protein and is not activated by superoxide, or inhibited by GDP [155]. The uncoupling in the transgenic mice is thus described as an artifact of supraphysiological expression. In another model of UCP3 overexpression in mice mRNA levels and mitochondrial protein levels are 15–20-fold normal [158]. The overall metabolic impact of overexpression in this model is less marked. When UCP3 overexpressor mice are fed a low fat chow, there is no difference in body weight or metabolic rate, compared to control mice. However, following four weeks of high fat diet feeding, overexpressors gain less weight and are leaner than controls [158]. While whole body oxygen consumption rate (expressed per gram body weight) is increased in overexpressors, the effect is absent when rates are expressed at the level of the whole body. Given that control mice have significantly higher adiposity levels than UCP3 overexpressors, comparisons of metabolic rates at the level of the whole body (not normalized to total body weight) are preferable [159].
In a recently published study, the metabolic effects of physiological transgenic increases in the expression of muscle UCP3 in mice were explored [160]. In congenic mice overexpressing the human UCP3 protein at levels approximately 230% of wild-type mice, and in UCP3 knockout mice, we assessed the amounts and activities of key proteins involved in muscle fatty acid uptake and oxidation. In muscle of UCP3 overexpressors, plasma membrane fatty acid binding protein (FABPpm) content is significantly increased compared with wild-type mice. While hormone-sensitive lipase activity is unchanged across the genotypes, there are increases in carnitine palmitoyltransferase-I, β-hydroxyacylCoA dehydrogenase and citrate synthase activities, and decreases in intramuscular triacylglycerol in muscle of overexpressors. Muscle mitochondrial content is equal across the genotype. Inconsistent with a role for UCP3 in uncoupled oxidative phosphorylation are the observations of increased high-energy phosphates and total muscle carnitine and CoA in UCP3 overexpressor compared with wild-type mice. Moreover whole body energy expenditure (VO2) is unchanged. The respiratory exchange ratio is decreased in the overexpressor mice, indicative of increased whole body fat oxidation. Altogether, the findings indicate an increased capacity for fat oxidation in the absence of any detectable increases in thermogenesis in UCP3 overexpressor mice. Findings from UCP3 knockout mice demonstrate few differences compared with wild-type mice, consistent with unknown potential compensatory mechanisms.
Physiological induction of UCP3 expression: Lack of effect on basal proton leak
The results of earlier studies in which physiological interventions were used to explore the possible function of UCP3 also are inconsistent with UCP3 functioning as an uncoupler of oxidative phosphorylation. Cadenas and colleagues explored the relationship between UCP2 and UCP3 expression and mitochondrial proton conductance in rat skeletal muscle [161]. A 24h fast increases UCP2 and UCP3 mRNA levels more than 5-fold and 4-fold, respectively, and UCP3 protein levels by 2-fold. Mitochondrial proton conductance is however unchanged. Similarly the effects of fasting on resting metabolic rate, respiratory quotient, muscle UCP3 expression, and mitochondrial proton leak were examined in wild-type and UCP3 knockout mice [154]. In wild-type mice, fasting causes 4-fold and 2-fold elevation of UCP3 and UCP2 mRNA, respectively, but does not significantly affect the kinetics of mitochondrial proton leak. Resting metabolic rates decrease with fasting in both groups. UCP3 knockout mice have higher respiratory quotients than wild-type mice in fed resting states, consistent with impaired fatty acid oxidation. Altogether, results show that the fasting-induced increases in UCP2 and UCP3 do not correlate with increased basal mitochondrial proton leak but support a possible role for UCP3 in fatty acid metabolism. It should be kept in mind that the in vitro incubation conditions used in both of these independent studies may have lacked key activators to reveal UCP3 function. On the other hand, some in vitro studies that have identified possible activators of UCP3 function (described below) have employed supraphysiological concentrations, opening the possibility for secondary effects or artefact. Regardless, both of the above studies show that changes in UCP2 and UCP3 levels are not associated with any alterations in basal proton leak in muscle mitochondria.
Given the originally hypothesized role for UCP3 in thermogenic uncoupling, one of the earliest identified paradoxes was indeed the increased expression of UCP3 in muscle during fasting, a situation where there is decreased energy expenditure in muscle. While expression patterns in muscle overall are consistent with the latter, expression is highest in glycolytic and mixed oxidative glycolytic fibre types and lowest in mitochondria-rich oxidative fibres. Moreover the greatest increases in expression induced by fasting occur in the glycolytic fibres rather than in the mitochondria-rich oxidative fibre types. Thus, there is increased expression in muscle when fatty acid supply exceeds oxidation capacity.
Altogether, findings from animal studies show that UCP3 is associated with protection from insulin resistance, and with increased fatty acid oxidation, decreased intramuscular triglyceride, and decreased reactive oxygen species production (Fig. (3)).
Fig. 3.
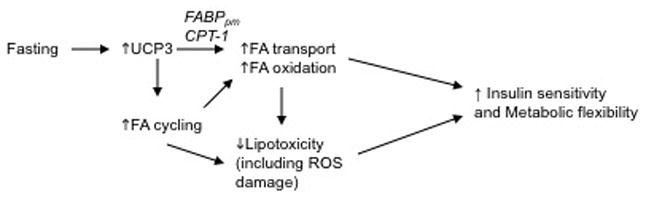
Hypothesized role of UCP3 in maintaining insulin sensitivity. In healthy individuals, fasting or eating of a high fat diet will induce UCP3 expression in skeletal muscle. Increased UCP3 is associated with an increase in enzymes associated with fatty acid transport (FABPpm) and oxidation (CPT-1) and with increased fatty acid recycling, which can increase the efficiency of oxidative metabolism. The increase in whole body fat oxidation will reduce overall adiposity and intramuscular triglyceride stores, which are associated with improved glucose tolerance. During the development of T2DM, a maladaptive response of UCP3 occurs, such that the glucolipotoxic environment lead to down-regulation of UCP3 expression.
In vitro studies – Illuminating the potential mechanisms of UCP3 action
Several hypothesized mechanisms of UCP3 action have been proposed. Generally they fall within one or two categories: 1) UCP3, when activated by superoxide or a peroxidized fatty acid, causes a mitochondrial proton leak that, in turn, acts in a negative feedback loop to mitigate further mitochondrial reactive oxygen species production, or 2) UCP3 acts as a fatty acid anion exporter to either facilitate fatty acid oxidation, or to remove potentially damaging fatty acid anions that have entered mitochondria independent of the carnitine palmitoyl transferase system. These mechanisms are further described below.
Several activators of UCP3-mediated proton conductance have been identified in studies of UCP3-containing liposomes and in mitochondria isolated from wild-type and knockout mice. Amongst the identified activators, fatty acids were the first, and they are shown to activate both UCP2 and UCP3 in liposomes [114,117]. Retinoate and its analogues are also identified as activators [113]. Further studies identify ubiquinone [117], superoxide [162], and most recently, the reactive alkenal 4-HNE [125] (see Fig. (1)). As is common with many in vitro studies into characteristics of molecular function, the latter findings, while greeted with widespread interest and enthusiasm are open to questions regarding physiological relevance. Concentrations of the activators often far-exceeded physiological concentrations. Moreover the criterion of purine nucleotide inhibition as a sine qua non of uncoupling protein activity is fundamentally perplexing given that physiological levels of the purine nucleotides equal or exceed those used in the in vitro studies. However, it is proposed that fatty acids may act in vivo to reduce the inhibition by purine nucleotides [163].
Others have proposed that UCP3 and fatty acids are more directly linked. Two hypotheses have been advanced proposing that UCP3 acts as a mitochondrial fatty acid anion exporter [151,164]. Schrauwen and colleagues hypothesize that when fatty acid supply exceeds oxidation capacity, there is increased entry of fatty acid into the mitochondria by flip-flop, resulting in the accumulation of non-esterified fatty acid in the matrix. The function of UCP3 is proposed as an exporter of the non-metabolizable non-esterified fatty [164]. This hypothesis does not link UCP3 with improved capacity for fatty acid oxidation; instead it advances a mechanism for efflux of fatty acid anions that cannot be used in the matrix for fuel.
The other hypothesis does link UCP3 function with an improved capacity for fatty acid oxidation [151]. It proposes that UCP3 functions in conjunction with a mitochondrial thioesterase when fatty acid supply exceeds oxidation. Mitochondrial thioesterase-1 (MTE-1) is a matrix enzyme that cleaves CoA units from long chain fatty acyl-CoA molecules, and is thought to liberate CoA to support high rates of Krebs cycle and β-oxidation pathway activities [165]. Non-esterified fatty acid anions in the matrix require reactivation outside of the matrix by acyl CoA synthetases (e.g., ACS5, found on the mitochondrial outer membrane) before they can be oxidized, and it is proposed that UCP3 fulfills the role of fatty acid anion export. Such a fatty acid cycle would theoretically allow greater rates of fat oxidation by releasing sequestered CoA and minimize detrimental effects of the accumulation of non-esterified fatty acid in the matrix. While supporting data are mainly correlative, MTE-1 expression is significantly increased in UCP3 overexpressor mice [166], and there is a strong correlation between UCP3 and MTE-1 expression and activity levels [167]. The recent findings of Bezaire and colleagues [160], described above, demonstrate that a consequence of physiological (but constitutive) increases in UCP3 in muscle lead to increased potential for fatty acid uptake, transport into the mitochondria and flux through the Krebs cycle and β-oxidation pathways. That CoA and carnitine levels are also elevated in the UCP3 overexpressor mice is consistent with augmented rates of fat oxidation. While the aim of the latter study was not to test mechanistic aspects of the hypothesis, the results are supportive.
Other studies have examined the effects of physiologic UCP3 overexpression on characteristics of fatty acid oxidation and mitochondrial uncoupling in L6 muscle cells. MacLellan and colleagues used an adenoviral construct to produce a 2.2–2.5-fold increase in UCP3 protein in L6 cells [168]. Increased expression of UCP3 protein results in increased palmitate oxidation in cells under normoglycemic or hyperglycemic conditions, while the chemical uncoupler, dinitrophenol, has no effect. Increased UCP3 does not modify glucose oxidation, while dinitrophenol or insulin treatment cause increases. Cellular oxygen consumption, assessed in situ using self-referencing microelectrodes is not altered by the increases in UCP3 protein, while dinitrophenol causes increases. Moreover there are no changes in mitochondrial membrane potential. Consistent with the results from many of the above described studies of UCP2 and UCP3 function, there are also significant reductions in reactive oxygen species production [168].
Concluding statements
The evidence collected since the discovery of the novel uncoupling proteins, UCP2 and UCP3 in 1997 clearly differentiate the functions of these molecules from those of the archetypal UCP1. Moreover, the distinct tissue localization of UCP2 and UCP3 is suggestive of distinctive physiological roles for them. Data obtained mainly from systems designed to over- or under-express the uncoupling proteins support the hypothesis that both UCP2 and UCP3 can modulate cellular metabolism. In the pancreatic β-cell, such modulation results in reduction of glucose-stimulated insulin secretion by UCP2 whereas in the skeletal muscle, UCP3 appears to have a role in regulating fatty acid oxidation. Both UCP2 and UCP3 may function to decrease reactive oxygen species generation. The role of UCPs in the pathophysiology of chronic metabolic diseases such as T2DM is highly dependent upon the specific UCP and its tissue of expression. Interesting, up-regulation of UCP2 in pancreatic islets may be a precursor to development of insulin insufficiency in T2DM while, in contrast, an increase in UCP3 expression in skeletal muscle could help to prevent the development of insulin resistance. Therefore, if the UCPs are to have any therapeutic relevance, strategies are required that incorporate acknowledgement of the need to up-regulate UCP3 while decreasing activity of UCP2, tissue-specifically. In the case of UCP2, the treatment should not impact expression or activity in other tissues, like the brain, where it appears to confer cytoprotective effects.
Acknowledgments
Research done by the authors has been supported by the Canadian Institutes of Health Research (CBC, MEH) and the Canadian Diabetes Association (CBC, MEH).
Abbreviations
- C/EBP
CCAAT enhancer binding protein
- HNF
hepatic nuclear factor
- 4-HNE
4-hydroxynonenal
- IL
interleukin
- MTE-1
mitochondrial thioesterase-1
- MUFA
monounsaturated fatty acid
- PPAR
peroxisome proliferator-activated receptor
- SREBP
sterol response element binding protein
- TNF
tumor necrosis factor
- T2DM
type 2 diabetes mellitus
- UCP
uncoupling protein
References
- 1.El-Assaad W, Buteau J, Peyot M-L, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology. 2003;144:4154–4163. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- 2.Bougneres P. Genetics of obesity and type 2 diabetes: Tracking pathogenic traits during the predisease period. Diabetes. 2002;51:S295–303. doi: 10.2337/diabetes.51.2007.s295. [DOI] [PubMed] [Google Scholar]
- 3.Poitout V, Robertson RP. Minireview: Secondary β-cell failure in type 2 diabetes--a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 4.Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-coA signaling, lipid partitioning, and glucolipotoxicity: Role in β-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51:S405–413. doi: 10.2337/diabetes.51.2007.s405. [DOI] [PubMed] [Google Scholar]
- 5.Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587. doi: 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- 6.Fatehi-Hassanabad Z, Chan CB. Transcriptional regulation of lipid metabolism by fatty acids: A key determinant of pancreatic beta-cell function. Nutr Metab. 2005;2 doi: 10.1186/1743-7075-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerrish K, Cissell MA, Stein R. The role of hepatic nuclear factor 1alpha and PDX-1 in transcriptional regulation of the pdx-1 gene. J Biol Chem. 2001;276:47775–47784. doi: 10.1074/jbc.M109244200. [DOI] [PubMed] [Google Scholar]
- 8.Das UN. Is obesity an inflammatory condition? Nutrition. 2001;17:953–966. doi: 10.1016/s0899-9007(01)00672-4. [DOI] [PubMed] [Google Scholar]
- 9.Trayhurn P, Wood IS. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347–355. doi: 10.1079/bjn20041213. [DOI] [PubMed] [Google Scholar]
- 10.Hirosumi J, Tuncman G, Chang L, et al. A central role for jnk in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 11.Xu A, Wang Y, Keshaw H, et al. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havel PJ. Update on adipocyte hormones: Regulation of energy balance and carbohydrate/lipid metabolism. Diabetes. 2004;53:S143–151. doi: 10.2337/diabetes.53.2007.s143. [DOI] [PubMed] [Google Scholar]
- 13.Randle PJ, Garland PB, Hales CN, et al. The glucose fatty acid cycle: Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 14.Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1 associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowell B, Shulman G. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 17.Dobbins RL, Szczepaniak LS, Bentley B, et al. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50:123–130. doi: 10.2337/diabetes.50.1.123. [DOI] [PubMed] [Google Scholar]
- 18.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1a-responsive genes involved in oxidative phosphorylation are corrdinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 20.Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koshkin V, Bikopoulos G, Chan CB, Wheeler MB. The characterization of mitochondrial permeability transition in clonal pancreatic β-cells: Multiple modes and regulation. J Biol Chem. 2004;279:41368–41376. doi: 10.1074/jbc.M406914200. [DOI] [PubMed] [Google Scholar]
- 22.Jezek P, Zackova M, Ruzicka M, Skobisova E, Jaburek M. Mitochondrial uncoupling proteins--facts and fantasies. Physiol Res. 2004;53 (Suppl 1):S199–S211. [PubMed] [Google Scholar]
- 23.Bouillaud F, Couplan E, Pecqueur C, Ricquier D. Homologues of the uncoupling protein from brown adipose tissue (UCP1): UCP2, UCP3, BMCP1 and UCP4. Biochim Biophys Acta. 2001;1504:107–119. doi: 10.1016/s0005-2728(00)00241-3. [DOI] [PubMed] [Google Scholar]
- 24.Nedergaard J, Cannon B. The ‘novel’ ‘uncoupling’ proteins UCP2 and UCP3: What do they really do? Pros and cons for suggested functions. Exp Physiol. 2003;88:65–84. doi: 10.1113/eph8802502. [DOI] [PubMed] [Google Scholar]
- 25.Stuart JA, Harper JA, Brindle KM, Brand MD. Uncoupling protein 2 from carp and zebrafish, ectothermic vertebrates. Biochim Biophys Acta. 1999;1413:50–54. doi: 10.1016/s0005-2728(99)00081-x. [DOI] [PubMed] [Google Scholar]
- 26.Raimbault S, Dridi S, Denjean F, et al. An uncoupling protein homologue putatively involved in facultative muscle thermogenesis in birds. Biochem J. 2001;353:441–444. doi: 10.1042/0264-6021:3530441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 2000;342:161–179. [PMC free article] [PubMed] [Google Scholar]
- 28.Jezek P. Possible physiological roles of mitochondrial uncoupling proteins--UCPn. Int J Biochem Cell Biol. 2002;34:1190–1206. doi: 10.1016/s1357-2725(02)00061-4. [DOI] [PubMed] [Google Scholar]
- 29.Enerback S, Jacobsson A, Simpson EM, et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- 30.Fleury C, Neverova M, Collins S, et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- 31.Gimeno RE, Dembski M, Weng X, et al. Cloning and characterization of an uncoupling protein homolog: A potential molecular mediator of human thermogenesis. Diabetes. 1997;46:900–906. doi: 10.2337/diab.46.5.900. [DOI] [PubMed] [Google Scholar]
- 32.Boss O, Samec S, Paoloni-Giacobino A, et al. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997;408:39–42. doi: 10.1016/s0014-5793(97)00384-0. [DOI] [PubMed] [Google Scholar]
- 33.Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lowell BB. UCP3: An uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Comm. 1997;235:79–82. doi: 10.1006/bbrc.1997.6740. [DOI] [PubMed] [Google Scholar]
- 34.Bouillaud F, Weissenbach J, Ricquier D. Complete cDNA-derived amino acid sequence of rat brown fat uncoupling protein. J Biol Chem. 1986;261:1487–1490. [PubMed] [Google Scholar]
- 35.Jacobsson A, Stadler U, Glotzer MA, Kozak LP. Mitochondrial uncoupling protein from mouse brown fat Molecular cloning, genetic mapping, and mRNA expression. J Biol Chem. 1985;260:16250–16254. [PubMed] [Google Scholar]
- 36.Lin CS, Klingenberg M. Isolation of the uncoupling protein from brown adipose tissue mitochondria. FEBS Lett. 1980;113:299–303. doi: 10.1016/0014-5793(80)80613-2. [DOI] [PubMed] [Google Scholar]
- 37.Ricquier D, Lin C, Klingenberg M. Isolation of the GDP binding protein from brown adipose tissue mitochondria of several animals and amino acid composition study in rat. Biochem Biophys Res Commun. 1982;106:582–589. doi: 10.1016/0006-291x(82)91150-0. [DOI] [PubMed] [Google Scholar]
- 38.Lin CS, Klingenberg M. Characteristics of the isolated purine nucleotide binding protein from brown fat mitochondria. Biochemistry. 1982;21:2950–2956. doi: 10.1021/bi00541a023. [DOI] [PubMed] [Google Scholar]
- 39.Garlid KD, Jaburek M, Jezek P. The mechanism of proton transport mediated by mitochondrial uncoupling proteins. FEBS Lett. 1998;438:10–14. doi: 10.1016/s0014-5793(98)01246-0. [DOI] [PubMed] [Google Scholar]
- 40.Klingenberg M. Uncoupling protein--a useful energy dissipator. J Bioenerg Biomembr. 1999;31:419–430. doi: 10.1023/a:1005440221914. [DOI] [PubMed] [Google Scholar]
- 41.Cannon B, Nedergaard J. The biochemistry of an inefficient tissue: Brown adipose tissue. Essays Biochem. 1985;20:110–164. [PubMed] [Google Scholar]
- 42.Rial E, Poustie A, Nicholls DG. Brown-adipose-tissue mitochondria: The regulation of the 32000-Mr uncoupling protein by fatty acids and purine nucleotides. Eur J Biochem. 1983;137:197–203. doi: 10.1111/j.1432-1033.1983.tb07815.x. [DOI] [PubMed] [Google Scholar]
- 43.Ricquier D, Bouillaud F. The mitochondrial uncoupling protein: Structural and genetic studies. Prog Nucleic Acid Res Mol Biol. 1997;56:83–108. doi: 10.1016/s0079-6603(08)61003-x. [DOI] [PubMed] [Google Scholar]
- 44.Strieleman PJ, Schalinske KL, Shrago E. Fatty acid activation of the reconstituted brown adipose tissue mitochondria uncoupling protein. J Biol Chem. 1985;260:13402–13405. [PubMed] [Google Scholar]
- 45.Winkler E, Klingenberg M. Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J Biol Chem. 1994;269:2508–2515. [PubMed] [Google Scholar]
- 46.Florez-Duquet M, McDonald RB. Cold-induced thermoregulation and biological aging. Physiol Rev. 1998;78:339–358. doi: 10.1152/physrev.1998.78.2.339. [DOI] [PubMed] [Google Scholar]
- 47.Kozak LP, Britton JH, Kozak UC, Wells JM. The mitochondrial uncoupling protein gene. Correlation of exon structure to transmembrane domains. J Biol Chem. 1988;263:12274–12277. [PubMed] [Google Scholar]
- 48.Tu N, Chen H, Winnikes U, et al. Structural organization and mutational analysis of the human uncoupling protein-2 (hUCP2) gene. Life Sci. 1999;64:PL41–50. doi: 10.1016/s0024-3205(98)00555-4. [DOI] [PubMed] [Google Scholar]
- 49.Zhang CY, Baffy G, Perret P, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 50.Cortez-Pinto H, Yang SQ, Lin HZ, et al. Bacterial lipopolysaccharide induces uncoupling protein-2 expression in hepatocytes by a tumor necrosis factor-alpha-dependent mechanism. Biochem Biophys Res Commun. 1998;251:313–319. doi: 10.1006/bbrc.1998.9473. [DOI] [PubMed] [Google Scholar]
- 51.Paradis E, Clavel S, Bouillaud F, Ricquier D, Richard D. Uncoupling protein 2: A novel player in neuroprotection. Trends Mol Med. 2003;9:522–525. doi: 10.1016/j.molmed.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Horvath TL, Diano S, Barnstable C. Mitochondrial uncoupling protein 2 in the central nervous system: Neuromodulator and neuroprotector. Biochem Pharmacol. 2003;65:1917–1921. doi: 10.1016/s0006-2952(03)00143-6. [DOI] [PubMed] [Google Scholar]
- 53.Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004;364:1786–1788. doi: 10.1016/S0140-6736(04)17402-3. [DOI] [PubMed] [Google Scholar]
- 54.Kopecky J, Rossmeisl M, Flachs P, et al. Energy metabolism of adipose tissue--physiological aspets and target in obesity treatment. Physiol Res. 2004;53 (Suppl 1):S225–S232. [PubMed] [Google Scholar]
- 55.Hesselink MK, Mensink M, Schrauwen P. Human uncoupling protein-3 and obesity: An update. Obes Res. 2003;11:1429–1443. doi: 10.1038/oby.2003.192. [DOI] [PubMed] [Google Scholar]
- 56.Saleh MC, Wheeler MB, Chan CB. Uncoupling protein-2: Evidence for its function as a metabolic regulator. Diabetologia. 2002;45:174–187. doi: 10.1007/s00125-001-0737-x. [DOI] [PubMed] [Google Scholar]
- 57.Marx J. Unraveling the causes of diabetes. Science. 2002;296:686–689. doi: 10.1126/science.296.5568.686. [DOI] [PubMed] [Google Scholar]
- 58.Bouchard C, Perusse L, Chagnon YC, Warden C, Ricquier D. Linkage between markers in the vicinity of the uncoupling protein 2 gene and resting metabolic rate in humans. Hum Mol Genet. 1997;6:1887–1889. doi: 10.1093/hmg/6.11.1887. [DOI] [PubMed] [Google Scholar]
- 59.Astrup A, Toubro S, Dalgaard LT, et al. Impact of the V/V 55 polymorphism of the uncoupling protein 2 gene on 24-h energy expenditure and substrate oxidation. Int J Obes Relat Metab Disord. 1999;23:1030–1034. doi: 10.1038/sj.ijo.0801040. [DOI] [PubMed] [Google Scholar]
- 60.Buemann B, Schierning B, Toubro S, et al. The association between the Val/Ala-55 polymorphism of the uncoupling protein 2 gene and exercise efficiency. Int J Obes Relat Metab Disord. 2001;25:467–471. doi: 10.1038/sj.ijo.0801564. [DOI] [PubMed] [Google Scholar]
- 61.Walder K, Norman RA, Hanson RL, et al. Association between uncoupling protein polymorphisms (UCP2-UCP3) and energy metabolism/obesity in Pima Indians. Hum Mol Genet. 1998;7:1431–1435. doi: 10.1093/hmg/7.9.1431. [DOI] [PubMed] [Google Scholar]
- 62.Kubota T, Mori H, Tamori Y, et al. Molecular screening of uncoupling protein 2 gene in patients with noninsulin-dependent diabetes mellitus or obesity. J Clin Endocrinol Metab. 1998;83:2800–2804. doi: 10.1210/jcem.83.8.4994. [DOI] [PubMed] [Google Scholar]
- 63.Urhammer SA, Dalgaard LT, Rensen TIA, et al. Mutational analysis of the coding region of the uncoupling protein 2 gene in obese NIDDM patients: Impact of a common amino acid polymorphism on juvenile and maturity onset forms of obesity and insulin resistance. Diabetologia. 1997;40:1227–1230. doi: 10.1007/s001250050811. [DOI] [PubMed] [Google Scholar]
- 64.Otabe S, Clement K, Rich N, et al. Mutation screening of the human UCP 2 gene in normoglycemic and NIDDM morbidly obese patients: Lack of association between new UCP 2 polymorphisms and obesity in French Caucasians. Diabetes. 1998;47:840–842. doi: 10.2337/diabetes.47.5.840. [DOI] [PubMed] [Google Scholar]
- 65.Yanovski JA, Diament AL, Sovik KN, et al. Associations between uncoupling protein 2, body composition, and resting energy expenditure in lean and obese African American, white, and Asian children. Am J Clin Nutr. 2000;71:1405–1420. doi: 10.1093/ajcn/71.6.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cassell PG, Neverova M, Janmohamed S, et al. An uncoupling protein 2 gene variant is associated with a raised body mass index but not type II diabetes. Diabetologia. 1999;42:688–692. doi: 10.1007/s001250051216. [DOI] [PubMed] [Google Scholar]
- 67.Evans D, Wolf AM, Nellessen U, et al. Association between polymorphisms in candidate genes and morbid obesity. Int J Obes Relat Metab Disord. 2001;25 (Suppl 1):S19–21. doi: 10.1038/sj.ijo.0801690. [DOI] [PubMed] [Google Scholar]
- 68.Bulotta A, Ludovico O, Coco A, et al. The common −866G/A polymorphism in the promoter region of the UCP-2 gene is associated with reduced risk of type 2 diabetes in Caucasians from Italy. J Clin Endocrinol Metab. 2005;90:1176–1180. doi: 10.1210/jc.2004-1072. [DOI] [PubMed] [Google Scholar]
- 69.D’Adamo M, Perego L, Cardellini M, et al. The −866A/A genotype in the promoter of the human uncoupling protein 2 gene is associated with insulin resistance and increased risk of type 2 diabetes. Diabetes. 2004;53:1905–1910. doi: 10.2337/diabetes.53.7.1905. [DOI] [PubMed] [Google Scholar]
- 70.Krempler F, Esterbauer H, Weitgasser R, et al. A functional polymorphism in the promoter of UCP2 enhances obesity risk but reduces type 2 diabetes risk in obese middle-aged humans. Diabetes. 2002;51:3331–3335. doi: 10.2337/diabetes.51.11.3331. [DOI] [PubMed] [Google Scholar]
- 71.Sasahara M, Nishi M, Kawashima H, et al. Uncoupling protein 2 promoter polymorphism 866G/A affects its expression in β-cells and modulates clinical profiles of Japanese type 2 diabetic patients. Diabetes. 2004;53:482–485. doi: 10.2337/diabetes.53.2.482. [DOI] [PubMed] [Google Scholar]
- 72.Reis AF, Dubois-Laforgue D, Bellanne-Chantelot C, Timsit J, Velho G. A polymorphism in the promoter of UCP2 gene modulates lipid levels in patients with type 2 diabetes. Mol Genet Metab. 2004;82:339–344. doi: 10.1016/j.ymgme.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 73.Sesti G, Cardellini M, Marini MA, et al. A common polymorphism in the promoter of UCP2 contributes to the variation in insulin secretion in glucose-tolerant subjects. Diabetes. 2003;52:1280–1283. doi: 10.2337/diabetes.52.5.1280. [DOI] [PubMed] [Google Scholar]
- 74.Ji Q, Ikegami H, Fujisawa T, et al. A common polymorphism of uncoupling protein 2 gene is associated with hypertension. J Hypertens. 2004;22:97–102. doi: 10.1097/00004872-200401000-00018. [DOI] [PubMed] [Google Scholar]
- 75.Dalgaard LT, Andersen G, Larsen LH, et al. Mutational analysis of the UCP2 core promoter and relationships of variants with obesity. Obes Res. 2003;11:1420–1427. doi: 10.1038/oby.2003.191. [DOI] [PubMed] [Google Scholar]
- 76.Mancini FP, Sabatino L, Colantuoni V, et al. Variants of uncoupling protein-2 gene and obesity: Interaction with peroxisome proliferator-activated receptor-©2. Clin Endocrinol. 2003;59:817–822. doi: 10.1046/j.1365-2265.2003.01926.x. [DOI] [PubMed] [Google Scholar]
- 77.Le Fur S, Le Stunff C, Dos Santos C, Bougneres P. The common −866 G/A polymorphism in the promoter of uncoupling protein 2 is associated with increased carbohydrate and decreased lipid oxidation in juvenile obesity. Diabetes. 2004;53:235–239. doi: 10.2337/diabetes.53.1.235. [DOI] [PubMed] [Google Scholar]
- 78.Wang H, Chu WS, Lu T, et al. Uncoupling protein-2 polymorphisms in type 2 diabetes, obesity, and insulin secretion. Am J Physiol Endocrinol Metab. 2004;286:E1–7. doi: 10.1152/ajpendo.00231.2003. [DOI] [PubMed] [Google Scholar]
- 79.Cho YM, Ritchie MD, Moore JH, et al. Multifactor-dimensionality reduction shows a two-locus interaction associated with type 2 diabetes mellitus. Diabetologia. 2004;47:549–554. doi: 10.1007/s00125-003-1321-3. [DOI] [PubMed] [Google Scholar]
- 80.Brown JE, Thomas S, Digby JE, Dunmore SJ. Glucose induces and leptin decreases expression of uncoupling protein-2 mRNA in human islets. FEBS Lett. 2002;513:189–192. doi: 10.1016/s0014-5793(02)02296-2. [DOI] [PubMed] [Google Scholar]
- 81.Chan CB, De Leo D, Joseph JW, et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: Mechanism of action. Diabetes. 2001;50:1302–1310. doi: 10.2337/diabetes.50.6.1302. [DOI] [PubMed] [Google Scholar]
- 82.Lameloise N, Muzzin P, Prentki M, Assimacopoulos-Jeannet F. Uncoupling protein 2: A possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes. 2001;50:803–809. doi: 10.2337/diabetes.50.4.803. [DOI] [PubMed] [Google Scholar]
- 83.Chan CB, Macdonald PE, Saleh MC, et al. Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes. 1999;48:1482–1486. doi: 10.2337/diabetes.48.7.1482. [DOI] [PubMed] [Google Scholar]
- 84.Zhou YT, Shimabukuro M, Koyama K, et al. Induction by leptin of uncoupling protein-2 and enzymes of fatty acid oxidation. Proc Natl Acad Sci USA. 1997;94:6386–6390. doi: 10.1073/pnas.94.12.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laybutt DR, Sharma A, Sgroi DC, et al. Genetic regulation of metabolic pathways in beta-cells disrupted by hyperglycemia. J Biol Chem. 2002;277:10912–10921. doi: 10.1074/jbc.M111751200. [DOI] [PubMed] [Google Scholar]
- 86.Kassis N, Bernard C, Pusterla A, et al. Correlation between pancreatic islet uncoupling protein-2 (UCP2) mRNA concentration and insulin status in rats. Int J Exp Diabetes Res. 2000;1:185–193. doi: 10.1155/EDR.2000.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joseph JW, Koshkin V, Saleh MC, et al. Free fatty acid-induced β-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279:51049–51056. doi: 10.1074/jbc.M409189200. [DOI] [PubMed] [Google Scholar]
- 88.Joseph JW, Koshkin V, Zhang CY, et al. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002;51:3211–3219. doi: 10.2337/diabetes.51.11.3211. [DOI] [PubMed] [Google Scholar]
- 89.Shimabukuro M, Zhou YT, Lee Y, Unger RH. Induction of uncoupling protein-2 mRNA by troglitazone in the pancreatic islets of Zucker diabetic fatty rats. Biochem Biophys Res Commun. 1997;237:359–361. doi: 10.1006/bbrc.1997.7140. [DOI] [PubMed] [Google Scholar]
- 90.Wang MY, Shimabukuro M, Lee Y, et al. Adenovirus-mediated overexpression of uncoupling protein-2 in pancreatic islets of Zucker diabetic rats increases oxidative activity and improves beta-cell function. Diabetes. 1999;48:1020–1025. doi: 10.2337/diabetes.48.5.1020. [DOI] [PubMed] [Google Scholar]
- 91.Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V. Induction of uncoupling protein 2 mRNA in beta-cells is stimulated by oxidation of fatty acids but not by nutrient oversupply. Endocrinology. 2002;143:1371–1377. doi: 10.1210/endo.143.4.8717. [DOI] [PubMed] [Google Scholar]
- 92.Patane G, Anello M, Piro S, et al. Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-gamma inhibition. Diabetes. 2002;51:2749–2756. doi: 10.2337/diabetes.51.9.2749. [DOI] [PubMed] [Google Scholar]
- 93.Winzell MS, Svensson H, Enerback S, et al. Pancreatic beta-cell lipotoxicity induced by overexpression of hormone-sensitive lipase. Diabetes. 2003;52:2057–2065. doi: 10.2337/diabetes.52.8.2057. [DOI] [PubMed] [Google Scholar]
- 94.Ito E, Ozawa S, Takahashi K, et al. PPAR-gamma overexpression selectively suppresses insulin secretory capacity in isolated pancreatic islets through induction of UCP-2 protein. Biochem Biophys Res Commun. 2004;324:810–814. doi: 10.1016/j.bbrc.2004.08.238. [DOI] [PubMed] [Google Scholar]
- 95.Tordjman K, Standley KN, Bernal-Mizrachi C, et al. PPARalpha suppresses insulin secretion and induces UCP2 in insulinoma cells. J Lipid Res. 2002;43:936–943. [PubMed] [Google Scholar]
- 96.Medvedev AV, Robidoux J, Bai X, et al. Regulation of the uncoupling protein-2 gene in INS-1 beta-cells by oleic acid. J Biol Chem. 2002;277:42639–42644. doi: 10.1074/jbc.M208645200. [DOI] [PubMed] [Google Scholar]
- 97.Eto K, Yamashita T, Matsui J, et al. Genetic manipulations of fatty acid metabolism in β-cells are associated with dysregulated insulin secretion. Diabetes. 2002;51:S414–420. doi: 10.2337/diabetes.51.2007.s414. [DOI] [PubMed] [Google Scholar]
- 98.Yamashita T, Eto K, Okazaki Y, et al. Role of uncoupling protein-2 up-regulation and triglyceride accumulation in impaired glucose-stimulated insulin secretion in a beta-cell lipotoxicity model overexpressing sterol regulatory element-binding protein-1c. Endocrinology. 2004;145:3566–3577. doi: 10.1210/en.2003-1602. [DOI] [PubMed] [Google Scholar]
- 99.Roduit R, Morin J, Masse F, et al. Glucose down-regulates the expression of the peroxisome proliferator-activated receptor-alpha gene in the pancreatic beta-cell. J Biol Chem. 2000;275:35799–35806. doi: 10.1074/jbc.M006001200. [DOI] [PubMed] [Google Scholar]
- 100.Krauss S, Zhang CY, Scorrano L, et al. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β cell dysfunction. J Clin Invest. 2003;112:1831–1842. doi: 10.1172/JCI19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang H, Antinozzi PA, Hagenfeldt KA, Maechler P, Wollheim CB. Molecular targets of a human HNF1〈 mutation responsible for pancreatic β-cell dysfunction. EMBO J. 2000;19:4257–4264. doi: 10.1093/emboj/19.16.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaestner KH. The hepatocyte nuclear factor 3 (HNF3 or Foxa) family in metabolism. Trends Endocrinol Metabol. 2000;11:281–285. doi: 10.1016/s1043-2760(00)00271-x. [DOI] [PubMed] [Google Scholar]
- 103.Lantz KA, Vatamaniuk MZ, Brestelli JE, et al. Foxa2 regulates multiple pathways of insulin secretion. J Clin Invest. 2004;114:512–520. doi: 10.1172/JCI21149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang H, Gauthier BR, Hagenfeldt-Johansson KA, Iezzi M, Wollheim CB. Foxa2 (HNF3beta) controls multiple genes implicated in metabolism-secretion coupling of glucose-induced insulin release. J Biol Chem. 2002;277:17564–17570. doi: 10.1074/jbc.M111037200. [DOI] [PubMed] [Google Scholar]
- 105.Seufert J, Weir GC, Habener JF. Differential expression of the insulin gene transcriptional repressor CCAAT/enhancer-binding protein beta and transactivator islet duodenum homeobox-1 in rat pancreatic beta cells during the development of diabetes mellitus. J Clin Invest. 1998;101:2528–2539. doi: 10.1172/JCI2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee F-YJ, Li Y, Yang EK, et al. Phenotypic abnormalities in macrophages from leptin-deficient, obese mice. Am J Physiol Cell Physiol. 1999;276:C386–394. doi: 10.1152/ajpcell.1999.276.2.C386. [DOI] [PubMed] [Google Scholar]
- 107.Merial C, Bouloumie A, Trocheris V, Lafontan M, Galitzky J. Nitric oxide-dependent downregulation of adipocyte UCP-2 expression by tumor necrosis factor-alpha. Am J Physiol Cell Physiol. 2000;279:C1100–1106. doi: 10.1152/ajpcell.2000.279.4.C1100. [DOI] [PubMed] [Google Scholar]
- 108.Masaki T, Yoshimatsu H, Chiba S, et al. Tumor necrosis factor-alpha regulates in vivo expression of the rat UCP family differentially. Biochim Biophys Acta. 1999;1436:585–592. doi: 10.1016/s0005-2760(98)00173-8. [DOI] [PubMed] [Google Scholar]
- 109.Memon RA, Hotamisligil GS, Wiesbrock SM, et al. Upregulation of uncoupling protein 2 mRNA in genetic obesity: Lack of an essential role for leptin, hyperphagia, increased tissue lipid content, and TNF-〈. Biochim Biophys Acta. 2000;1484:41–50. doi: 10.1016/s1388-1981(99)00195-x. [DOI] [PubMed] [Google Scholar]
- 110.Busquets S, Alvarez B, Van Royen M, et al. Increased uncoupling protein-2 gene expression in brain of lipopolysaccharide-injected mice: Role of tumour necrosis factor-〈? Biochim Biophys Acta. 2001;1499:249–256. doi: 10.1016/s0167-4889(00)00126-9. [DOI] [PubMed] [Google Scholar]
- 111.Lee FY, Li Y, Zhu H, et al. Tumor necrosis factor increases mitochondrial oxidant production and induces expression of uncoupling protein-2 in the regenerating mice liver. Hepatology. 1999;29:677–687. doi: 10.1002/hep.510290320. [DOI] [PubMed] [Google Scholar]
- 112.Li LX, Yoshikawa H, Egeberg KW, Grill V. Interleukin-1beta swiftly down-regulates UCP-2 mRNA in beta-cells by mechanisms not directly coupled to toxicity. Cytokine. 2003;23:101–107. doi: 10.1016/s1043-4666(03)00204-7. [DOI] [PubMed] [Google Scholar]
- 113.Rial E, Gonzalez-Barroso M, Fleury C, et al. Retinoids activate proton transport by the uncoupling proteins UCP1 and UCP2. Embo J. 1999;18:5827–5833. doi: 10.1093/emboj/18.21.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jaburek M, Varecha M, Gimeno RE, et al. Transport function and regulation of mitochondrial uncoupling proteins 2 and 3. J Biol Chem. 1999;274:26003–26007. doi: 10.1074/jbc.274.37.26003. [DOI] [PubMed] [Google Scholar]
- 115.Chavin KD, Yang S, Lin HZ, et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem. 1999;274:5692–5700. doi: 10.1074/jbc.274.9.5692. [DOI] [PubMed] [Google Scholar]
- 116.Fink BD, Hong YS, Mathahs MM, et al. UCP2-dependent proton leak in isolated mammalian mitochondria. J Biol Chem. 2002;277:3918–3925. doi: 10.1074/jbc.M107955200. [DOI] [PubMed] [Google Scholar]
- 117.Echtay KS, Winkler E, Frischmuth K, Klingenberg M. Uncoupling proteins 2 and 3 are highly active H(+) transporters and highly nucleotide sensitive when activated by Coenzyme Q (ubiquinone) Proc Natl Acad Sci USA. 2001;98:1416–1421. doi: 10.1073/pnas.98.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Krauss S, Zhang C-Y, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci USA. 2002;99:118–122. doi: 10.1073/pnas.012410699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hong Y, Fink BD, Dillon JS, Sivitz WI. Effects of adenoviral overexpression of uncoupling protein-2 and −3 on mitochondrial respiration in insulinoma cells. Endocrinology. 2001;142:249–256. doi: 10.1210/endo.142.1.7889. [DOI] [PubMed] [Google Scholar]
- 120.Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic β-cells exposed to free fatty acids. J Biol Chem. 2003;278:19709–19715. doi: 10.1074/jbc.M209709200. [DOI] [PubMed] [Google Scholar]
- 121.Klingenberg M, Echtay KS. Uncoupling proteins: The issues from a biochemist point of view. Biochim Biophys Acta. 2001;1504:128–143. doi: 10.1016/s0005-2728(00)00242-5. [DOI] [PubMed] [Google Scholar]
- 122.Nakazaki M, Kakei M, Ishihara H, et al. Association of upregulated activity of KATP channels with impaired insulin secretion in UCP1-expressing insulinoma cells. J Physiol (Lond) 2002;540:781–789. doi: 10.1113/jphysiol.2001.013048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Joseph JW, Chan CB, Wheeler MB. UCP2 knockout mouse islets have lower consumption and faster oscillations of beta cell oxygen. Can J Diabetes. 2005;29:19–26. [Google Scholar]
- 124.Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002;277:47129–47135. doi: 10.1074/jbc.M208262200. [DOI] [PubMed] [Google Scholar]
- 125.Echtay KS, Esteves TC, Pakay JL, et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. Embo J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shimabukuro M, Koyama K, Chen G, et al. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci USA. 1997;94:4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang H, Maechler P, Antinozzi PA, et al. The transcription factor SREBP-1c is instrumental in the development of beta-cell dysfunction. J Biol Chem. 2003;278:16622–16629. doi: 10.1074/jbc.M212488200. [DOI] [PubMed] [Google Scholar]
- 128.Winzell MS, Svensson H, Enerback S, et al. Pancreatic beta-cell lipotoxicity induced by overexpression of hormone-sensitive lipase. Diabetes. 2003;52:2057–2065. doi: 10.2337/diabetes.52.8.2057. [DOI] [PubMed] [Google Scholar]
- 129.Rubi B, Antinozzi PA, Herrero L, et al. Adenovirus-mediated overexpression of liver carnitine palmitoyltransferase I in INS1E cells: Effects on cell metabolism and insulin secretion. Biochem J. 2002;364:219–226. doi: 10.1042/bj3640219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Antinozzi PA, Ishihara H, Newgard CB, Wollheim CB. Mitochondrial metabolism sets the maximal limit of fuel-stimulated insulin secretion in a model pancreatic beta cell. A survey of four fuel secretagogues. J Biol Chem. 2002;277:11746–11755. doi: 10.1074/jbc.M108462200. [DOI] [PubMed] [Google Scholar]
- 131.Arsenijevic D, Onuma H, Pecqueur C, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 132.Bai Y, Onuma H, Bai X, et al. Persistent nuclear factor-| ℜ activation in UCP2-/- mice leads to enhanced nitric oxide and inflammatory cytokine production. J Biol Chem. 2005;280:19062–19069. doi: 10.1074/jbc.M500566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V. Uncoupling protein-2 participates in cellular defense against oxidative stress in clonal beta-cells. Biochem Biophys Res Commun. 2001;282:273–277. doi: 10.1006/bbrc.2001.4577. [DOI] [PubMed] [Google Scholar]
- 134.Murphy MP, Echtay KS, Blaikie FH, et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: Studies using a mitochondria-targeted spin trap derived from 〈-phenyl-n-tert-butylnitrone. J Biol Chem. 2003;278:48534–48545. doi: 10.1074/jbc.M308529200. [DOI] [PubMed] [Google Scholar]
- 135.Goglia F, Skulachev VP. A function for novel uncoupling proteins: Antioxidant defense of mitochondrial matrix by translocating fatty acid peroxides from the inner to the outer membrane leaflet. FASEB J. 2003;17:1585–1591. doi: 10.1096/fj.03-0159hyp. [DOI] [PubMed] [Google Scholar]
- 136.Jaburek M, Miyamoto S, Di Mascio P, Garlid KD, Jezek P. Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. J Biol Chem. 2004;279:53097–53102. doi: 10.1074/jbc.M405339200. [DOI] [PubMed] [Google Scholar]
- 137.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 138.Pecqueur C, Couplan E, Bouillaud F, Ricquier D. Genetic and physiological analysis of the role of uncoupling proteins in human energy homeostasis. J Mol Med. 2001;79:48–56. doi: 10.1007/s001090000150. [DOI] [PubMed] [Google Scholar]
- 139.Krook A, Digby J, O’Rahilly S, Zierath JR, Wallberg-Henriksson H. Uncoupling protein 3 is reduced in skeletal muscle of NIDDM patients. Diabetes. 1998;47:1528–1531. doi: 10.2337/diabetes.47.9.1528. [DOI] [PubMed] [Google Scholar]
- 140.Schrauwen P, Hesselink MKC, Blaak EE, et al. Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2001;50:2870–2873. doi: 10.2337/diabetes.50.12.2870. [DOI] [PubMed] [Google Scholar]
- 141.Russell AP, Gastaldi G, Bobbioni-Harsch E, et al. Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: A case of good vs. bad lipids? FEBS Letters. 2003;551:104–106. doi: 10.1016/s0014-5793(03)00875-5. [DOI] [PubMed] [Google Scholar]
- 142.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 143.Khalfallah Y, Fages S, Laville M, Langin D, Vidal H. Regulation of uncoupling protein-2 and uncoupling protein-3 mRNA expression during lipid infusion in human skeletal muscle and subcutaneous adipose tissue. Diabetes. 2000;49:25–31. doi: 10.2337/diabetes.49.1.25. [DOI] [PubMed] [Google Scholar]
- 144.Schrauwen P, Hesselink MKC. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 145.Brand MD. The proton leak across the mitochondrial inner membrane. Biochim Biophys Acta. 1990;1018:128–133. doi: 10.1016/0005-2728(90)90232-s. [DOI] [PubMed] [Google Scholar]
- 146.Harper ME, Brand MD. The quantitative contributions of mitochondrial proton leak and ATP turnover reactions to the changed respiration rates of hepatocytes from rats of different thyroid status. J Biol Chem. 1993;268:14850–14860. [PubMed] [Google Scholar]
- 147.Rolfe DF, Brand MD. The physiological significance of mitochondrial proton leak in animal cells and tissues. Biosci Rep. 1997;17:9–16. doi: 10.1023/a:1027327015957. [DOI] [PubMed] [Google Scholar]
- 148.Stuart JA, Cadenas S, Jekabsons MB, Roussel D, Brand MD. Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochim Biophys Acta. 2001;1504:144–158. doi: 10.1016/s0005-2728(00)00243-7. [DOI] [PubMed] [Google Scholar]
- 149.Dulloo AG, Samec S. Uncoupling proteins: Their roles in adaptive thermogenesis and substrate metabolism reconsidered. Br J Nutr. 2001;86:123–139. doi: 10.1079/bjn2001412. [DOI] [PubMed] [Google Scholar]
- 150.Argyropoulos G, Harper ME. Uncoupling proteins and thermoregulation. J Appl Physiol. 2002;92:2187–2198. doi: 10.1152/japplphysiol.00994.2001. [DOI] [PubMed] [Google Scholar]
- 151.Himms-Hagen J, Harper ME. Physiological role of UCP3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: An hypothesis. Exp Biol Med. 2001;226:78–84. doi: 10.1177/153537020122600204. [DOI] [PubMed] [Google Scholar]
- 152.Gong D-W, Monemdjou S, Gavrilova O, et al. Lack of obesity and normal response to fasting and thyroid hormone in mice lacking uncoupling protein-3. J Biol Chem. 2000;275:16251–16257. doi: 10.1074/jbc.M910177199. [DOI] [PubMed] [Google Scholar]
- 153.Vidal-Puig AJ, Grujic D, Zhang CY, et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. 2000;275:16258–16266. doi: 10.1074/jbc.M910179199. [DOI] [PubMed] [Google Scholar]
- 154.Bezaire V, Hofmann W, Kramer JKG, Kozak LP, Harper M-E. Effects of fasting on muscle mitochondrial energetics and fatty acid metabolism in UCP3(−/−) and wild-type mice. Am J Physiol Endocrinol Metab. 2001;281:E975–982. doi: 10.1152/ajpendo.2001.281.5.E975. [DOI] [PubMed] [Google Scholar]
- 155.Cadenas S, Echtay KS, Harper JA, et al. The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. J Biol Chem. 2002;277:2773–2778. doi: 10.1074/jbc.M109736200. [DOI] [PubMed] [Google Scholar]
- 156.Argyropoulos G, Brown AM, Willi SM, et al. Effects of mutations in the human uncoupling protein 3 gene on the respiratory quotient and fat oxidation in severe obesity and type 2 diabetes. J Clin Invest. 1998;102:1345–1351. doi: 10.1172/JCI4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Clapham JC, Arch JR, Chapman H, et al. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature. 2000;406:415–418. doi: 10.1038/35019082. [DOI] [PubMed] [Google Scholar]
- 158.Son C, Hosoda K, Ishihara K, et al. Reduction of diet-induced obesity in transgenic mice overexpressing uncoupling protein 3 in skeletal muscle. Diabetologia. 2004;47:47–54. doi: 10.1007/s00125-003-1272-8. [DOI] [PubMed] [Google Scholar]
- 159.Himms Hagen J. On raising energy expenditure in ob/ob mice. Science. 1997;276:1132–1133. doi: 10.1126/science.276.5315.1132. [DOI] [PubMed] [Google Scholar]
- 160.Bezaire V, Spriet LL, Campbell S, et al. Constitutive UCP3 overexpression at physiological levels increases mouse skeletal muscle capacity for fatty acid transport and oxidation. FASEB J. 2005;19:977–979. doi: 10.1096/fj.04-2765fje. [DOI] [PubMed] [Google Scholar]
- 161.Cadenas S, Buckingham JA, Samec S, et al. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Letters. 1999;462:257–260. doi: 10.1016/s0014-5793(99)01540-9. [DOI] [PubMed] [Google Scholar]
- 162.Echtay KS, Roussel D, St-Pierre J, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 163.Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. 2004;279:38236–38248. doi: 10.1074/jbc.M402375200. [DOI] [PubMed] [Google Scholar]
- 164.Schrauwen P, Saris WHM, Hesselink MKC. An alternative function for human uncoupling protein 3: Protection of mitochondria against accumulation of nonesterified fatty acids inside the mitochondrial matrix. FASEB J. 2001;15:2497–2502. doi: 10.1096/fj.01-0400hyp. [DOI] [PubMed] [Google Scholar]
- 165.Alexson SE, Nedergaard J. A novel type of short- and medium-chain acyl-CoA hydrolases in brown adipose tissue mitochondria. J Biol Chem. 1988;263:13564–13571. [PubMed] [Google Scholar]
- 166.Moore GB, Himms-Hagen J, Harper ME, Clapham JC. Overexpression of UCP-3 in skeletal muscle of mice results in increased expression of mitochondrial thioesterase mRNA. Biochem Biophys Res Commun. 2001;283:785–790. doi: 10.1006/bbrc.2001.4848. [DOI] [PubMed] [Google Scholar]
- 167.Moreno M, Lombardi A, De Lange P, et al. Fasting, lipid metabolism, and triiodothyronine in rat gastrocnemius muscle: Interrelated roles of uncoupling protein 3, mitochondrial thioesterase, and Coenzyme Q. FASEB J. 2003;17:1112–1114. doi: 10.1096/fj.02-0839fje. [DOI] [PubMed] [Google Scholar]
- 168.MacLellan JD, Gerrits MF, Gowing A, et al. Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes. 2005;54:2343–2350. doi: 10.2337/diabetes.54.8.2343. [DOI] [PubMed] [Google Scholar]