

Abstract
Purpose
A Phase I study to define toxicity and recommend a Phase II dose of the HSP90 inhibitor alvespimycin (17-DMAG; 17-dimethylaminoethylamino-17-demethoxygeldanamycin). Secondary endpoints included evaluation of pharmacokinetic profile, tumor response and definition of a biologically effective dose (BED).
Patients and Methods
Patients with advanced solid cancers were treated with weekly, intravenous (IV) 17-DMAG. An accelerated titration dose escalation design was used. The maximum tolerated dose (MTD) was the highest dose at which ≤ 1/6 patients experienced dose limiting toxicity (DLT). Dose de-escalation from the MTD was planned with mandatory, sequential tumor biopsies to determine a BED. Pharmacokinetic and pharmacodynamic assays were validated prior to patient accrual.
Results
Twenty five patients received 17-DMAG (range 2.5 to 106 mg/m2). At 106mg/m2 of 17-DMAG 2/4 patients experienced DLT, including one treatment related death. No DLT occurred at 80mg/m2. Common adverse events were gastrointestinal, liver function changes and ocular. AUC and Cmax increased proportionally with 17-DMAG doses ≤ 80mg/m2. In peripheral blood mononuclear cells significant (p <0.05) HSP72 induction was detected (≥ 20mg/m2) and sustained for 96 hours (≥ 40mg/m2). Plasma HSP72 levels were greatest in the two patients who experienced DLT. At 80mg/m2 client protein (CDK4, LCK) depletion was detected and tumor samples from 3/5 patients confirmed HSP90 inhibition. Clinical activity included complete response (castration refractory prostate cancer, CRPC 124 weeks), partial response (melanoma, 159 weeks) and stable disease (chondrosarcoma, CRPC and renal cancer for 28, 59 and 76 weeks respectively).
Conclusion
The recommended Phase II dose of 17-DMAG is 80mg/m2 weekly, IV.
INTRODUCTION
The molecular chaperone HSP90 ensures correct folding and function of numerous client proteins [1-4] including the androgen receptor and oncogenic kinases such as BRAF (for an up to date list visit website of Dr Didier Picard *). HSP90 inhibition targets client proteins for proteasomal destruction [3]. The resulting combined effect on multiple oncogenic client proteins, their associated biochemical pathways, and hallmark cancer traits [5] forms the basis for the observed anticancer activity [6-10].
HSP90 inhibition results in a well-characterized, mechanism-based change in expression of specific proteins [11, 12]. Depletion of client proteins (e.g. CDK4, ERBB2 or LCK) together with induction of certain heat shock proteins (e.g. HSP72, the inducible isoform of HSP70) constitute a molecular signature of HSP90 inhibition that can be measured as a pharmacodynamic endpoint [13-15].
The HSP90 inhibitor alvespimycin (17-dimethylaminoethylamino-17-demethoxygeldanamycin; 17-DMAG) exhibits reduced metabolic liability, lower plasma protein binding, increased water solubility higher oral bioavailability and superior antitumor activity compared to tanespimycin (17-allylamino-17-demethoxy geldanamycin; 17-AAG), the first HSP90 inhibitor in clinical trials [10].
Selectivity of HSP90 inhibitors for tumor over normal tissue was demonstrated [6, 16] and, like 17-AAG, 17-DMAG is retained longer in tumor than in normal tissue [17]. We postulated that obtaining a biologically effective dose (BED) lower than the MTD may be possible.
The primary objective was evaluation of drug safety and recommendation of a phase II dose. Secondary objectives were to investigate the pharmacokinetic and pharmacodynamic properties, define a BED and evaluate tumor response.
Other phase I studies of 17-DMAG performed concurrently utilized different schedules and administration routes [18-20]. Pre-clinical studies confirmed anti-cancer activity of 17-DMAG using a variety of dosing schedules [8]. We proposed a weekly schedule also based on experience with 17-AAG, for which weekly administration was convenient, deliverable with manageable toxicity and showed potential clinical activity whereas schedules with increased dosing frequency (e.g. daily) were more toxic [21, 22]. This present study is to our knowledge the only one to incorporate pharmacodynamic assays validated before patient accrual [23-25]. Additionally, the 3+3 design facilitated investigation of the pharmacokinetic profile of 17-DMAG and evidence of target inhibition was obtained.
PATIENTS AND METHODS
Study design
A phase I trial of weekly IV 17-DMAG was performed with dose escalation (to determine MTD) and planned subsequent dose de-escalation (to define a BED).
The starting dose was 2.5 mg/m2, approximately 1/10th the dose lethal (LD10) to dogs [7]. The study design incorporated an accelerated dose escalation scheme [26]. Toxicities were assessed using NCI-CTCAE version 3.0. Dose limiting toxicities (DLT) were defined as any of the following causally related to 17-DMAG within the first 28 days of treatment: absolute neutrophil count < 0.5×109/l for > 5 days or with associated fever; platelet count < 25×109/l; any other non-hematological toxicity (≥ Grade 3) except nausea, vomiting, diarrhea, rash, arthralgia or myalgia without appropriate prophylactic measures or alopecia (grade 1 or 2); or toxicity that prevented completion of 4 weeks 17-DMAG treatment. Patients who did not complete four weeks 17-DMAG for reasons other than toxicity were replaced.
Cohorts of three patients were entered and dose doubling performed until ≥ Grade 2 toxicity occurred. Further dose escalations were limited to 50%, in event of Grade 2 toxicity or 33% following ≥ Grade 3 toxicity.
After observing DLT, the cohort increased to 6 patients maximum. The maximum administered dose (MAD) was that at which ≥ 2/6 patients experienced DLT. The MTD was the previous dose level tested at which ≤ 1/6 patients experienced DLT.
The first patient at each dose level completed two weeks of 17-DMAG prior to other patients being treated. No delay was mandated between treating the second and subsequent patients.
Pre and post 17-DMAG tumor biopsies were planned. Once MTD was determined, additional patients with biopsiable disease were entered, initially at MTD level, to yield five, paired, pre and post dose biopsies per dose cohort. Detection of HSP90 inhibition (HSP72 induction with either CDK4 and/or ERBB2 depletion) in tumor from ≥ 4/5 patients allowed dose de-escalation to the prior dose level. A BED was defined as the lowest dose at which the HSP90 inhibition was detected in tumor samples from ≥ 4/5 patients.
The study was conducted under a Clinical Trial Authorisation (CTA, number 21106/0224/001) sponsored by Cancer Research UK, and monitored by the Cancer Research UK Drug Development Office (DDO). The study was managed and conducted in accordance with the principles of Good Clinical Practice and according to Cancer Research-UK DDO's Standard Operating Procedures. Two centres participated, the Royal Marsden NHS Foundation Trust, Sutton, UK and the Belfast City Hospital, Belfast, N. Ireland, UK. The protocol was reviewed by the Cancer Research UK Central Internal Review Board, the NCI, the Metropolitan Multi-centre Research Ethics Committee (Southampton) and clinical research committees of both institutions. The trial was registered on the NCI Clinical Trials Registry (NCT 00248521). Patients gave informed, written consent prior to study entry with additional consent for tumor biopsies.
Inclusion and exclusion criteria
Patients, aged ≥ 18 years, with histologically/cytologically confirmed solid tumors refractory to available therapy were entered. Prior treatment, radiotherapy (except for palliative reasons), endocrine therapy, immunotherapy or chemotherapy, was completed at least four weeks (six weeks for nitrosoureas and mitomycin-C) prior to 17-DMAG. All toxic manifestations of previous treatments had resolved (except alopecia or peripheral neuropathy CTCAE Grade 1 allowed). Concomitant use of bisphosphonates, erythropoietin or LHRH analogues in patients with castration resistant prostate cancer (CRPC) and a rising PSA were allowed. ECOG performance status was 0/1 and patients' life expectancy estimated to exceed 12 weeks. Adequate organ function was defined as: ANC > 1.5×109/l, platelets ≥ 100×109/l, haemoglobin ≥ 9.0 g/dl, serum creatinine within normal limits (WNL) or calculated creatinine clearance WNL, plasma bilirubin WNL, ALT /AST ≤ 1.5 × ULN. All patients agreed to use appropriate contraception.
Exclusion criteria were pregnancy, lactation, prior therapy with 17-AAG (there was no restriction on prior treatment with any tyrosine kinase inhibitor or monoclonal antibody), active treatment with another anti-cancer investigational agent, known CNS metastases, uncontrolled intercurrent illness, active second malignancy, patients known to be hepatitis B, C or HIV positive, left bundle branch block, serious ventricular dysrhythmia, symptomatic pulmonary disease requiring medication, moderate/severe dry eye syndrome or corneal disease.
Drug administration
17-DMAG was supplied by the NCI and Kosan Biosciences. The final concentration for intravenous administration was 0.1-1.0 mg/mL in 0.9% saline or 5% dextrose. Drug was administered over one hour, every week, continuously and one cycle was defined as 4 weeks of treatment.
Dose adjustments
Dose reductions to the previous dose tested were made for patients who experienced DLT or toxicity risking patient safety. Patients were allowed re-treatment at full dose on days 8, 15 or 22 of a cycle where ANC > 1.0×109/l, platelets > 75×109/l and other drug-related toxicity had resolved to ≤ Grade 1 (allowing alopecia, nausea, vomiting or diarrhea if appropriate prophylactic or therapeutic measures not undertaken).
Pharmacokinetic sampling and analysis
Plasma concentrations of 17-DMAG were analyzed using high performance liquid chromatography-mass spectroscopy (HPLC-MS). During the first course of 17-DMAG blood samples were taken prior to, during (30 and 60 minutes after infusion commenced) and 5, 15, 30, 60, and 90 minutes, 2, 4, 6, 8, 16, 24, 48, 72 and 96 hours after the end of infusion. Blood samples (5 mL) were collected into heparinized tubes and stored on ice until centrifuged at 252g for five minutes at 4°C to obtain plasma which was stored at −80°C until analyzed.
The analytical method was validated prior to trial recruitment [27]. Pharmacokinetics were analyzed using a non-compartmental model (model 202), with constant infusion input for plasma using WinNonLin software® version 5.2. Dose proportionality was assessed by linear regression.
Pharmacodynamic sampling and analyses
Western Blotting
Blood samples were collected into BD Vacutainer™ tubes for analysis pre-dose, end of infusion and 1, 8, 24, 48 and 96 hours after 17-DMAG. A further sample was taken 24 hours after the 5th weekly infusion. Peripheral blood mononuclear cells (PBMC) were separated using the Ficoll Hypaque method and stored at −80°C. Tumor biopsies were taken before and 24 hours after first 17-DMAG dose, snap frozen and stored at −80°C. Samples were lysed and analyzed using previously reported methods [15, 22, 24]; full method details are in supplementary data. Prior to study recruitment, measurement of HSP72, CDK4 and ERBB2 protein expression by western blotting [24, 28] were validated as fit for purpose [23] to measure HSP90 inhibition in tumor or PBMC samples following 17-DMAG administration. The validation package addressed sample acquisition, storage and stability as well as assay specificity and inter- and intra-assay variation and included experiments designed to replicate study conditions in relevant tissues (human PBMC and human tumor xenografts) [24, 25, 29]. LCK was also detected by western blot but considered as a research endpoint. Assay validation was assessed independently by Cancer Research UK DDO and passed audit inspection by the UK Medicines Healthcare & Regulatory Authority (MHRA).
According to the validated and audited method, results from each time-point were compared visually to pre-treatment levels for each protein of interest and scored from 0-5 (0 no signal, 1 barely visible after 10 minutes exposure, 2 less than positive control but visible, 3 equivalent to positive control, 4 greater than positive control and 5 over exposed). A pharmacodynamic effect was recorded if a one point change was observed (client proteins down or HSP72 up); see also supplementary Figure 1. Tumor biopsy results were verified by two blinded, experienced assessors. Additional quantification was performed, although not externally validated, using ImageQuant™ software and protein levels were normalized to corresponding GAPDH control.
ELISA
Blood samples were collected pre-dose and 24 hours after 17-DMAG for HSP72 measurement in plasma and PBMC by ELISA / Dissociation Enhanced Lanthanide Fluorescent Immunoassay (DELFIA) format. PBMC were separated as above and stored at -80C until assay. Analytical methods [30] are available as supplementary data. Descriptive statistics and histograms were used. HSP72 was expressed as the change in HSP72 measured per unit of total protein or plasma. Mean change for each cohort was compared to mean change for the first cohort and analyzed for statistical significance (p ≤ 0.05) using a one-tailed t-test.
Characterization of response
Tumors were assessed before 17-DMAG and eight weekly using RECIST criteria version 1.0 [31], CA125 [32] or PSA [33] criteria. All responses (CR or PR) were confirmed with repeat measurements not less than four weeks apart and were reviewed by an independent clinician and radiologist.
RESULTS
Demographics
Between February 2006 and April 2008, 25 patients were recruited to the study and all received at least one 17-DMAG dose (Table 1). The male: female ratio was 14:11, with median age of 58 (range 38-78) years. Malignant melanoma (7/25) was the commonest histological subtype. All patients had an ECOG performance status of 0 or 1.
Table 1.
Summary of patient demographic details.
| Number | ||
|---|---|---|
|
| ||
| Patients treated | 25 | |
|
| ||
| Male: Female ratio | 14:11 | |
|
| ||
| Age, years | ||
| Median | 58 | |
| Range | 38-78 | |
|
| ||
| Performance status | ||
| 0 | 9 | |
| 1 | 16 | |
|
| ||
| Tumor type | ||
| Melanoma | 7 | |
| Prostate | 3 | |
| Breast | 3 | |
| Soft tissue sarcoma | 3 | |
| Pancreas | 2 | |
| Colon | 2 | |
| Cervix | 2 | |
| Kidney | 1 | |
| Uterine | 1 | |
| Cholangiocarcinoma | 1 | |
|
| ||
| Prior therapy | ||
| Surgery | 23 | |
| Chemotherapy (prior lines) | - 1 | 5 |
| - 2 | 9 | |
| - ≥3 | 9 | |
| Molecularly targeted agent | 10 | |
| Radiotherapy | 9 | |
Dose escalation and de-escalation
The starting dose was 2.5 mg/m2 which doubled incrementally to 80mg/m2 except for one single larger escalation from 5 to 20mg/m2 (based on safety data from parallel 17-DMAG studies).
In the first cohort, one patient experienced grade 3 lymphopenia and at 5mg/m2 grade 3 hyponatremia was detected in one patient. Both events occurred after completion of cycle one, not influencing dose escalation. One additional patient was added in the 5mg/m2 and 80mg/m2 cohorts to replace patients who progressed early. Further Grade 2 toxicity related to 17-DMAG was not reported until 80mg/m2 (fatigue, vomiting, blurred vision and dry eye in two patients).
The next dose level was 106 mg/m2 (chosen in light of toxicity data from parallel 17-DMAG studies). DLT occurred (2/4 patients), which was Grade 3 fatigue and hypoalbuminemia in one patient. The fourth patient in this cohort, with malignant melanoma, experienced rapid (within 24 hours of treatment) onset Grade 4 AST rise, Grade 3 diarrhea with Grade 2 nausea, vomiting, fever and anorexia. Subsequent Grade 4 hypotension and Grade 3 dehydration, hyponatremia, acidosis with creatinine elevation preceded anuric renal failure by day four post treatment. Dialysis was commenced; however, the patient died 5 days following the last dose of 17-DMAG. An autopsy request was declined, cause of death was assessed as related to 17-DMAG. Two other patients were treated at 106mg/m2; one died 16 days after receiving 17-DMAG following a gastro-intestinal hemorrhage, subsequent pulmonary edema and myocardial infarction. Endoscopy (gastroscopy and colonoscopy) confirmed that colonic infiltration by tumor caused the hemorrhage and subsequent events were not attributed to 17-DMAG. Rapid disease progression necessitated removal and replacement of the third patient in this cohort.
Four additional patients were entered at 80mg/m2 to generate five evaluable pre- and post-17-DMAG tumor biopsies. The criteria for further dose de-escalation were not met; therefore the study was declared complete and closed. No DLT occurred in eight patients who received 80 mg/m2 17-DMAG.
Toxicity
17-DMAG was well tolerated at doses ≤ 80mg/m2. Common adverse events (AEs) of nausea, vomiting, fatigue and liver enzyme disturbances were low grade and reversible (Table 2). Four patients experienced ten ocular AEs related to 17-DMAG, comprising blurred vision (three), dry eye (three), keratitis (two), conjunctivitis or ocular surface disease (two). Most (9/10) events occurred at ≥ 80mg/m2 and all were ≤ Grade 2; 2 patients required a dose reduction. At 106mg/m2, severe toxicities were encountered including one treatment related death.
Table 2.
Summary listing of all adverse events related to 17-DMAG.
| System | Adverse event | Grade |
||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
|
| ||||||
| Blood/bone marrow | Lymphopenia | 2 | - | 1 | - | - |
| Thrombocytopenia | 3 | - | 1 | - | - | |
| Raised neutrophils | 2 | - | - | - | - | |
| Raised leucocytes | 1 | - | - | - | - | |
| Low hematocrit | 1 | - | - | - | - | |
| Low red cell count | 1 | - | - | - | - | |
| Eosinophilia | 1 | - | - | - | - | |
| Low eosinophils | 1 | - | - | - | - | |
|
| ||||||
| Cardiac | Hypotension | - | - | - | 1 | - |
| Hypertension | 3 | - | - | - | - | |
| Sinus tachycardia | 1 | - | - | - | - | |
|
| ||||||
| Constitutional | Fatigue | - | 2 | 1 | - | - |
| Alopecia | 2 | - | - | - | - | |
| Fever | 2 | 1 | - | - | - | |
| Weight loss | 1 | - | - | - | - | |
|
| ||||||
| Gastrointestinal | Vomiting | 2 | 4 | - | - | - |
| Nausea | 5 | 1 | - | - | - | |
| Diarrhea | 5 | 1 | 1 | - | - | |
| Dehydration | 1 | - | 1 | - | - | |
| Anorexia | 1 | 1 | - | - | - | |
| Heartburn | 1 | - | - | - | - | |
|
| ||||||
| Metabolic/ laboratory | Hepatic | |||||
| AST | 4 | - | 1 | 1 | - | |
| ALT | 2 | 1 | 1 | - | - | |
| γGT | - | - | 1 | - | - | |
| ALP | 2 | 1 | - | - | - | |
| Raised bilirubin | 1 | - | - | - | - | |
| Non-hepatic | ||||||
| Hyponatraemia | 3 | - | 1 | - | - | |
| Acidosis | - | - | 1 | - | - | |
| Creatinine | - | - | 1 | - | - | |
| Hypokalaemia | 2 | - | - | - | - | |
| Hypergylcaemia | 2 | - | - | - | - | |
| Raised amylase | 1 | - | - | - | - | |
| Raised CK | 1 | - | - | - | - | |
| Hyperuricaemia | 1 | - | - | - | - | |
| Hypocalcaemia | 1 | - | - | - | - | |
| High protein | 1 | - | - | - | - | |
| Low urea | 1 | - | - | - | - | |
|
| ||||||
| Ocular/ visual | Blurred vision | 2 | 2 | - | - | - |
| Dry eye | 1 | 2 | - | - | - | |
| Keratitis | - | 2 | - | - | - | |
| Eye (herpes simplex) | - | 1 | - | - | - | |
| Ocular surface disease | 2 | - | - | - | - | |
|
| ||||||
| Pain | Joint | 5 | 2 | - | - | - |
| Headache | 1 | - | - | - | - | |
| Limb | 1 | - | - | - | - | |
| Myalgia | 2 | - | - | - | - | |
| Abdomen | 1 | - | - | - | - | |
|
| ||||||
| Renal | Renal failure | - | - | - | - | 1 |
| Proteinuria | 3 | - | - | - | - | |
| Oliguria | 1 | - | - | - | - | |
|
| ||||||
| Other | Limb edema | 2 | - | - | - | - |
| Dry mouth | 3 | - | - | - | - | |
| Dizziness | 1 | - | - | - | - | |
| Depression | 1 | - | - | - | - | |
| Neuropathy | 1 | - | - | - | - | |
| Vivid dreams | 1 | - | - | - | - | |
| Nightmares | 1 | - | - | - | - | |
| Rash | 1 | - | - | - | - | |
| Voice change | 1 | - | - | - | - | |
| Muscle spasm | 1 | - | - | - | - | |
| Hand foot syndrome | 1 | - | - | - | - | |
Pharmacokinetics of 17-DMAG
Table 3 summarizes the pharmacokinetic data for each cohort. At the MTD, 80mg/m2, plasma 17-DMAG concentration exceeded 63nM (mean IC50 for 17-DMAG in the NCI 60 human tumor cell line panel) for >24 hours in all patients (Figure 1A). At this dose the mean volume of distribution was 385 L, mean clearance 18.9 L/hr and mean peak concentration (Cmax) 2680 nmol/L. Both the area under the curve (AUC) and Cmax of 17-DMAG increased proportional to drug dose ≤ 80mg/m2 (r2 values 0.88 and 0.75 respectively). Including the 106mg/m2 AUC data decreased the r2 values suggesting a non-linear relationship between 17-DMAG dose and AUC (Figure 1B).
Table 3.
Summary of plasma pharmacokinetic data listing maximum drug concentration (Cmax), time to Cmax (Tmax), half life, area under concentration curve (AUC0-∞), clearance and volume at steady state (Vss) for each dose cohort of 17-DMAG (all data quoted to at least 2 or 3 significant figures.
| Dose (mg/m2) |
Number of patients |
Maximum concentration Cmax (nM) |
Time to Cmax Tmax(hr) |
Half life (hr) |
Area under the curve (0-∞) AUC (nM hr) |
Clearance (L hr−1) |
|---|---|---|---|---|---|---|
|
2.5 (CV %) |
3 | 64.1 ± 19.7 (30.7) |
0.76 ± 0.29 (38.2) |
25.2 ± 12.9 (51.3) |
581 ± 236 (40.5) |
13.1 ± 4.7 (36.1) |
|
5.0 (CV %) |
4 | 85.4 ± 55.2 (64.6) |
0.80 ± 0.35 (43.8) |
18.4 ± 7.50 (40.8) |
864 ± 651 (75.3) |
51.5 ± 68.2 (132) |
|
20.0 (CV %) |
5* | 480 ± 176 (36.6) |
0.97 ± 0.27 (27.7) |
28.7 ± 14.2 (49.3) |
3730 ± 2040 (54.6) |
20.7 ± 11.3 (54.5) |
|
40.0 (CV %) |
3 | 957 ± 248 (25.9) |
0.88 ± 0.33 (38.0) |
21.5 ± 4.3 (19.7) |
6510 ± 3220 (49.5) |
19.3 ± 9.3 (48.2) |
|
80.0 (CV %) |
8 | 2680 ± 1320 (49.4) |
0.65 ± 0.27 (41.5) |
27.8 ± 7.1 (25.4) |
12700 ± 2870 (22.65) |
18.9 ± 5.7 (30.5) |
|
106.0 (CV %) |
4 | 3750 ± 1370 (36.5) |
0.79 ± 0.32 (36.5) |
24.7 ± 1.8 (7.1) |
32300 ± 16300 (50.5) |
12.8 ± 6.2 (48.6) |
Figure 1.

The pharmacokinetics of 17-DMAG given over 1 hour IV, weekly, to patients with advanced solid tumors. A). Time-courses of concentration against time for individual patients treated with 80mg/m2 (left panel) or 106mg/m2 (right panel) of 17-DMAG. The mean IC50 in the NCI human tumor cell line panel is marked with a dotted line on both graphs. B). Graphs with Cmax or AUC plotted against 17-DMAG dose, linear regression value (r2) shown above each plot. For AUC two graphs are: centre, all dose levels (r2 = 0.66) with two patients who experienced DLT marked (arrows); and left, data from 106mg/m2 have been excluded (r2 = 0.88).
Pharmacodynamics of 17-DMAG
Using western blotting, transient HSP72 induction (< 24 hours) was detected in PBMCs at doses of 17-DMAG ≥ 5 mg/m2. Doses ≥ 20mg/m2 were required to achieve sustained HSP72 induction up to 96 hours post 17-DMAG (Figure 2A). Measured by ELISA (DELFIA), see Figure 2B and C, baseline HSP72 expression varied in both PBMC (mean 1.5, range < LLD – 3.3 fmole/μg protein extract) and plasma (mean 76, range < LLD – 702) fmole/ml. HSP72 induction was detected in PBMC from patients treated at ≥ 20 mg/m2 (Figure 2B). Mean HSP72 expression 24 hours after 17-DMAG was significantly different compared with 2.5 mg/m2 at 20, 80 and 106 mg/m2 dose levels (p = 0.01, 0.02 and 0.03 respectively). Mean plasma HSP72 did not differ between dose levels (Figure 2C). The highest HSP72 plasma levels post 17-DMAG of 1250 and 5610 fmole/ml were observed in 2 patients with DLT, compared to a mean 86 ± 140 fmole/ml in all other patients.
Figure 2.

Pharmacodynamic changes in inducible HSP70 (HSP72) observed in PBMC and plasma. A). For each dose level of 17-DMAG, a western blot from a representative patient is shown. Samples were taken pre-dose (0), end of infusion (E), then 1, 8, 24, 48 and 96 hours after 17-DMAG; an additional sample was taken 24 hours after the fifth weekly infusion of 17-DMAG (#2). An HT29 human adenocarcinoma colon tumor sample is included as a positive control (+) and equal protein loading is confirmed by corresponding GAPDH expression. HSP72 levels were also detected in duplicate samples taken before and 24 hours after 17-DMAG dose. B). Histogram plotting HSP72 expression changes in PBMC as measured by ELISA. Values are the difference between pre- and post treatment samples expressed as mean ± SD. Values marked (*) were significantly, p < 0.05, different to the mean HSP72 expression change in the first dose level. C). Histogram showing HSP72 expression change in plasma samples. Values are change in HSP72 24 hours after 17-DMAG, expressed as mean ± SD. There was no statistically different (p > 0.05) change in mean HSP72 level compared to that of the first dose cohort.
In PBMC, early LCK induction, as seen with 17-AAG [22], followed by later depletion was observed in individual patients exposed to 17-DMAG ≥ 40mg/m2 (Supplementary Figure 2) CDK4 depletion was demonstrated in PBMC in some patients treated at ≥ 80 mg/m2 (Supplementary Figure 2).
The complete HSP90 inhibition pharmacodynamic signature (HSP72 induction with depletion of CDK4) was detected in PBMC from 2/8 patients at 80 mg/m2 and in 2/3 patients at 106 mg/m2 respectively (Supplementary Table 2).
HSP72 was induced in 4/5 tumors 24 hrs after an 80mg/m2 dose and client protein depletion (CDK4 or ERBB2) was detected in 3/5. Overall, HSP90 inhibition was detected in 3/5 patients. In the single set of samples available, HSP90 inhibition was confirmed in tumor following 106 mg/m2 17-DMAG (Figure 3).
Figure 3.
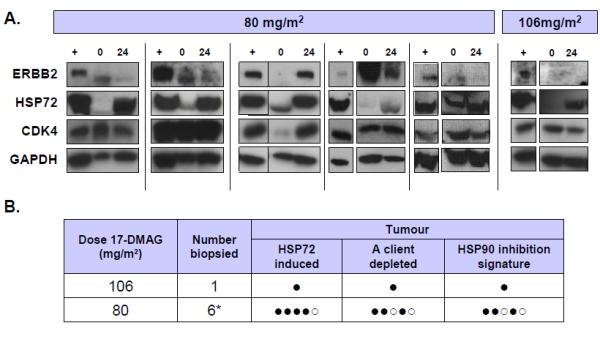
Pharmacodynamic changes following 17-DMAG administration in tumor samples. A). Expression of ERBB2, inducible HSP70 (HSP72) and CDK4 detected by western blots from patients treated with 80 or 106mg/m2 of 17-DMAG. GAPDH is included as a loading control. Samples are pre dose (0), 24 hours after 17-DMAG (24) or HT29 human colon adenocarcinoma positive control (+). B). Table summarizing pharmacodynamic changes in tumor. For each patient samples are marked positive (●) or negative (○) for induction of HSP70 and/ or depletion of a client protein (CDK4 or ERBB2). If both changes were detected then the sample was positive for detecting the molecular signature of HSP90 inhibition. * One sample set did not pass quality control.
Efficacy
Twenty patients were evaluable for tumor response. Nine patients had progressive disease (PD), four within the first treatment cycle. Prolonged stable disease (SD) > 6 months occurred in three patients, with chondrosarcoma (5mg/m2 escalated to 20mg/m2), CRPC (20mg/m2) and clear cell renal cancer (80mg/m2) on study for 28, 59 and 76 weeks respectively. Another patient, with CRPC, had a complete response (CR) confirmed by CT and PSA measurements. Previous treatment included bicalutamide and radical radiotherapy to the prostate, LHRH antagonist and bicalutamide withdrawal. At this time he had lymph node metastasis and was treated with 17-DMAG at 5mg/m2, then escalated to 20mg/m2 and remained on treatment for 124 weeks before PD (Figure 4A).
Figure 4.

Selected patient case histories. A). Graph of PSA changes in a patient with prostate adenocarcinoma treated with 17-DMAG. Time-points marked are: initial diagnosis and commencement of bicalutamide (1), radical radiotherapy (2), LHRH antagonist (3), bicalutamide withdrawal (4) and starting 17-DMAG (5), PSA and CT confirmed CR (6) and progression of disease (7). B). CT scans from a patient with metastatic melanoma at commencement of study and 30 months after starting 17-DMAG. Prior therapy had been adjuvant interferon and combination chemotherapy with dacarbazine and sorafenib. Patient received 159 weeks of 17-DMAG prior to PD.
A patient with metastatic melanoma, treated at 40mg/m2, had a partial response (PR) and was on treatment for 159 weeks before PD (Figure 4B). Prior treatment was adjuvant interferon, followed by combination chemotherapy (dacarbazine and sorafenib) on diagnosis of metastases. Progression of known intra-pulmonary and lymph node metastasis preceded trial entry.
Discussion
The MTD of weekly 17-DMAG was 80 mg/m2 IV. Nausea, vomiting, fatigue and liver enzyme disturbances were the commonest toxicities, all low grade and reversible at doses ≤ 80 mg/m2. A significant number of patients experienced ocular AEs and prophylactic lubricating eye drops were recommended with doses ≥ 80mg/m2.
DLT (at 106mg/m2) occurred in two patients and included: a drug related death (Grade 5 renal failure), Grade 4 AST rise and hypotension, Grade 3 dehydration, hyponatremia, acidosis, creatinine elevation, fatigue, diarrhea and hypoalbuminamia.
Pharmacokinetic studies showed that both Cmax and AUC (0-∞) increased proportionately with dose ≤ 80 mg/m2 (Figure 1). The two patients with DLTs had the highest drug exposures (Figure 1C). Increased drug exposure due to non-linear pharmacokinetics at 106 mg/m2 may explain the adverse toxicity and the narrow therapeutic window observed.
In PBMC sustained induction (at least 96 hours) of HSP72 was detected following 17-DMAG (≥ 20 mg/m2). Mean HSP72 levels 24 hours after 17-DMAG (≥ 20mg/m2) were significantly increased (p < 0.05) as measured by ELISA. Preliminary data suggest high plasma HSP72 levels might be a pharmacodynamic toxicity marker. CDK4 depletion was detected after ≥ 80 mg/m2 17-DMAG and modulation of LCK was detected at doses ≥ 40 mg/m2. As defined by the molecular signature of client protein depletion and HSP72 induction, HSP90 was inhibited in tumor samples from 3/5 patients taken 24 hours after 80 mg/m2 17-DMAG.
Clinical activity was observed across a range of dose levels including CRPC (CR), melanoma (PR), renal cancer, CRPC and chondrosarcoma (> 6 months stable disease). The CR occurred following anti-androgen withdrawal; however marked, durable (> 1-year) responses are rarely reported in this context [34-36]. A hypothesis to explain this activity is that androgen receptor stability and function are known to be dependent on HSP90 [37], similar to other oncogenic client proteins such as ERBB2 [38], EGFR [39] or BRAF [40, 41]. Other investigators have reported CR in patients with refractory acute myeloid leukemia (AML) [42] as well as prolonged (> 6 months) stable disease [43-45].
Studies employing alternative 17-DMAG schedules have been reported [42, 44-46] although pharmacodynamic studies were only informative in a study of AML patients [42]. In our study, although HSP90 inhibition was confirmed in 3/5 patients at MTD, pre-defined criteria to select a BED might have been suboptimal. Validating western blotting as fit for purpose [23, 47] limited the protein panel analyzed and practical limitations restricted sampling to one time-point. It remains challenging to balance acceptable scientific rigor (i.e. validation) with the currently limited knowledge of molecular biology, especially which client protein(s) is/are critical for tumorigenesis in an individual tumor, given the range of HSP90 client proteins and differential sensitivity to HSP90 inhibition.
Clinical benefit was observed over a range of dose-levels and robust definition of BED would aid dose and schedule selection for future studies. The challenges to defining a BED should not deter investigators from future efforts [48]. Combination studies of HSP90 inhibitors have enjoyed early success in clinical trials, e.g. HSP90 inhibition with trastuzumab in breast cancer [49, 50] or bortezomib in myeloma [51]. Use of BED in combination studies potentially minimizes toxicity and requires thorough pharmacokinetic and pharmacodynamic measurements [25, 48, 52-54].
Our data support further evaluation of HSP90 inhibitors. However, at this time there are no Phase II or III studies using the weekly schedule of 17-DMAG that we are aware of. Future studies of 17-DMAG should consider using alternative schedules or administration routes to minimize side effects in light of the severe toxicity observed at the highest dose level tested.
Statement of translational relevance.
Multiple critical oncogenic signaling pathways are disrupted by inhibition of the molecular chaperone HSP90. Interesting hints of clinical activity have been reported in early phase studies with agents such as 17-AAG. The 17-AAG analogue 17-DMAG was chosen for its superior pharmaceutical and therapeutic properties. In this phase I study we utilized a novel study design which aimed to define a biologically effective dose (BED) by incorporating a dose de-escalation phase after defining the maximum tolerated dose (MTD). BED was assessed by measurement of HSP90 inhibition in tumor tissue. Previously determined as fit for purpose, i.e. robust and validated commensurate with the stage of clinical drug development, western blot or ELISA assays were used to measure HSP72 and client proteins. We demonstrated evidence of clinical activity (including complete and partial responses) as well as target inhibition in tumor at MTD. Although the strict criteria did not allow us to define a BED lower than MTD, the study design proved to be robust and provided a valid template for defining BED in future studies of molecularly targeted novel anticancer therapy.
Supplementary Material
Supplementary Table 1: ELISA performance characteristics summary
Supplementary Table 2: Summary of detected pharmacodynamic changes in PBMCs for each dose level the number of patients treated is shown. For each patient samples are marked positive (●), negative (○) or no result (x) for induction of HSP70 and/ or depletion of a validated client protein (CDK-4 or ERB B2). If both changes were detected then the sample was positive for detecting the signature of HSP90 inhibition. * Cohort included 2 patients that underwent intra-patient dose escalation.
Supplementary Figure 1: Scoring system for western blotting. SKMEL-28, human melanoma cells treated in vitro with pharmacologically relevant concentrations of 17-DMAG (1,3,5 & 10 × IC50) as indicated. ERBB2, CDK4 and HSP72 expression was measured by western blotting. Results were quantified according the following system; 0 – no signal visible following 10 minutes exposure, 1 - signal just visible after 10 minutes exposure, 2 – visible but less than positive control, 3 – equal to positive, 4- greater than positive control, 5- over-exposed. An increase (HSP72) or decrease (ERBB2 or CDK4) of one point was considered a pharmacodynamic effect. Confirmation of HSP90 inhibition required induction of HSP72 combined with depletion of either ERBB2 and/or CDK4. The HT29 human colon adenocarcinoma cell line was included as positive control (Con).
Supplementary Figure 2: Pharmacodynamic changes in HSP90 client proteins (CDK4 or LCK) observed in PBMC. A). Expression of LCK is shown from representative patient's western blots with corresponding GAPDH as a loading control. Data are shown for dose levels of 40, 80 and 106mg/m2 17-DMAG. Samples were taken pre-dose (0), end of infusion (E), then 1, 8, 24, 48 and 96 hours after 17-DMAG. B). Expression of CDK4 is shown from representative western blots with corresponding GAPDH as a loading control. Data are shown for dose levels of 80 and 106mg/m2 17-DMAG.
Acknowledgements
Research Support:
The trial was conducted under the sponsorship and management of Cancer Research-UK's Drug Development Office (study number CRUKD/06/052). Supported by Cancer Research-UK programme grant numbers C309/A8274, C212/A5720, C212/A7324, C212/A7324 and C212/A11342. Paul Workman is a Cancer Research-UK Life Fellow and Simon Pacey was the grateful recipient of a Cancer Research-UK New Agents Committee Clinical Research Fellowship. We acknowledge funding to NIHR Biomedical Research Centre and Department of Health and Cancer Research UL funding to the Experimental Cancer Research Centre.
The authors would like to acknowledge the contribution of the staff at: the Drug Development Unit (Royal Marsden Hospital, Sutton, UK), the Belfast Experimental Cancer Medicine Centre (N. Ireland Cancer Clinical Trials Unit at Queens University Belfast and Belfast City Hospital, Northern Ireland, UK) and the Drug Development Office of Cancer Research UK (London, UK). In particular, we acknowledge Daleen Lopez- Begg, Georgia Wilson, Bee Ayite, Bronagh McClory, Ruth Hall, Mairead Devine, Lesley Curwen, Robert Beecham and Pia Somugompely.
Most of all, we thank our patients, their families and friends for their support and participation in our early clinical trials.
Disclaimers:
Professor Paul Workman and his team received research funding on the development of HSP90 inhibitors from Vernalis Ltd and intellectual property from this programme was licensed to Vernalis Ltd and Novartis. Pacey, Judson, deBono, Banerji, Raynaud, Moreno-Farre, Aherne, Hardcastle and Workman are/were employees of The Institute of Cancer Research which has a commercial interest in HSP90 inhibitors under development by Novartis Ltd. Paul Workman has been a consultant to Novartis. Dr Raynaud has undertaken consultancy work for Elara Pharma. 17-DMAG was supplied by the NCI and Kosan Biosciences Ltd.
Footnotes
Reference List
- 1.Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562:11–5. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 2.Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–69. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 3.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem.J. 2008;410:439–53. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 4.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat.Rev.Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann.N.Y.Acad.Sci. 2007;1113:202–16. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 7.Glaze ER, Lambert AL, Smith AC, Page JG, Johnson WD, McCormick DL, et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: potential clinical relevance. Cancer Chemother.Pharmacol. 2005;56:637–47. doi: 10.1007/s00280-005-1000-9. [DOI] [PubMed] [Google Scholar]
- 8.Hollingshead M, Alley M, Burger AM, Borgel S, Pacula-Cox C, Fiebig HH, et al. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother.Pharmacol. 2005;56:115–25. doi: 10.1007/s00280-004-0939-2. [DOI] [PubMed] [Google Scholar]
- 9.Smith V, Sausville EA, Camalier RF, Fiebig HH, Burger AM. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models. Cancer Chemother.Pharmacol. 2005;56:126–37. doi: 10.1007/s00280-004-0947-2. [DOI] [PubMed] [Google Scholar]
- 10.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat.Rev.Cancer. 2010;10:537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Workman P. How much gets there and what does it do?: The need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Curr Pharm Des. 2003;9:891–902. doi: 10.2174/1381612033455279. [DOI] [PubMed] [Google Scholar]
- 12.Clarke PA, Hostein I, Banerji U, Stefano FD, Maloney A, Walton M, et al. Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene. 2000;19:4125–33. doi: 10.1038/sj.onc.1203753. [DOI] [PubMed] [Google Scholar]
- 13.Lundgren K, Zhang H, Brekken J, Huser N, Powell RE, Timple N, et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol.Cancer Ther. 2009;8:921–9. doi: 10.1158/1535-7163.MCT-08-0758. [DOI] [PubMed] [Google Scholar]
- 14.Solit DB, Zheng FF, Drobnjak M, Munster PN, Higgins B, Verbel D, et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8:986–93. [PubMed] [Google Scholar]
- 15.Banerji U, Walton M, Raynaud F, Grimshaw R, Kelland L, Valenti M, et al. Pharmacokinetic-pharmacodynamic relationships for the heat shock protein 90 molecular chaperone inhibitor 17-allylamino, 17-demethoxygeldanamycin in human ovarian cancer xenograft models. Clin.Cancer Res. 2005;11:7023–32. doi: 10.1158/1078-0432.CCR-05-0518. [DOI] [PubMed] [Google Scholar]
- 16.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 17.Eiseman JL, Lan J, Lagattuta TF, Hamburger DR, Joseph E, Covey JM, et al. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005;55:21–32. doi: 10.1007/s00280-004-0865-3. [DOI] [PubMed] [Google Scholar]
- 18.Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-Kaye M, et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur.J Cancer. 2009 doi: 10.1016/j.ejca.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaherty KT, Gore L, Avadhani A, Leong S, Harlacker K, Zhong Z, et al. Phase 1, pharmacokinetic (PK) and pharmacodynamic (PD) study of oral alvespimycin (A; KOS-1022; 17-DMAG): Two different schedules in patients with advanced malignancies. J Clin Oncol. 2007;25 [Google Scholar]
- 20.Murgo AJ, Kumar S, Gardner ER, Figg W, Chen X, Yancey M, et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) administered twice weekly. J.Clin.Oncol. 2007;25:3566. [Google Scholar]
- 21.Pacey S, Banerji U, Judson I, Workman P. Hsp90 inhibitors in the clinic. Handb.Exp.Pharmacol. 2006:331–58. doi: 10.1007/3-540-29717-0_14. [DOI] [PubMed] [Google Scholar]
- 22.Banerji U, O'Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J.Clin.Oncol. 2005;23:4152–61. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 23.Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm.Res. 2006;23:312–28. doi: 10.1007/s11095-005-9045-3. [DOI] [PubMed] [Google Scholar]
- 24.Pacey S, Walton M, Eisen T, Gore M, Judson I, Workman P. Validation and use of western blotting to measure pharmacodynamic endpoints in Cancer Research UK clinical trials of Heat Shock Protein (HSP90) inhibitors. National Cancer Research Institute (NCRI) Cancer Conference; Birmingham, UK.: 2006. Abstract. [Google Scholar]
- 25.Sarker D, Pacey S, Workman P. Use of pharmacokintetic/ pharamcodynamic biomarkers to support rational cancer drug development. Biomarkers in Medicine. 2007;1:399–417. doi: 10.2217/17520363.1.3.399. [DOI] [PubMed] [Google Scholar]
- 26.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl.Cancer Inst. 1997;89:1138–47. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- 27.Moreno-Farre J, Asad Y, Pacey S, Workman P, Raynaud FI. Development and validation of a liquid chromatography/tandem mass spectrometry method for the determination of the novel anticancer agent 17-DMAG in human plasma. Rapid Commun.Mass Spectrom. 2006;20:2845–50. doi: 10.1002/rcm.2668. [DOI] [PubMed] [Google Scholar]
- 28.Pacey S, Gore M, Chao D, Banerji U, Larkin J, Sarker S, et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs. 2010 doi: 10.1007/s10637-010-9493-4. [DOI] [PubMed] [Google Scholar]
- 29.Pacey S, Gore M, Chao D, Banerji U, Larkin J, Sarker S, et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs. 2010 doi: 10.1007/s10637-010-9493-4. [DOI] [PubMed] [Google Scholar]
- 30.Hardcastle A, Boxall K, Richards J, Tomlin P, Sharp S, Clarke P, et al. Solid-phase immunoassays in mechanism-based drug discovery: their application in the development of inhibitors of the molecular chaperone heat-shock protein 90. Assay.Drug Dev.Technol. 2005;3:273–85. doi: 10.1089/adt.2005.3.273. [DOI] [PubMed] [Google Scholar]
- 31.Therasse P. Evaluation of response: new and standard criteria. Ann.Oncol. 2002;13(Suppl 4):127–9. doi: 10.1093/annonc/mdf649. [DOI] [PubMed] [Google Scholar]
- 32.Rustin GJ. Use of CA-125 to assess response to new agents in ovarian cancer trials. J Clin Oncol. 2003;21:187s–93s. doi: 10.1200/JCO.2003.01.223. [DOI] [PubMed] [Google Scholar]
- 33.Bubley GJ, Carducci M, Dahut W, Dawson N, Daliani D, Eisenberger M, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 34.Scher HI, Kelly WK. Flutamide withdrawal syndrome: its impact on clinical trials in hormone-refractory prostate cancer. J Clin Oncol. 1993;11:1566–72. doi: 10.1200/JCO.1993.11.8.1566. [DOI] [PubMed] [Google Scholar]
- 35.Small EJ, Srinivas S. The antiandrogen withdrawal syndrome. Experience in a large cohort of unselected patients with advanced prostate cancer. Cancer. 1995;76:1428–34. doi: 10.1002/1097-0142(19951015)76:8<1428::aid-cncr2820760820>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 36.Small EJ, Carroll PR. Prostate-specific antigen decline after casodex withdrawal: evidence for an antiandrogen withdrawal syndrome. Urology. 1994;43:408–10. doi: 10.1016/0090-4295(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 37.Fang Y, Fliss AE, Robins DM, Caplan AJ. Hsp90 regulates androgen receptor hormone binding affinity in vivo. J Biol Chem. 1996;271:28697–702. doi: 10.1074/jbc.271.45.28697. [DOI] [PubMed] [Google Scholar]
- 38.Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, et al. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–8. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 39.Shimamura T, Li D, Ji H, Haringsma HJ, Liniker E, Borgman CL, et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008;68:5827–38. doi: 10.1158/0008-5472.CAN-07-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc.Natl.Acad.Sci.U.S.A. 2006;103:57–62. doi: 10.1073/pnas.0609973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Rocha DS, Friedlos F, Light Y, Springer C, Workman P, Marais R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686–91. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- 42.Lancet JE, Gojo I, Burton M, Quinn M, Tighe SM, Kersey K, et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010 doi: 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- 43.Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-Kaye M, et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur.J Cancer. 2009 doi: 10.1016/j.ejca.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaherty KT, Gore L, Avadhani A, Spratlin JL, Harlacker K, Zhong Z, et al. First use of an oral Hsp90 inhibitor in patients (Pts) with solid tumors: Alvespimycin (A) administered QOD or QD. J Clin Oncol. 2008;26:2502. [Google Scholar]
- 45.Ramanathan RK, Egorin MJ, Erlichman C, Remick SC, Ramalingam SS, Naret C, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J.Clin.Oncol. 2010;28:1520–6. doi: 10.1200/JCO.2009.25.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-Kaye M, et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur.J.Cancer. 2010;46:340–7. doi: 10.1016/j.ejca.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JW, Weiner RS, Sailstad JM, Bowsher RR, Knuth DW, O'Brien PJ, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: a conference report. Pharm.Res. 2005;22:499–511. doi: 10.1007/s11095-005-2495-9. [DOI] [PubMed] [Google Scholar]
- 48.Banerji U, de Bono J, Judson I, Kaye S, Workman P. Biomarkers in early clinical trials: the committed and the skeptics. Clin Cancer Res. 2008;14:2512–4. doi: 10.1158/1078-0432.CCR-07-5224. [DOI] [PubMed] [Google Scholar]
- 49.Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J.Clin.Oncol. 2007;25:5410–7. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 50.Miller K, Rosen S, Modi S, Schneider J, Roy J, Chap L, et al. Phase I trial of alvespimycin (KOS-1022; 17-DMAG) and trastuzumab (T) J.Clin.Oncol. 2007;25:1115. [Google Scholar]
- 51.Richardson PG, Chanab-Khan A, Lonial S, Krishnan A, Carroll M, Cropp GF, et al. A Multicenter Phase 1 Clinical Trial of Tanespimycin (KOS-953) + Bortezomib in Relpased Refractory Mutliple Myeloma. J.Clin.Oncol. 2007;25 [Google Scholar]
- 52.Workman P. Challenges of PK/PD measurements in modern drug development. Eur J Cancer. 2002;38:2189–93. doi: 10.1016/s0959-8049(02)00395-7. [DOI] [PubMed] [Google Scholar]
- 53.Workman P. Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther. 2003;2:131–8. [PubMed] [Google Scholar]
- 54.Ratain MJ, Glassman RH. Biomarkers in phase I oncology trials: signal, noise, or expensive distraction? Clin Cancer Res. 2007;13:6545–8. doi: 10.1158/1078-0432.CCR-07-2133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: ELISA performance characteristics summary
Supplementary Table 2: Summary of detected pharmacodynamic changes in PBMCs for each dose level the number of patients treated is shown. For each patient samples are marked positive (●), negative (○) or no result (x) for induction of HSP70 and/ or depletion of a validated client protein (CDK-4 or ERB B2). If both changes were detected then the sample was positive for detecting the signature of HSP90 inhibition. * Cohort included 2 patients that underwent intra-patient dose escalation.
Supplementary Figure 1: Scoring system for western blotting. SKMEL-28, human melanoma cells treated in vitro with pharmacologically relevant concentrations of 17-DMAG (1,3,5 & 10 × IC50) as indicated. ERBB2, CDK4 and HSP72 expression was measured by western blotting. Results were quantified according the following system; 0 – no signal visible following 10 minutes exposure, 1 - signal just visible after 10 minutes exposure, 2 – visible but less than positive control, 3 – equal to positive, 4- greater than positive control, 5- over-exposed. An increase (HSP72) or decrease (ERBB2 or CDK4) of one point was considered a pharmacodynamic effect. Confirmation of HSP90 inhibition required induction of HSP72 combined with depletion of either ERBB2 and/or CDK4. The HT29 human colon adenocarcinoma cell line was included as positive control (Con).
Supplementary Figure 2: Pharmacodynamic changes in HSP90 client proteins (CDK4 or LCK) observed in PBMC. A). Expression of LCK is shown from representative patient's western blots with corresponding GAPDH as a loading control. Data are shown for dose levels of 40, 80 and 106mg/m2 17-DMAG. Samples were taken pre-dose (0), end of infusion (E), then 1, 8, 24, 48 and 96 hours after 17-DMAG. B). Expression of CDK4 is shown from representative western blots with corresponding GAPDH as a loading control. Data are shown for dose levels of 80 and 106mg/m2 17-DMAG.