

Abstract
Schizophrenia is a remarkably complex disorder with a multitude of behavioral and biological perturbations. Cognitive deficits are a core feature of this disorder, and involve abnormalities across multiple domains, including memory, attention, and perception. The complexity of this debilitating illness has led to a view that the key to unraveling its pathophysiology lies in deconstructing the clinically-defined syndrome into pathophysiologically distinct intermediate phenotypes. Accumulating evidence suggests that one of these intermediate phenotypes may involve phospholipid signaling abnormalities, particularly in relation to arachidonic acid (AA). Our data show relationships between levels of AA and performance on tests of cognition for schizophrenia patients, with defects in AA signaling associated with deficits in cognition. Moreover, dopamine may moderate these relationships between AA and cognition. Taken together, cognitive deficits, dopaminergic neurotransmission, and bioactive lipids have emerged as related features of schizophrenia. Existing treatment options for cognitive deficits in schizophrenia do not specifically target lipid-derived signaling pathways; understanding these processes could inform efforts to identify novel targets for treatment innovation.
Keywords: Schizophrenia, Neurocognitive deficits, Phospholipids, Arachidonic acid, Eicosanoids, Dopamine, Review
2. INTRODUCTION
Schizophrenia is one of the world’s leading causes of disability (1). Despite the extensive research its etiology remains very poorly understood. Part of the difficulty arises from the striking diversity of the documented abnormalities that a theory of etiology must explain, including the cognitive deficits that are a core feature of the illness, as well as the myriad biochemical abnormalities that influence neurodevelopment, neurotransmission, and neuromodulation. The deficits in cognition produce substantial functional disability, which is reflected in high rates of unemployment and reduced financial competence. Given the range and severity of cognitive impairment, efforts are now underway to identify novel targets for pharmacological enhancement. The purpose of this review is to integrate recent conceptual and empirical advances that implicate cognitive disorder in schizophrenia as an outcome of compromises to interacting neurochemical systems. We first summarize the rationale for identifying cognitive disorder as a target for treatment innovation, including the rationale for focusing on specific domains of cognition. We next present the background for our hypothesized linkage of cognitive deficits, abnormalities in membrane phospholipids, and alterations in monoamine transmission (Figure 1). The theoretical framework advanced is directed toward identifying novel targets that can lead to more successful treatment of cognitive disorder in schizophrenia.
Figure 1.
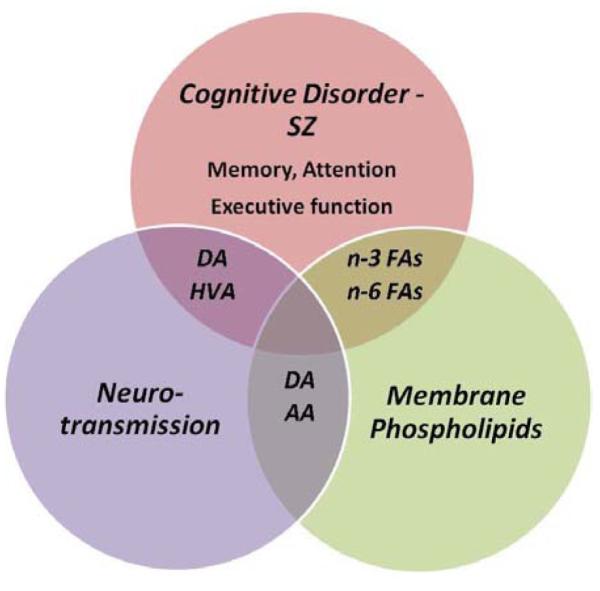
Overview of cognitive disorder in schizophrenia as a function of membrane phospholipids and monoamine neurotransmission. Abbreviations: SZ, schizophrenia; DA, dopamine; HVA, homovanillic acid; PUFAs, polyunsaturated fatty acids; AA, arachidonic acid.
3. COGNITION, NEUROTRANSMISSION, AND MEMBRANE PHOSPHOLIPIDS IN SCHIZOPHRENIA
Cognitive deficits are a core feature of schizophrenia, involving abnormalities across the domains of memory, attention, perception, and speed of processing. More than 70% of this patient population is estimated to be neuropsychologically impaired (2), with 98% of patients falling below expected achievement levels based on their premorbid level of intellectual functioning and the level of education attained by their parents (3). Cognitive impairment contributes to the functional disability observed for this patient population (4-6), which is reflected in an estimated 80% unemployment rate (7). Findings from studies completed during the last decade show that cognitive deficits have consequences for fairly specific aspects of patients’ functional outcome, including success in job rehabilitation placement, job tenure and number of hours worked, and social skills (review by Green et al. (8)). To complicate patients’ real-world challenges, cognitive deficits further contribute to deficiencies in financial competence by producing problems in basic money skills, use of banks, financial judgment, and understanding about their own income and expenses (9). Given the high rate and severity of impairment, efforts are currently underway to identify novel pharmacological targets for cognitive enhancement (10-12), and to create training programs for selected cognitive processes (13).
3.1. Memory impairment in schizophrenia
Memory impairment is among the most common and severe of the cognitive deficits associated with schizophrenia. Deficits in memory function occur across variations in clinical states and course, appearing in neuroleptic-naïve first-episode patients as well as patients with chronic illness under differing pharmacological regimens. Memory impairment also appears in the unaffected relatives of patients suggesting an association with increased risk for manifest illness. Moreover, impairment in this cognitive domain involves disturbance to a broad range of memory systems and processes. Understanding about the breadth of this impairment for this patient population has progressed as investigators have increasingly adopted conceptual and technical advances made in the areas of neuropsychology and cognitive neuroscience.
3.1.1. Systems of memory
Although a summary of the scientific study of memory is beyond the scope of this review, it is important to note that contemporary divisions of memory into systems, subsystems, and processes have been traced to early discussions by Endel Tulving in 1972 (14) and Elizabeth Warrington in 1979 (15) (for critical treatments of this topic see Schacter and Tulving (16) and Schacter et al. (17)). Recent investigations of memory in schizophrenia have included a fine-grained partitioning of global memory into semantic, working, and episodic systems, as well as declarative and nondeclarative forms. Semantic memory represents a person’s cumulative knowledge about the world, such as the meanings of words, defining features of concepts like ‘table’, name of the first president of the United States, and so on. In this regard, the semantic system provides a dynamic record of a person’s ongoing learning and experience. Working memory involves the temporary storage and manipulation of information that is being processed during the performance of tasks, and, ultimately, is linked to longer-term memory and other complex cognition, such as comprehension and reasoning (18-19). Episodic memory involves the recollection of events and episodes from a person’s life. It is also important to emphasize the distinction, advanced most prominently by Larry Squire and his colleagues, regarding the conscious remembering of facts and events, termed “declarative memory,” which is contrasted to memory for content and processes that are out of one’s awareness, or unconscious, and referred to as “nondeclarative memory” (20). The integrity of these memory systems has been examined for schizophrenia using strategies developed in neuropsychology and cognitive neuroscience.
3.1.2. Memory deficits in schizophrenia patients
3.1.2.1. Verbal declarative memory
Deficits in verbal learning and verbal declarative memory are among the most common and severe of the cognitive impairments associated with schizophrenia (21-24), and contribute to the high rate of functional disability in this disorder (6, 25). The tasks used to investigate this ability have included immediate and delayed recall and recognition of word lists, number series, and brief stories. The qualitative review provided by Cirillo and Seidman (22) showed that impairment in verbal declarative memory was observed for schizophrenia patients in 92% of the 110 studies examined. In the quantitative review conducted by Heinrichs and Zakzanis (23), effect sizes were examined from 204 studies in which the performance of schizophrenia and control groups was compared across multiple cognitive domains, including memory, attention, executive function, language, motor skill, and general intelligence. Results showed the largest mean effect size was obtained for global verbal memory. Similarly, the meta-analysis conducted by Aleman et al. (21), which was based on 70 studies of verbal memory performance for schizophrenia and healthy control groups, determined the largest mean effect size was obtained for total verbal memory recall. Moreover, memory for information that is represented in linguistic codes has emerged as important for etiological considerations in that deficits in reading and listening comprehension are observed in individuals long before their first diagnosis of schizophrenia (26-27). Abnormalities also appear in the speech communications of schizophrenia patients and their nonpsychotic monozygotic co-twins (28), as well as their nonpsychotic parents and siblings (29-30).
3.1.2.2. Semantic memory
Disturbance to the semantic memory system is one of the most prominent and enduring hypotheses advanced to explain the disorganized thinking that is a hallmark feature of schizophrenia (31). This clinical symptom, referred to as formal thought disorder, is detected most easily in the tangential and incoherent speech of many patients. Studies of semantic memory in schizophrenia have largely focused on semantic priming, which involves aspects of both verbal declarative memory and nondeclarative memory. The typical or normal semantic priming response involves differentiation of pairs of words on the basis of their meaning relationship, with semantically related (dog cat) and unrelated (truck coin) pairs eliciting the largest response difference. Exaggerated or ‘hyperpriming’ is believed to cause the disorganized thought of schizophrenia, and the influence of a wide range of linguistic and cognitive factors has been examined to test this hypothesis. One common approach has involved the use of different stimulus onset asynchronies (SOAs) that are created by varying the exposure duration and presentation rate of consecutive stimuli. Priming elicited under short SOAs is believed to reflect unconscious and automatic spread of activation within the semantic network. An exaggerated or overly broad spread of automatic activation is predicted to occur under short SOAs for patients who exhibit formal thought disorder. Longer SOAs are believed to enable the recruitment of attention resources that underlie conscious processing and executive function, such as working memory.
Several reviews have now been conducted that indicate variability of semantic priming in schizophrenia. In the qualitative review provided by Minzenberg and colleagues (32), schizophrenia patients showed variable semantic priming, based on behavioral response times (RTs), under experimental conditions designed to elicit automatic activation, or unconscious, processing. In contrast, more consistent disturbance involving reduced priming was observed for patients under conditions of controlled attention and conscious processing. Results from a recent meta-analysis by Doughty and Done (33) were generally consistent with the conclusions of Minzenberg et al. in showing the largest effect sizes for semantic tasks that are correlated with executive function. Variability of semantic priming was also observed in the quantitative review conducted by Pomarol-Cloret et al. (34) that involved 36 semantic priming studies in which response times were compared between schizophrenia patients and healthy controls. The pooled effect size based on patients as a whole was not significant, and the data were characterized by significant heterogeneity. Significant effect sizes were observed, however, for the comparison between patients diagnosed with thought disorder and controls, and SOA influenced the direction of these effects. Positive effect sizes, indicating increased priming for patients, were determined for short stimulus onset asynchronies (SOAs ≤ 400 ms) and negative effect sizes, indicating reduced priming for patients, were found for long SOAs (> 400 ms).
Electrophysiological studies of semantic priming in schizophrenia indicate a pattern of variability that is similar to the findings for patients’ behavior responding. The N400 component of the event-related brain potential (ERP) is the measure used most frequently in these studies. The term ‘N400’ represents its negative deflection in amplitude, relative to a pre-stimulus baseline, beginning at approximately 200 ms and peaking around 400 ms following the onset of a stimulus. Our knowledge about this endogenous brain response is based on several decades of work beginning with its discovery by Kutas and Hillyard (35). This ERP component is believed both to index semantic integration processes, and to reflect the effort required to access information from long-term memory (36). This electrophysiological response reflects the degree to which a word fits its proximal context, as seen in the N400s generated to sentences and pairs of words. For example, the pair ‘dog - cat’ typically elicits smaller N400 amplitude than does the word pair ‘dog - pencil’. The interpretation of such stimuli as meaningful and relevant further depends on access to the representations stored within the semantic network.
Based on their recent qualitative review, Kuperberg et al. (37) concluded that variability of N400 semantic priming is observed in schizophrenia patients under conditions of automatic or spreading activation, and consistency in disturbance of N400 priming is seen under conditions that demand attention resources. Additional considerations include the psycholinguistic characteristics of semantic stimuli and the nature of the behavior task, which some authors have suggested may influence the integrity of priming response in schizophrenia patients relative to controls. In particular, Mathalon et al. (38) proposed that variability across N400 studies of automatic priming in schizophrenia is due to differences in the classes of relations employed, which activate differentially the gradient of connections within the semantic network; direct (birthday → cake), indirect (birthday → [cake] → pie), and categorical (animal → cat and lion → tiger) relationships. Kreher et al. (39) suggested that the implicit or explicit nature of the behavior task can shape the priming response. Finally, our group has argued that activation of semantic memory in schizophrenia is influenced by neurochemistry dynamics that are modulated by pharmacological regimen (40-42) and abnormalities in cell membrane integrity and fluidity (43) (see Sections 3.4. and 3.5).
3.1.3. Memory deficits in nonaffected family members of patients
Memory deficits are observed in schizophrenia patients across differences in clinical status and clinical course, occurring in neuroleptic-naïve patients during their first episode of schizophrenia (44) and in stable, unmedicated patients with chronic illness (40-41, 45). The memory impairments that are reliably observed for patients diagnosed with schizophrenia disorder are also observed in their unaffected family members (46-52) and non-psychotic monozygotic co-twins (53-54). The pattern emerging from these studies is suggestive of an association between cognitive impairment and increased risk for developing schizophrenia due to a biological relationship to an individual with manifest illness. Moreover, heritability estimates for candidate cognitive markers, including verbal declarative memory and semantic memory, suggest that these measures may be informative in genetics studies of schizophrenia (55-56). Although recent challenges have been made to the assumption that a simpler genetic architecture may underlie biological markers (endophenotypes) than for the clinical phenotype itself (e.g., the meta-analysis of genetic association studies conducted by Flint and Munafo (57)), strong agreement remains regarding the utility of stable neurocognitive (48) and neurophysiological (58) traits for the purpose of identifying liability genes. Discovery of biochemical pathways that influence putative neurocognitive and neurophysiological markers therefore presents a unique opportunity to shed further light on the etiology of this complex clinical syndrome.
3.2. Association between cognition and neurochemistry
Neurochemical theories of schizophrenia have a long history. The dopamine (DA) hypothesis, in particular, is one of the most prominent and long-lived, and has recently undergone revision and renewed interest (e.g., increased striatal dopamine activity as the ‘final common pathway’ to psychosis (59)). Alternative models have focused on glutamatergic (60) and γ-aminobutyric acid– glutamatergic interaction (61) mechanisms. Phospholipid hypotheses of schizophrenia etiology are attractive because they can potentially explain many of the disparate alterations that accompany this debilitating disease (62-66). Rather than replacing neurotransmission-based theories, phospholipids models can extend the two classes of theories by integrating the effects of both. The n-3 polyunsaturated fatty acid (PUFA)/dopamine hypothesis, for example, proposes that the effects of PUFAs on behavior are mediated through monoamine neurotransmission (67-69). The viability of this non-additive assumption receives support from animal studies demonstrating that chronic deficiency in n-3 PUFA influences dopamine-related structures and function in frontal cortex (70) and ventral striatum (71). Importantly, dopamine D2 receptors can be coupled to the activation of phospholipase A2 (PLA2) and the release of n-6 PUFA arachidonic acid from cell membrane phospholipids (72). Functional linkages discovered among deficits in cognition and abnormalities within phospholipids and monoamine pathways could shed new light on the biochemical cause(s) of cognitive disorder in schizophrenia. To the extent that existing treatment options do not specifically target lipid-derived signaling pathways, such discoveries could also suggest novel targets for treatment innovation.
3.2.1. Cognition and the dopamine hypothesis
Dysregulation of dopamine is considered by a number of contemporary theorists to underlie cognitive disturbance in schizophrenia, beginning with a seminal paper by Cohen and Servan-Schreiber (73) in which this neurotransmitter was considered a primary determinant of the processing of context. In the more recent formulation by Kapur (74), dopamine is assumed to mediate a person’s interpretation of the salience of information. One prominent idea is that dopamine dsyregulation, in particular increased striatal dopamine activity, represents a ‘final common pathway’ that causes aberrant processing of stimulus meaning and relevance, which, in turn, results in the development of psychotic symptoms (59).
The importance of dopamine to cognition has been demonstrated by studies in which dopamine levels and activity were modulated using different types of pharmacological compounds. Following administration of amphetamine, for example, cognitive performance improved and was associated with the magnitude of change in regional cerebral blood flow (rCBF) for schizophrenia patients receiving antipsychotic medication (75). Amphetamine also improved accuracy and reduced response latency for healthy controls and medicated schizophrenia patients during their performance of tasks that capture working memory, attention, and language production processes (76). Administration of the dopamine precursor L-3,4-dihydroxyphenylalanine (L-DOPA) to healthy volunteers produced more focused activation of semantic memory, reflected in reduced indirect priming (77), and increased the rate of learning and long-term retention of an artificial vocabulary (78). Comparison of the relative influences of the dopamine D1/D2 receptor agonist pergolide and the D2 receptor agonist bromocriptine showed that only pergolide decreased indirect priming in healthy volunteers (79), suggesting the importance of D1 receptor activity for this semantic memory function. Enhancement of dopamine transmission has also been determined to benefit working memory performance in populations that experience dopamine depletion, including aged monkeys (80-81) and patients with Parkinson’s disease (82).
While the influence of dopamine on cognition is clearly complex (83) and may not provide a complete account for the full range of cognitive deficits in schizophrenia (e.g.: α2-Noradrenergic effects on the early components of the event-related potential during auditory information processing (84-86)), a growing body of evidence indicates that striatal dopamine activity, which is altered in patients (87-90), plays an important role in cognitive abilities that are influenced by prefrontal cortex function. In a series of studies involving dopamine D2 receptor transgenic mice, Kellendonk and Simpson and their colleagues (91-92) determined that overexpression of D2 receptors in the striatum during development caused persistent abnormalities in executive function, including working memory and conditional associative learning. Using high pressure liquid chromatography (HPLC) methodology, those investigators also found decreased dopamine turnover in the prefrontal cortex of these D2 transgenic mice.
3.2.2. Possible roles of polyunsaturated fatty acids (PUFAs) in cognition
Lipids comprise approximately 50% of the dry weight of the human brain, and the majority of lipids (46-70%) are phospholipids (93). The major phospholipids in neuronal membranes consist of phosphatidylethanolamine, phosphatidylcholine, phosphatidylserine, phosphatidylinositol, and sphingomyelin. Alterations in these core phospholipids have been reliably observed for schizophrenia patients (reviewed by Fenton et al. (94) and Skosnik and Yao (65)). While saturated and monounsaturated fatty acids can be synthesized de novo in mammals, the precursors for the n-3 and n-6 polyunsaturated fatty acid (PUFA) series must be obtained from dietary sources. An interdependent relationship holds between the n-3 and n-6 series; dietary deficiency of n-3 fatty acids produces decreased levels of the n-3 docosahexaenoic acid in brain and other organs, and a reciprocal increase in the n-6 docosapentanoic acid (reviewed by Fedorova and Salem (95)).
Investigations of the influence of polyunsaturated fatty acids (PUFAs) on cognition have followed two general tracks: (i) animal studies, in which dietary intake of fatty acids is manipulated, either by inducing dietary deficiency or dietary enhancement, and cognitive performance is evaluated as a function of concentrations of fatty acids in brain tissue; (ii) evaluation of human cognition as a function of dietary fatty acids and phospholipids in red blood cell (RBC) and plasma. Tables 1 – 3 summarize studies identified from public databases (MEDLINE, The Cochrane Library) that have been published since previous reviews of this literature appeared. Previous reviews include: an exhaustive review by Innis (96) addressing the role of essential fatty acids in mammalian growth and CNS development; recent reviews of the effects of PUFA supplementation on neurodevelopment and physical growth during infancy and childhood by Hoffman et al. (97), Ryan et al. (98), and Simmer et al. (99); an extensive review by Federova and Salem (95) of animal studies up to 2006; reviews of clinical studies involving treatment with PUFAs by Freeman et al. (100), McNamara and Carlson (101), Peet (102), and Richardson (103).
Table 1.
Influence of bioactive lipids on cognition: Animal studies
| Study | Study aims and design | Sample | Variables | Results |
|---|---|---|---|---|
| G. W. Arendash et al. (121) |
Effect of diet enriched with n-3 FAs on cognition and brain tissue (cortical FAs; hippocampal Aβ) in animals varying in genetic risk for cognitive impairment. |
2 genotypes: (1) 17 double transgenic (Tg) APP + PS1 mice (2) 17 nontransgenic (NT) (normal) mice |
IV: 50% of each genotype group randomly assigned to either: (1) 13% n-3 FA diet (n-6:n-3 ratio = 3.8 to 1) vs. (2) Standard diet Diets administered beginning at 2 mos of age (young adulthood). NT mice received diet for 6 mos; Tg mice received diet for 7.5 mos. DV: (1) Performance on cognitive test battery administered during final 6 weeks of diet: open-field activity, balance beam, string agility, Y-maze, elevated-plus maze, Morris water maze, circular platform, platform recognition, radial arm water maze. (2) Aβ levels in hippocampus; FA levels in frontal cortex |
A. No effect of diet on cognitive performance for either genotype (n-3 FA diet = standard diet). B. Relationship between FAs and cognition: (1) Significant association between cortical levels of n- 6 FAs and cognition for both genotypes (Tg and NT mice): ↑ n-6 FAs ↓ cognitive performance (2) No association between cortical levels of n-3 FAs and cognitive performance for either genotype. C. Relationship between diet and cortical levels of FAs differed between genotypes: (1) Tg mice: No effect of n-3 FA diet on cortical FA levels. (2) NT mice: n-3 FA diet: ↑ cortical levels of n-3 FAs ↓ cortical levels of n-6 FAs. D. Effect of diet on Aβ levels: Tg mice: No effect of n-3 FA diet on hippocampal Aβ level, but significant association between hippocampal Aβ level and cognitive impairment. |
| R. C. S. Barcelos et al. (138) |
Effect of supplementation with n-3 FAs on oxidative stress and abnormalities of involuntary movement and cognition induced by administration of neuroleptic drugs. |
Male Wistar rats, 3- mos age |
IV: (1) Dietary Supplementation (a) n-3 FA [fish oil with 20% EPA, 6% DHA, 0.4% α-linolenic acid, 1.2% DPA] vs. (b) control (tap water) (2) Antipsychotic drug [depot haloperidol vs. fluphenazine] DV: (1) Behavior tests (orofacial dyskinesia, Morris water maze, catalepsy) (2) Lipid peroxidation (LP) TBARS in hippocampus, substantia nigra Experiment 1 (haloperidol): For 8 wks, 2 diet groups (14 rats each) administered either: (1) Fish oil in tap water, or (2) Tap water only After 4 wks, 2 diet groups subdivided and administered (im) one of following conditions for remainder of protocol: C (water + soybean oil) FO (fish oil + soybean oil) H (water + haloperidol) FO + H (fish oil + haloperidol) Experiment 2 (fluphenazine): 2 diet groups (14 rats each) administered same protocol as Experiment 1, but treated with fluphenazine instead of haloperidol. |
A. Antipsychotic drugs produced movement abnormalities (stereotypy, catalepsy), which were reduced by n-3 FA supplementation. B. Antipsychotic-induced impairment of spatial memory (water-maze task) was reduced by n-3 FA supplementation. C. Effects of n-3 FA supplementation on lipid peroxidation (TBARS) in haloperidol and fluphenazine-treated rats: (1) Haloperidol: ↑ TBARS in plasma and hippocampus, which were ↓ by n-3 FA supplementation. (2) Fluphenazine: ↑ TBARS in plasma and substantia nigra, which were ↓ by n-3 FA supplementation. |
| I. Fedorova et al. (71) |
Effect of dietary deficiency of n-3 FAs on spatial learning and levels of FAs and monoamine neurotransmitters in brain tissue. |
21 Female Long- Evans rats obtained on 3rd day of pregnancy, and male pups. |
IV: (1) 2 n-3 FA diet groups: (a) n-3 adequate (3.8% of total FAs) vs. (b) n-3 deficient (0.05% of total FAs). Otherwise, all rats received same basal dietary composition. (2) Diets administered to pregnant dams during gestation and lactation; pups weaned to same diet as their dams. (3) Only male pups tested on behavior tasks, which began at 8 weeks of age. DV: (1) Behavior: (a) Spontaneous locomotor activity (open-field test) (b) Anxiety-related responses (elevated- plus maze) (c) Spatial performance – escape response accuracy and latency (Barnes circular maze) (2) Brain tissue: (a) FAs at end of study when pups were 17 weeks old. (b) Neurotransmitters and metabolites obtained from additional sample of 7-wk- old rats fed n-3 FA deficient vs. n-3 FA adequate diets (n=6/diet group): noradrenaline (NA), dopamine (DOPAC, HVA), serotonin (5-HT, 5-HIAA) |
A. 58% reduction in total brain DHA in rats fed n-3 FA deficient diet, compared to rats fed n-3 FA adequate diet. B. No effect of diet on spontaneous locomotor activity or anxiety responses. C. ↓ Spatial learning in rats fed n-3 FA deficient diet, compared with rats fed n-3 FA adequate diet. Level of impairment increased as task difficulty increased. D. Relationship between n-3 FA diet and monoamine transmitters: Compared to n-3 FA adequate diet, n-3 FA deficient diet produced: (1) 50% reduction in DA levels in ventral striatum; (2) ↑ DOPAC in frontal cortex and hypothalamus; (3) ↑ DA turnover (DOPAC/DA and HVA/DA ratios) in frontal cortex and hypothalamus. |
| S. Holguin et al. (104) |
Effect of dietary supplementation (DHA, UMP) on cognitive performance and brain phosphatidylcholine (PC) synthesis in rats exposed to enriched or impoverished rearing environments. Diets and rearing environments administered for 1 mo beginning at weaning. |
Male Sprague Dawley rats, 4 wks age |
IV: (1) Rearing environment: Rats assigned to either enriched (EC) or impoverished (IC) rearing condition. (2) Diet: Subgroups of EC- and IC-exposed rats administered one of following dietary conditions: (a) control diet (b) control diet + UMP (0.5%) in diet (c) control diet + DHA (300 mg/kg) daily by gavage (d) control diet + UMP (0.5%) in diet + DHA (300 mg/kg) daily by gavage All diet regimens included choline chloride. DV: (1) Cognitive tasks categorized as hippocampus- or striatal- dependent memory (hidden- vs. visible-platform versions of Morris water maze, respectively). (2) Phospholipid content of brain tissue |
A. Effect of diet on cognitive performance differed by rearing condition and task. (1) IC rats: (a) Either DHA or UMP produced ↑ performance on hippocampus-dependent task (hidden version- water maze); (b) Co-administration of DHA and UMP further ↑ performance on hippocampus- dependent task. (2) EC rats: No effect of diet on performance of hippocampus-dependent task. (3) Neither diet nor rearing environment affected performance on striatal- dependent task (visible version-water maze). B. Effect of diet on brain levels of phosphatides: Co-administration of UMP + DHA ↑ brain levels of PC, PE, SM, PS, PI, and total phospholipid levels. C. Effect of rearing environment on brain tissue: EC environment ↑ brain levels of PC, PS, PI, and total phospholipids, compared with IC environment. |
| K-F. Ng and S. M. Innis (152) |
Effect of dietary polyunsaturated FAs (PUFAs) and dopamine challenge on fear responses and exploratory behavior (elevated plus maze) in neonatal piglets. |
12 Male Yorkshire piglets (6 per diet condition). |
IV: (1) 2 diet conditions – beginning at age 1 day: (a) low PUFA diet [1.2% linoleic acid; 0.05% α-linolenic acid] (b) high PUFA diet [10.7% linoleic acid; 1.1% α-linolenic acid; 0.3% AA; 0.3% DHA] (2) Dopamine (DA) challenge (randomized cross-over design): (a) DA precursor (L-Dopa with Carbidopa) (b) DA D2 receptor blocker (sulperide). DV: (1) From age 18-22 days, behavior in elevated plus maze (number and type of arm entries, time spent in arms) recorded before and after administration of each DA challenge condition. (2) On day 26, home cage behavior recorded (stereotyped responses; biting; head thrusting; activity level). (3) Brains removed at age 30 days. Total lipids extracted from frontal cortex and phospholipids (PC, PE, PS, PI) separated. |
A. Effect of diet on frontal cortex FAs: Compared to high-PUFA diet, low- PUFA diet ↓ DHA by 30, 22, and 40% in frontal cortex PE, PS, and PC, respectively. B. Effect of diet on maze behavior: Compared to high-PUFA diet, low-PUFA diet produced: ↓ open and total arm entries; ↑ time spent in closed arms. C. Effects of diet on maze behavior differed under each type of DA challenge: (1) L-Dopa ↑overall number of arm entries in piglets fed low-PUFA diet. (2) L-Dopa ↑ number of closed arm entries in piglets fed high-PUFA diet. (3) Sulperide did not affect maze behavior for the 2 diet groups. D Trend effect of diet on home cage behavior: Piglets fed low-PUFA diet showed ↑ number of stereotyped responses (snout-rubbing). |
| A. L. Petursdottir et al. (105) |
Effect of dietary supplementation with n-3 PUFAs on brain phospholipid DHA status, learning, and memory in aged senescence- accelerated prone mice, which develop learning and memory impairments at age 8- 12 mos. |
20 SAMP8 (senescence- accelerated) mice |
IV: At age 10 mos., mice randomly assigned to: (1) Low-DHA diet (4.4% total n-3 PUFA and 0.3% DHA) vs. (2) High-DHA diet (30.3% total n-3 PUFA and 14.3% DHA). Diets maintained for 8 wks DV: Behavior training/testing began at age 12 mos.: (a) Acquisition and retention in T-maze foot shock avoidance paradigm (b) Phospholipid DHA status in hippocampus and amygdala |
Compared to mice fed low- DHA diet, mice fed high- DHA diet showed: ↑ acquisition (learning) and retention (memory) in T- maze foot shock avoidance; ↑ proportion of DHA in PC and PE of both hippocampus and amygdala. |
Abbreviations for Table 1: ↑, increase; ↓, decrease; AA, Arachidonic acid; DA, Dopamine; DHA, Docosahexaenoic acid; DPA, Docosapentaenoic acid; DV, Dependent variable; EPA, Eicosapentaenoic acid; FA, Fatty Acid; IM, Intramuscular; IV, Independent variable; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PI, Phosphatidylinositol; PS, Phosphatidylserine; PUFA, Polyunsaturated fatty acid; SM, Sphingomyelin; TBARS, Thiobarbituric acid reactive substances; UMP, Uridine monophosphate.
Table 3.
Influence of bioactive lipids on cognition: Clinical studies of psychosis
| Study | Study aims and design | Sample | Variables | Results |
|---|---|---|---|---|
| G. P. Amminger et al. (115) |
Effect of n-3 PUFAs on rate of progression to first-episode psychotic disorder in young individuals with subclinical psychosis. Randomized, double- blind, placebo-controlled trial conducted from 2004 to 2007 |
81 individuals (13 – 25 yrs) at ultra- high risk for psychotic disorder |
IV: Random assignment (stratified by depression symptom score) to 12-week treatment trial of: (1) n-3 PUFA (1.2 g/day marine fish oil: 700 mg/day EPA, 480 mg/day DHA, 220 mg/day Other n-3 PUFAs including α-linolenic acid) vs. (2) placebo (coconut oil) DV: (1) Conversion to psychotic disorder determined by assessments conducted weekly (first 4 wks), and then at 8 and 12 wks, and 6 and 12 mos. (2) n-6 to n-3 ratio at baseline and 12- wks (fasting erythrocyte FA composition). |
A. Cumulative rates of conversion to psychotic disorder at 12 mos.: (1) 4.9% in n-3 PUFA group (2) 27.5% in placebo group. B. Compared to placebo, n-3 PUFAs improved functioning and reduced positive, negative, and general symptoms. C. For n-3 PUFA group, magnitude of change in n-6 to n-3 ratio from baseline to 12 wks was significantly associated with improvement in functioning between baseline and 12 wks. |
| G. E. Berger et al. (116) |
Effect of n-3 FA augmentation on efficacy and tolerability of treatment with antipsychotic drugs in patients with first-episode psychosis (FEP). Randomized, double- blind, placebo-controlled trial conducted from 2000 to 2003 |
80 FEP patients, which included 53 (76.8%) patients meeting criteria for non-affective psychosis |
IV: 12-weeks augmentation protocol: (1) 2 g/d ethyl-eicosapentaenoic acid (E-EPA) vs. (2) placebo oil Both E-EPA and placebo were added on to flexible doses of atypical antipsychotics (risperidone, olanzapine, or quetiapine). Additional medications allowed: benzodiazepines, chlorpromazine, zuclopenthixol, zolpidem, SSRIs. DV: Clinical assessments conducted at 5 time points (baseline and weeks 3, 6, 9, 12): (1) Symptom change scores and time to first response (2) Tolerability measures and cumulative antipsychotic dose |
A. No difference between E- EPA and placebo groups for symptom change scores at 12 weeks. B. Time to first response was shorter in patients diagnosed with non-affective psychosis and receiving E-EPA. C. Patients receiving E-EPA required lower doses of antipsychotic medications between weeks 4 and 6, compared to patients receiving placebo. D. Patients receiving E-EPA showed fewer extrapyramidal side effects during the initial 9 wks, compared to patients receiving placebo. |
| R. Condray et al. (43) |
Association between RBC membrane FAs and cognition in schizophrenia patients. Repeated measures design, double-blind placebo-replacement. |
37 chronic schizophrenia patients (mean age = 38 yrs) tested during haloperidol maintenance therapy (n=30) and placebo replacement (n=21). (Different n’s for medicated and placebo phases due to electrophysiological artifact.) |
DV: (1) RBC membrane FAs (venous blood draw after overnight fast) (2) Cognition: Electrophysiological measure of semantic memory (N400 component of event-related brain potential: N400- Unrelated minus N400-Related words) |
A. Semantic memory was associated with AA and total PUFAs for patients during unmedicated (placebo) phase: ↑ N400 semantic priming ↓ AA and Total PUFAs B. Semantic memory was not significantly associated with PUFAs for patients during haloperidol maintenance therapy. |
Abbreviations for Table 3: ↑, increase; ↓, decrease; AA, Arachidonic acid; DHA, Docosahexaenoic acid; DV, Dependent variable; EPA, Eicosapentaenoic acid; FA, Fatty Acid; IV, Independent variable; PUFA, Polyunsaturated fatty acid; RBC, Red Blood Cell.
3.2.2.1. n-3 PUFAs
3.2.2.1.1. Animal studies
The cumulative evidence from several decades of animal work shows relationships among dietary levels of n-3 fatty acids (Figure 2A), levels of fatty acids in brain tissue, and performance on many, although not all, tasks of learning and memory (review by Fedorova and Salem (95)). This work also suggests that dietary deficiency of n-3 fatty acids may increase vulnerability to stress, which, in turn, could influence cognitive performance (95). Table 1 summarizes animal studies published since the 2006 review by Fedorova and Salem. The focus of recent studies has concerned the interplay among cognition, biochemistry, genes, and environment, and the study by Holguin et al. (104) provides a striking example of this direction. Those investigators varied rearing environment (enriched versus impoverished) and dietary intake of compounds required for phosphatide synthesis (DHA and uridine monophosphate, UMP) for rats beginning at one month of age. Learning and memory were evaluated with tasks that tap hippocampal- and striatal-dependent forms of memory (hidden- versus visible-versions of Morris water maze, respectively). Results showed interaction effects involving cognitive task, diet, and rearing environment. Co-administration of DHA and UMP increased brain levels of phosphatides and phospholipids. The enriched rearing environment also increased brain levels of phosphatides and phospholipids. Moreover, dietary enhancement improved performance on the hippocampal-dependent memory task, but only for rats reared under the impoverished environment condition. In contrast, neither diet nor rearing environment affected performance on the striatal-dependent memory task.
Figure 2.

n-3 (A) and n-6 (B) polyunsaturated fatty acid pathways. Adapted from Conklin et al. (130).
Recent findings also show cognitive outcome is influenced by biochemistry that is moderated by genetically-based vulnerability or risk for disorder. In the study by Petursdottir et al. (105), for example, the influence of dietary supplementation with DHA on cognition was examined for senescence-accelerated prone (SAMP8) mice that develop impairment in learning and memory at 8-12 months of age. At 10 months of age, mice received either a low-DHA or high-DHA diet for two months. Mice were 12 months old at the start of behavior training and retention testing in a T-maze foot-shock paradigm. Mice receiving the high-DHA diet required fewer trials to learn and remember the shock-avoidance response, compared to mice fed the low-DHA diet. The mice fed the high-DHA diet also had higher proportions of DHA in hippocampal and amygdala phospholipids. These results suggest that dietary enhancement with DHA delayed cognitive decline in a susceptible genotype.
3.2.2.1.2. Human studies
A number of prospective studies have now been completed to determine the relationship between cognitive performance and dietary and biological levels of n-3 PUFAs for healthy individuals (recent studies summarized in Table 2; reviews of earlier work published through 2009 (97-99, 106-107)). The cumulative findings suggest that higher dietary intake and RBC and plasma levels of n-3 PUFAs are associated with optimal cognitive development in children (108-110) and reduced cognitive decline for aging and elderly populations (111-112), although negative results have also been reported for young children (99) and older adults (113-114).
Table 2.
Influence of bioactive lipids on cognition: Human studies
| Study | Study aims and design | Sample | Variables | Results |
|---|---|---|---|---|
| M. A. Beydoun et al. (111) |
Association between plasma n-3 FAs and cognitive decline in older adults. Prospective cohort design. |
2251 individuals (aged 50 – 65 yrs) residing in Minneapolis, MN during 1990-1992 and 1996-1998. |
DV: (1) Cognition assessed at 2 time points (3-yr interval): (a) verbal learning and memory (b) psychomotor (c) verbal fluency (d) global cognitive decline (composite score based on change scores for tests a- c) (2) Plasma FAs at study entry (saturated, monounsaturated, linoleic acid, α- linolenic acid, n-3 and n-6 FAs) |
Association between FAs and cognitive decline from baseline to 3 years: (1) ↑ global cognitive decline associated with: ↑ palmitic acid ↑ AA ↓ linoleic acid (2) ↓ decline in verbal fluency associated with: ↑ DHA + EPA |
| J. Colombo et al. (108) |
Association between levels of phospholipid DHA in mothers and infants at delivery, and the development of attention during infancy and toddlerhood. Longitudinal (prospective) follow- up design. |
70 infants recruited for follow-up from among 350 infants and mothers enrolled in study of effects of DHA supplementation on pregnancy outcomes (randomized, double-blind, controlled clinical trial). |
IV: (1) Supplemented mothers’ diet during last trimester of pregnancy by providing eggs with high- vs. standard-levels of DHA (135 mg vs. 35 mg DHA/egg, respectively). (2) Group membership (high- vs. low- DHA) determined by levels of DHA in RBC for mother and infant at delivery. DV: Laboratory measures (gaze duration, heart rate) of attention in children during: (a) Visual habituation task in infants at ages 4, 6, and 8 mos. (b) Single-object exploration and distractibility tasks in toddlers at ages 12 and 18 mos. |
Compared to children of mothers with low-DHA levels at delivery: (1) Infants of mothers with high-DHA levels at delivery showed accelerated development for visual attention at ages 4 and 6 mos. (shorter gaze duration during visual habituation task). (2) Toddlers of mothers with high-DHA levels at delivery showed more mature development of attention (longer single-object exploration; reduced distractibility). |
| E. Cyhlarova et al. (123) |
Association between literacy skills and n-3 and n-6 FAs in dyslexia. |
32 dyslexic adults, 20 non- dyslexic controls (mean ages = 32 and 34 yrs, respectively) |
DV: RBC levels of n-3 and n-6 FAs (venous blood draw after overnight fast) Literacy skills and intelligence: WRAT word reading, spelling; WAIS subtests (similarities, vocabulary, block design, picture arrangement, digit span, digit symbol) |
A. Dyslexic individuals and non-dyslexic controls did not differ for mean levels of RBC FAs. B. Both dyslexic and control groups: Word reading and total n-3 FAs positively correlated: ↑ total n-3 FAs ↑ reading score C. Dyslexic individuals only: (1) Word reading positively correlated with n-3 FAs: ↑ α-linolenic acid ↑ reading score (2) Word reading negatively correlated with n-6 FAs: ↑ adrenic acid ↓ reading score D. Control individuals only: Word reading positively correlated with n-3 FAs: ↑ DHA ↑ reading score |
| C. Dullemeijer et al. (112) |
Association between n-3 PUFAs and change in cognition over 3 years in older adults. Parent protocol: Randomized, placebo-controlled trial to determine effect of folic acid on cognitive performance, carotid intima-media thickness, hearing. Baseline assessments occurred during 2000 and 2001. |
819 male and female adults at baseline (ages 50-70 yrs) |
IV: Random assignment to folic acid supplementation (n=406) vs. placebo (n=413) DV: Data reported for individuals in placebo-arm only (n=404): (1) Change in performance from baseline to 3 yrs in 5 domains: (a) Memory (Word Learning Test) (b) Sensorimotor speed (Stroop Color-Word-Subtest I; Concept Shifting-Subtests O, A, B) (c) Complex speed (Stroop Color-Word- III; Concept Shifting- C) (d) Information-processing speed (Letter Digit Substitution) (e) Word fluency (Verbal Fluency Test) (2) Plasma n-3 PUFAs at baseline (venous blood after overnight fast): EPA, docosapentaeoic acid, DHA |
A. Cognitive performance declined from baseline to 3 yrs for speed-related responding: sensorimotor speed, complex speed, information-processing speed. B. n-3 PUFA concentrations at baseline were associated with decline in speed-related responding at 3 yrs: ↑ n-3 PUFAs at baseline ↓ decline over 3 yrs for sensorimotor speed and complex speed. |
| J. A. Dunstan et al. (109) |
Effects of antenatal n-3 PUFA supplementation on cognitive development in children at age 2.5 yrs. Randomized, double-blind placebo-controlled trial conducted between January 2000 and September 2001. |
72 infants of 98 women enrolled during pregnancy |
IV: From 20 weeks gestation to delivery, pregnant Caucasian women with allergic disease randomly assigned to receive: (1) n-3 FA supplementation (n=52; 4 g/day fish oil with 2.2 g/day DHA and 1.1 g/day EPA) vs. (2) placebo (n=46; 4 g/day olive oil with 2.7 g/day n-9 oleic acid). Both n-3 FA supplement and placebo included 3-4 mg/g oil α-tocopherol. DV: (1) n-3 and n-6 PUFAs in phospholipids of erythrocyte membranes in umbilical cord blood (2) Assessments of children at age 2.5 yrs on measures of infant growth and development (GMDS), receptive language (PPVT), and behavior (CBCL) |
A. Effect of antenatal supplementation with n-3 PUFA on FA composition of erythrocyte membranes in umbilical cord blood of neonates: ↑ n-3 PUFAs ↓ n-6 PUFAs B. Effects of antenatal supplementation with n-3 PUFA on child’s cognitive development at age 2.5 yrs: ↑ eye–hand coordination C. Association between cognition at age 2.5 yrs and PUFA levels in cord–blood erythrocytes: (1) Eye–hand coordination positively correlated with n-3 PUFA levels (EPA, DHA): ↑ n-3 PUFA levels ↑ eye–hand coordination. (2) Eye–hand coordination and performance scores negatively correlated with n-6 PUFA levels: ↑ AA levels ↓ eye–hand coordination and performance. |
| Y. Ishikura et al. (124) |
Effect of dietary supplementation with AA on auditory P300 in healthy older men. Double-blind crossover design |
25 healthy men (ages 57-68 yrs) |
IV: 3-month crossover protocol (1 mo/phase): Group A (n=13): AA, washout, placebo Group B (n=12): placebo, washout, AA AA supplement: 600 mg/day of AA-enriched triglyceride (240 mg AA); Placebo: olive oil (600 mg/day) DV: (1) EEG recorded from midline parietal electrode (Pz) to evaluate amplitude and latency of auditory P300 component of event-related potential elicited using an oddball paradigm. (2) Serum phospholipids measured before and after supplement and placebo periods. |
A. Significant effect of AA- supplementation on P300 response: ↓ P300 latency (mean Δ = −10.1 ms ± 15.9) ↑ P300 amplitude (mean Δ = 0.8 μV ± 1.7). B. Significant effect of AA- supplementation on serum phospholipids: ↑ serum AA (mean Δ = 37.7 μg/ml ± 26.2) ↓ serum EPA (mean Δ = −13.0 μg/ml ± 20.3). |
| J. L. Jacobson et al. (110) |
Relationship of cord-plasma DHA concentration to physical growth and cognitive development at ages 6 and 11 mos for Inuit infants living in Arctic Quebec. Developmental outcomes associated with PUFA intake from breast feeding were also evaluated. Prospective longitudinal cohort study, November 1995 to March 2001 |
109 Inuit infants and their mothers |
DV: (1) Biological samples: (a) Blood collected from severed umbilical cord and from mothers after delivery. Milk collected from breast- feeding mothers at 1-mo postnatal interview. (b) FA compositions of plasma phospholipids and total maternal milk lipids. (2) Perceptual, cognitive, and motor development assessed at ages 6 mos (Teller; FTII) and 11 mos (Teller; FTII; BSID-II) |
A. ↑ DHA concentration in umbilical cord positively associated with development: ↑ gestation length ↑ visual acuity (6 mos) ↑ novelty preference (6 mos) ↑ Mental development score (11 mos) ↑ Psychomotor development score (11 mos) B. Post-natal DHA intake from breast-feeding was not correlated with developmental outcome. |
| O. van de Rest et al. (114) |
Association between dietary intake of fatty fish and change in cognition over 6 years in elderly men. Prospective cohort study, the Veterans Affairs Normative Aging Study. Recruitment initiated in 1963 in Boston, MA area. |
451 men for whom complete set of measures available (mean age at baseline cognitive assessment = 68 yrs). |
DV: (1) Estimates of n-3 PUFA intake from dietary questionnaire (g of n-3 FAs per portion/day): dark-meat fish (1.37 g); canned tuna (0.69 g); shellfish (0.46 g); other fish (0.17 g) (2) Cognitive domains assessed at baseline and over 6 yrs followup: language, speed, attention, memory, and spatial copying |
A. Cross-sectional (baseline) analyses: Cognitive performance was not associated with dietary fatty fish or n-3 PUFA intake. B. Longitudinal analyses: Cognitive change over 6 yrs was not associated with dietary fatty fish or n-3 PUFA intake. |
| L. J. Whalley et al. (120) |
Influence of erythrocyte n-3 PUFA concentration on age- related cognitive decline in the presence or absence of the APOE ε4 allele. Prospective study of population- based sample (1936 birth cohort). Recruitment from November 1999 to February 2002. |
Original sample = 478 healthy volunteers (ages 63-66 yrs); Subsample = 120 individuals for whom cognitive assessments and FA data were available. |
DV: (1) Intelligence score at 11 yrs available from national database (2) At baseline, venous blood collected for (a) APOE genotyping and (b) FA composition of erythrocyte membranes (total n-3 PUFAs, EPA, DHA, n-6:n-3 ratio) (3) Cognition evaluated at ages 64 (baseline), 66, and 68 yrs: nonverbal reasoning (RPM), verbal declarative memory (AVLT), executive function (Uses of Common Objects), information processing speed and psychomotor performance (WAIS-R Digit Symbol), constructional ability (WAIS Block Design) |
A. Relationship between PUFA concentrations and intelligence: (1) For APOE ε4 noncarriers (absent), total n-3 PUFA at baseline (≈ 63 yrs) was positively correlated with general intelligence at ages 11 yrs and 63-65 yrs: ↑ n-3 PUFA at 63 yrs ↑ Intelligence score at ages 11 and 63 yrs. (2) Relationship between PUFA concentrations and intelligence was not significant for APOE ε4 carriers (present). B. Erythrocyte membrane PUFA content did not differ between groups based on APOE ε4 status. C. Verbal declarative memory (AVLT) score differed significantly between APOE ε4 allele groups at all time points: APOE ε4 allele noncarriers > carriers. |
Abbreviations for Table 2: ↑, increase; ↓, decrease; Δ, change; AA, Arachidonic acid; AVLT, Rey’s Auditory Verbal Learning Test; BSID-II, Bayley Scales of Infant Development, 2nd edition; CBCL, Child Behavior Checklist; CGI, Clinical Global Impression scale; DHA, Docosahexaenoic acid; DV, Dependent variable; EEG, Electroencephalogram; EPA, Eicosapentaenoic acid; FA, Fatty Acid; FTII, Fagan Test of Infant Intelligence; GMDS, Griffiths Mental Development Scales; IV, Independent variable; PPVT, Peabody Picture Vocabulary Test; PUFA, Polyunsaturated fatty acid; RBC, Red Blood Cell; RPM, Raven’s Progressive Matrices; Teller, Teller Visual Acuity Card Test; WAIS-R, Wechsler Adult Intelligence Scale–revised.
Intervention with n-3 PUFAs appears promising for schizophrenia, although the combined results from randomized, placebo-controlled trials indicate that illness stage and dose range moderate the clinical efficacy of this treatment. Table 3 details the results of recent clinical studies (reviews of earlier work are provided in Fenton et al. (94), Freeman et al. (100), and Peet (102)). In their study of individuals at ultra-high risk for psychosis, Amminger et al. (115) observed that 12 weeks of n-3 PUFA treatment (700 mg/day of EPA + 480 mg/day of DHA) produced a lower rate of conversion to psychosis at one year, compared to placebo. Berger et al. (116) determined that first-episode psychosis patients who received 12 weeks of ethyl eicosapentanoic-acid (2 g/day E-EPA) augmentation had a more rapid first clinical response, required lower doses of antipsychotic drugs, and exhibited fewer extrapyramidal side effects. In the study of patients with chronic schizophrenia by Fenton et al. (117), however, medicated outpatients receiving 16 weeks of E-EPA augmentation (3 g/day) did not differ from those receiving placebo with respect to improvement in clinical symptoms or cognitive performance at 16 weeks. The findings of Fenton et al. contrast to the results obtained by Peet and Horrobin (118) and Emsley et al. (119) for patients with persistent residual symptoms. Peet and Horrobin augmented outpatients’ antipsychotic drug regimen with one of four dose levels of ethyl eicosapentaenoate (0, 1, 2, or 4 g/day E-EPA) for 12 weeks, and observed the greatest clinical improvement for patients receiving clozapine and 2 g/day E-EPA. Similarly, Emsley et al. augmented patients’ antipsychotic regimen with 3 g/day of E-EPA for 12 weeks, and observed greater improvement in clinical symptoms and dyskinesia for the E-EPA group, compared to those receiving placebo.
As reflected in these combined studies, the role of PUFA status in cognition and mental health is not straightforward. Importantly, in parallel with the animal research, this rapidly growing literature includes recognition that the relationship between PUFA status and cognition in humans is moderated by biological risk for disorder. A noteworthy example is provided by the study of Whalley et al. (120), in which concentrations of erythrocyte n-3 PUFA were associated with better cognitive performance in older healthy individuals, but only in the absence of the APOE ε4 allele, thereby highlighting the combined influences of genetic liability and environmental factors for cognitive aging.
3.2.2.2. n-6 PUFAs
Converging evidence from animal and human clinical studies suggest that cortical levels of n-6 fatty acids (Figure 2B) may be more tightly coupled with cognition than are cortical levels of n-3 fatty acids, at least for some cognitive functions. The recent study by Arendash and colleagues (121) illustrates this specificity of linkage. Those investigators examined the effects on cognitive performance of a diet comparable to the 13% n-3 fatty acid diet of Greenland Eskimos (122). Alzheimer’s transgenic mice (Tg) and non-transgenic (NT) normal mice littermates were fed either a diet high in n-3 fatty acids or a standard diet from two to nine months of age. Learning and memory were evaluated during the final six weeks of the dietary condition. Diet influenced cortical levels of n-3 fatty acids differently for each genotype. Cortical levels of n-3 fatty acids were increased and n-6 fatty acids were decreased in NT mice fed the diet enriched with n-3 fatty acids. In contrast, the enriched n-3 fatty acid diet had no effect on cortical levels of fatty acids in Tg mice. Moreover, the type of PUFA was important for the relationship between fatty acids in frontal cortex and cognition. Cognitive performance was correlated with cortical levels of n-6 fatty acids for both Tg and NT mice. The direction of this relationship indicated that increases in cortical levels of n-6 fatty acids, including arachidonic acid, were associated with decreases in cognitive performance. However, cognition was not associated with cortical levels of n-3 fatty acids in either Tg or NT mice.
The pattern of association between arachidonic acid (AA) and cognitive performance that was observed by Arendesh and colleagues parallels the results reported for schizophrenia patients, dyslexic individuals, and children whose mothers received n-3 PUFA supplementation during pregnancy. Findings from our laboratory revealed significant inverse relationships between AA and reading, semantic memory, and general intelligence in unmedicated patients with chronic schizophrenia, with increases in RBC AA level associated with decreases in single-word reading accuracy, intelligence test score, and electrophysiological response to semantic memory activation (see Section 3.5). Inverse relationships between n-6 PUFAs and cognition have also been observed for individuals with dyslexia and children. Cyhlarova et al. (123) reported negative associations between n-6 fatty acid concentrations and word reading for dyslexic individuals. Dunstan et al. (109) reported an inverse correlation between level of AA in umbilical cord blood at delivery and cognitive performance in children at 30 months of age. Other findings, however, suggest positive associations between AA concentration and behavior. In addition to increases in serum level of AA, one-month of dietary supplementation with AA (240 mg/day) produced improvement in electrophysiological response (increased amplitude and reduced latency of P300) for healthy older men (124). Moreover, results from the E-EPA augmentation trial for schizophrenia that was conducted by Peet and Horrobin (118) included positive correlations between changes in RBC AA levels and clinical improvement from baseline to 12 weeks. Horrobin et al. (125) interpreted this pattern as due to the beneficial effect of lower doses of EPA (viz., the mid-range dose of 2 g/day that was associated with clinical improvement in Peet and Horrobin (118)), which produced elevated levels of AA, rather than reduced levels. The implication of these findings is that elevations of both EPA and AA may be beneficial for behavior.
3.2.2.3. Eicosanoids (Arachidonic acid cascade)
In addition to the formation of second messengers, the newly released AA from membrane phospholipids can be converted to a variety of biologically active metabolites, which are collectively referred to as eicosanoids, through the concerted reactions of cyclooxygenase (COX), lipoxygenases (LOX) and cytochrome P-450 (CYP) (see Figure 3). Eicosanoids can modulate neural cell function and thereby mediate several pathophysiological processes (126). Since AA is the major C20 PUFA in mammalian tissues, the prostaglandin-2 (PG2) and thromboxanes-2 (TX2) series are the predominant classes of eicosanoids (shown in Figure 3). Studies involving the inhibition of COX by nonsteroidal anti-inflammatory drugs have revealed the significance of PG2 in the regulation of nerve conduction, neurotransmitter release, inflammation, pain, fever, immune responses, apoptosis and psychosis (127). In schizophrenia, there are reduced levels of AA in membrane phospholipids (65, 128-130) that could conceivably lead to a decreased synthesis of eicosanoids. A deficiency of prostaglandins also has been previously related to schizophrenia (131). It is therefore likely that the reduced AA availability may explain, in part, a variety of clinical observations that are usually ignored by the receptor-based etiological hypotheses (64). For example, in schizophrenia there appears to be a lower risk of arthritis and other inflammatory diseases (132-134), and greater resistance to pain (131, 135). Remission of psychosis during fever has also been observed (131). This pattern of effects could be secondary to abnormal eicosanoid signaling.
Figure 3.

Arachidonic acid cascade. Abbreviations: COX, Cyclooxygenases; CYP, Cytochrome P-450; LOX, Lipoxygenase; DiHETE, Dihydroxyeicosatetraenate; EET, Epoxyeicosatrienate; ETT, Epoxytetraenate; HETE, Hydroxyeicosatetraenate; HHT, Hydroxyl-heptadecatrienate; HPETE, Hydroperoxyeicosatetraenate; LT, Leukotriene; LX, Lipoxin; Prostaglandin; Tx, Thromboxane.
Both the dopamine excess and cerebral perfusion abnormalities associated with schizophrenia could be accounted for by a deficiency in the release or response to prostaglandins. PGD2 and other PGs stimulate the production of cyclic AMP (cAMP) and thereby exert functional antagonism of dopamine-D2 receptors, which, in turn, modulate inhibition of cAMP synthesis (136-137). PGD2 therefore counteracts the biochemical and behavioral effects of dopamine in vitro and in vivo. Moreover, PGD2 administration blocks the behavioral effects of dopaminergic agents such as apomorphine, L-DOPA, and amphetamine, and induces catalepsy in a manner identical to dopamine-D2 receptor antagonists such as haloperidol (136-137). Deficient PGD2 signaling in the brain would therefore be expected to influence dopamine transmission. The adverse effect of dopaminergic agents on movement may also be moderated by neuroleptic-induced oxidative stress (138). A role for PGs as endogenous anti-dopaminergic agents is supported by the findings that inhibitors of PG synthesis can cause psychotic symptoms in some people and can worsen psychotic symptoms in schizophrenia patients (139-141).
3.3. Phospholipids signaling and schizophrenia
The behavioral and biological complexity of schizophrenia disorder has led to a view that the key to unraveling its pathophysiology lies in deconstructing the clinically-defined illness syndrome into pathophysiologically distinct intermediate phenotypes (142-143). We and others have reported evidence suggesting that one of these intermediate phenotypes may involve phospholipid signaling abnormalities. Utilizing varying types of samples (plasma, RBC, platelets, postmortem brain) and methodologies (31P MRS, platelet function, niacin-induced flushing), somewhat consistent patterns of decreased PUFAs and increased phospholipid turnover are apparent (62-66, 129, 144-145), particularly in relation to AA. An abnormality in phospholipid turnover could potentially explain many seemingly unrelated neurochemical observations in schizophrenia.
Figure 4 shows an adaptation of the model of phospholipids turnover that was previously proposed by our group (146), in which most of the known data in relation to phospholipids turnover and schizophrenia were integrated, and includes several mechanisms that could lead to increased phospholipid breakdown and AA release, such as decreased AA incorporation, and increased phospholipase activities (PLA2 and PLC) possibly resulting from increased oxidative stress and cytokine release. The resulting changes in AA level could then affect more ‘downstream’ processes, including neurodevelopment via GAP-43, neurotransmitter homeostasis, phosphatidylinositol signaling, and neuromodulatory actions of endocannabinoids (65-66).
Figure 4.

An overview of phospholipids turnover, polyunsaturated fatty acids and cognition in schizophrenia. Abbreviations: AODS, antioxidant defense system; NT, neurotransmitters; RO, reactive oxygen species; PC, phosphatidylcholine; PE, phosphatidyl-ethanolamine; PI, phosphatidylinositol; apoD, apolipoprotein D; DA, dopamine; PLA2, phospholipase A2; PLC, phospholipase C; IP3, inositol triphosphate; AA, arachidonic acid; DHA, docosahexaenoic acid, DAG, diacylglycerol; LOX, lipoxygenase; COX, cyclooxygenase; 2-AG, 2-arachidonoyl glycerol; GAP, growth-associated protein; PKC, phosphokinase C; CB, cannabinoid. Adapted from Skosnik and Yao (65).
3.4. Interactions between monoaminergic neurotransmission and PUFAs: A model for cognitive deficits in schizophrenia
Abnormalities in long-chain PUFAs produce alterations in monoaminergic transmission that are suggestive of an imbalance between cortical and subcortical dopamine activity. Findings from animal studies indicate that PUFA abnormalities, induced by chronic dietary deficiency of n-3 fatty acids, have a differential effect on monoamine transmission in mesocortical and mesolimbic dopamine pathways (reviewed by Chalon (67, 147)). Some of those alterations include lower concentration of endogenous dopamine and reduced density of D2 receptors in frontal cortex (148-149). The range of reduction to endogenous dopamine in frontal cortex was substantial; 40-75% less in the rats receiving a diet deficient in α-linolenic acid, the precursor of long-chain n-3 PUFAs (149). Dietary deficiency of α-linolenic acid also produced a decrease in dopamine release within frontal cortex following tyramine stimulation, suggesting reduced release of dopamine from the vesicular storage pool (150). Moreover, the number of vesicles in dopamine presynaptic terminals was reduced in frontal cortex of rats fed the diet deficient in α-linolenic acid (70). In combination, these data provide support for an association between alterations in n-3 PUFA concentrations and abnormalities of structure and function within the dopamine system.
Altered PUFA status and brain levels of monoamines are further associated with abnormalities in cognition and behavior. Fedorova et al. (71) observed moderate to profound impairments in spatial learning for rats that experienced diet-induced reductions in brain levels of DHA (58% reduction compared to controls). Importantly, the rats with deficient brain levels of DHA also had 50% reduction to dopamine levels in ventral striatum and enhanced levels of the dopamine metabolite DOPAC (3,4-dihydroxyphenylacetic acid) in frontal cortex. Chalon and colleagues (151) found activity level of rats was affected by the influence of dietary intake of n-3 PUFA on dopamine neurotransmission. Compared to rats fed a diet low in n-3 PUFA, those receiving dietary fish oil developed endogenous dopamine concentrations that were 40% greater in frontal cortex, increased binding to D2 receptors in frontal cortex, and decreased binding to D2 receptors in striatum. Moreover, ambulatory activity was reduced for the rats receiving dietary fish oil, which was interpreted as a possible consequence of striatal dopamine function on locomotion. Ng and Innis (152) examined the effects of dietary PUFAs and dopamine challenge on fear responses and exploratory behavior in neonatal piglets. Piglets fed a diet high in n-3 PUFAs for 30 days developed greater levels of DHA in frontal cortex and were more efficient during maze learning, compared to piglets fed a low n-3 PUFA diet. However, administration of the dopamine precursor L-Dopa abolished the dietary group difference in maze performance, supporting the importance of DHA to central dopamine function.
3.5. Hypothesis: The relationship between cognition and membrane phospholipids in schizophrenia is moderated by dopaminergic neurotransmission
Our group has proposed that semantic memory in schizophrenia is influenced by alterations in dopaminergic neurotransmission (40-42) and abnormalities in cell membrane phospholipids (43). We further hypothesize that the relationship between cognition and membrane phospholipids in schizophrenia is moderated by dopamine. Findings from our program that support linkages among these domains for schizophrenia include: (1) associations between RBC concentrations of the membrane phospholipid AA and cognition, including semantic memory, reading, and general intelligence; (2) association between semantic memory and dopamine turnover, measured by level of the dopamine metabolite homovanillic acid (HVA) in cerebrospinal fluid (CSF); (3) association between membrane phospholipids (RBC AA) and dopamine turnover (CSF HVA). These findings are summarized briefly in the following:
3.5.1. Association between AA and semantic memory, reading, and general intelligence
3.5.1.1. Semantic memory
Schizophrenia patients are characterized by abnormalities in their electrophysiological discrimination of semantic information (overview in Section 3.1.2.2.). In healthy individuals, the amplitude of the N400 component of the event-related brain potential is typically larger for semantically unrelated words (‘dog - pencil’) than for words that are meaningfully associated (‘dog - cat’). This differential response is referred to as the N400 semantic priming effect. We previously reported findings for the N400 semantic priming effect that was recorded for patients with chronic schizophrenia during their participation in a double-blind placebo-controlled protocol, in which the effects of haloperidol-only were compared to placebo (40-41). While healthy controls exhibited a clear differentiation of semantic relationships within the N400 time window (270-550 ms post-word onset), patients’ N400 priming response was severely reduced under the placebo condition and did not normalize during haloperidol maintenance therapy.
Linkage between cognition and membrane phospholipids was additionally examined for a subset of this sample of schizophrenia patients for whom both cognitive and neurochemistry data were available. N400 semantic priming effect – Figure 5 shows the relationship between arachidonic acid (AA) and semantic memory obtained for a subgroup (n=14) of unmedicated patients (originally reported in Condray et al. (43)). The inverse direction of this relationship indicates that increases in RBC concentrations of AA were associated with decreases in the N400 semantic priming effect (i.e., reduced electrophysiological discrimination of meaning). Reading skill and general intelligence – Results based on the same subgroup of unmedicated patients demonstrate that increases in RBC AA were also associated with decreases in single-word reading accuracy (AA and WRAT Reading score: Spearman’s rho = − 0.60, p < 0.05) and decreases in general intelligence score (AA and WAIS-R FIQ: Spearman’s rho = − 0.61, p < 0.05) (R. Condray, unpublished data).
Figure 5.
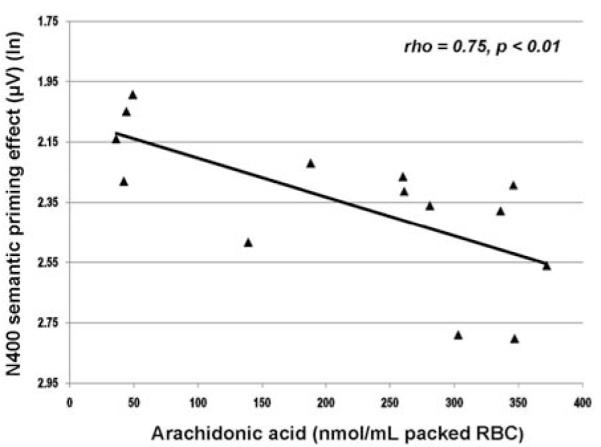
Relationship between arachidonic acid (20:4n-6) (nmol/mL packed RBC) and N400 semantic priming effect (μV) for unmedicated schizophrenia patients (n=14). N400 semantic priming effect reflects the difference between the mean area integration of N400 amplitudes (μV) recorded for semantically unrelated and related words over the 270 to 550 ms range at anterior midline scalp (Fz) [N400-unrelated words minus N400-related words]. Negativity is upward. Greater negativity in this difference measure reflects more typical (or normal) N400 semantic priming response. Abbreviation: ln, natural log. Reprinted from Condray et al. (43), with permission from Elsevier.
Taken together, these data suggest a coupling between AA and cognition in schizophrenia. We propose that the pattern of relationships observed, in which lower levels of AA were associated with better cognitive performance, are due to defects in the AA cascade. To understand this pattern, however, it is important to consider steps that occur during phospholipids biosynthesis, but that were not measured in our earlier work (see Section 3.2.2.3. and Figure 3). In addition to the formation of second messengers, the newly released AA from membrane phospholipids can be converted to a variety of biologically active metabolites, collectively referred to as eicosanoids, through the concerted reactions of COX, LOX, and CYP. As described above, eicosanoids can modulate neural cell function. Because AA is the major C20 PUFA in mammalian tissues, the prostaglandin-2 (PG2) and thromboxanes-2 series are the predominant classes of eicosanoids. As noted, studies involving the inhibition of COX by nonsteroidal anti-inflammatory drugs show the importance of PG2 in the regulation of nerve conduction, neurotransmitter release, inflammation, pain, fever, immune responses, apoptosis, and psychosis.
Based on the above considerations, the reduced levels of AA in membrane phospholipids observed for schizophrenia could conceivably lead to a decreased synthesis of eicosanoids, a possibility that receives support from the finding of reduced levels of prostaglandins for this patient group. Our findings of inverse correlations between patients’ cognitive performance and their levels of AA may be due to such defects in the AA cascade. Thus, one plausible explanation for these inverse relationships is that patients’ potential precursor pool of AA, which is available for conversion to PGD2 through COX, is limited. The better cognitive performance that was associated with lower levels of phospholipids AA may therefore indirectly reflect higher levels of free AA that lead to synthesis of PGD2. This interpretation requires careful empirical testing.
3.5.2. Association between semantic memory and dopamine turnover
Figure 6 presents the correlation obtained between HVA levels in cerebrospinal fluid and the N400 semantic priming response for patients evaluated during placebo replacement (R. Condray, unpublished data). The direction of this relationship indicates that increases in CSF level of HVA were associated with increases in N400 semantic priming (i.e., more typical or normal electrophysiological response to semantic activation). (Details for measurement of CSF data are provided in van Kammen et al. (153).)
Figure 6.
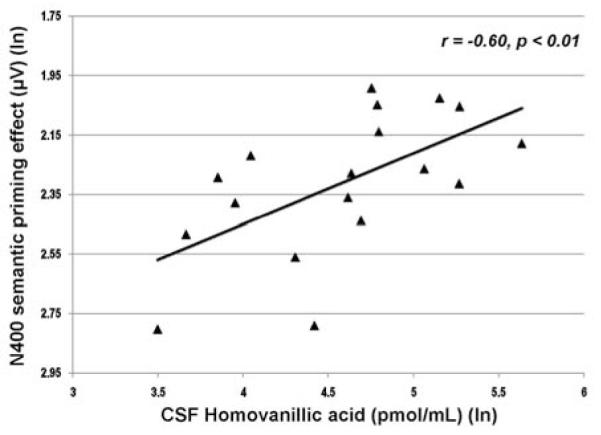
Relationship between CSF homovanillic acid (pmol/ml) and N400 semantic priming effect (μV) for unmedicated schizophrenia patients (n=18). N400 semantic priming effect reflects the difference between the mean area integration of N400 amplitudes (μV) recorded for semantically unrelated and related words over the 270 to 550 ms range at anterior midline scalp (Fz) [N400-unrelated words minus N400-related words]. Negativity is upward. Greater negativity in this difference measure reflects more typical (or normal) N400 semantic priming response. Abbreviation: ln, natural log.
3.5.3. Association between membrane phospholipids and dopamine turnover
Our hypothesis that dopaminergic neurotransmission plays an important role in the relationship between cognition and membrane phospholipids receives support from two additional aspects of our data: (a) As shown in Figure 7, concentrations of arachidonic acid and levels of HVA were significantly and negatively correlated for patients during placebo replacement (R. Condray, unpublished data), indicating that increases in CSF HVA were associated with decreases in RBC AA. This relationship is suggestive of linkage between dopamine neurotransmission and AA cascade for schizophrenia, although this possibility requires careful empirical testing. (b) AA concentration and semantic memory response were significantly correlated only for patients when unmedicated during placebo replacement; AA and semantic memory were not correlated when patients were receiving maintenance doses of haloperidol, a typical antipsychotic drug with high affinity for dopamine D2 receptors.
Figure 7.
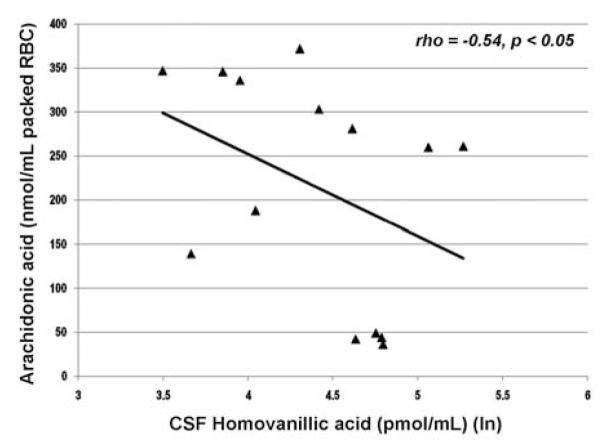
Relationship between CSF homovanillic acid (pmol/ml) and arachidonic acid (20:4n-6) (nmol/mL packed RBC) for unmedicated schizophrenia patients (n=14). Abbreviation: ln, natural log.
4. SUMMARY AND PERSPECTIVE
The cause(s) of the hypothesized interaction of PUFAs and monoamine neurotransmission have not been determined although the cumulative evidence suggests a high degree of functional interdependence. Cortical levels of phospholipids influence structure and transmission within dopaminergic pathways. Stimulation of dopamine D2 receptors can enhance the activation of PLA2 and subsequent release of arachidonic acid from membranephos pholipids. Arachidonic acid and dopamine are tightly coupled to cognition and behavior. In combination, it is therefore possible that such interconnected trajectories are compromised in schizophrenia due to the known abnormalities involving the n-6 family of phospholipids and PLA2, one of the enzymes key for phospholipids breakdown. As discussed above, PUFA dysregulation has been consistently observed for schizophrenia, primarily involving decreased levels of AA. Possible causes of this dysregulation include increased PLA2 activity and decreased incorporation of AA into membrane phospholipids (reviewed by Skosnik and Yao (65)).
Our model of the combined influence of membrane PUFAs and monoamine neurotransmission on cognition in schizophrenia (Figure 1) was adapted from the model that was proposed earlier by our group (Figure 4), in which the known data in relation to phospholipids turnover and schizophrenia were integrated (65, 146). As we have emphasized, that model included several mechanisms that can lead to increased phospholipid breakdown and AA release, such as decreased incorporation of AA into phospholipids, and increased phospholipase activities (PLA2 and PLC), possibly resulting from increased oxidative stress and cytokine release. Furthermore, the resulting changes in AA level could then influence more ‘downstream’ processes, including neurodevelopment via GAP-43, neurotransmitter homeostasis, phosphatidylinositol signaling, and neuromodulatory actions of endocannabinoids (65-66).
Our principal assumption for cognitive disorder in schizophrenia is related mostly to the effects of alterations in AA signaling and its interaction with the neurochemistry of monoamine transmission, particularly dopamine. We predict that AA also influences the neurochemistry of glutamate release, and circulating levels of the endocannabinoids anandamide and 2-arachidonoyl-glycerol (2-AG). In addition, alterations in AA may also affect the inflammatory response, which can then affect PLA2 release via cytokines, further exacerbating phospholipid turnover and AA release. Hence, in our current conceptualization, a biological abnormality involving AA could account for a portion of the deficits in neurocognition observed in schizophrenia.
5. ACKNOWLEDGEMENTS
This material is based upon work supported in part by the grants from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development [Merit Reviews (RC, JKY) and Senior Research Career Scientist Award (JKY), VA Pittsburgh Healthcare System], and the National Institutes of Health [MH50631 (RC) and MH58141 (JKY)]. The work was also supported with resources and use of facilities at the Western Psychiatric Institute and Clinic, University of Pittsburgh School of Medicine, and the VA Pittsburgh Healthcare System, Pittsburgh, PA. The data presented for schizophrenia patients were developed in collaboration with Daniel P. van Kammen, M.D., Ph.D, and Stuart R. Steinhauer, Ph.D. We are grateful to two anonymous reviewers who provided valuable recommendations. The funding institutions had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
6. REFERENCES
- 1.Murray CJL, Lopez AD. The global burden of disease: a comprehensive assessment of mortality and disability from diseases, injuries, and risk factors in 1990 and projected to 2020. Harvard University Press; Cambridge, MA: 1996. [Google Scholar]
- 2.Palmer BW, Heaton RK, Paulsen JS, Kuck J, Braff D, Harris JM, Zisook S, Jeste DV. Is it possible to be schizophrenic yet neuropsychologically normal? Neuropsychology. 1997;11:437–446. doi: 10.1037//0894-4105.11.3.437. [DOI] [PubMed] [Google Scholar]
- 3.Keefe RSE, Eesley CE, Poe MP. Defining a cognitive function decrement in schizophrenia. Biol Psychiatry. 2005;57:688–691. doi: 10.1016/j.biopsych.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Palmer BW, Dawes SE, Heaton RK. What do we know about neuropsychological aspects of schizophrenia? Neuropsychol Rev. 2009;19:365–384. doi: 10.1007/s11065-009-9109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowie CR, Leung WW, Reichenberg A, McClure MM, Patterson TL, Heaton RK, Harvey PD. Predicting schizophrenia patients’ real-world behavior with specific neuropsychological and functional capacity measures. Biol Psychiatry. 2008;63:505–511. doi: 10.1016/j.biopsych.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green MF. What are the functional consequences of neurocognitive deficit in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- 7.Shean GD. Recent developments in psychosocial treatments for schizophrenic patients. Expert Rev Neurother. 2007;7:817–827. doi: 10.1586/14737175.7.7.817. [DOI] [PubMed] [Google Scholar]
- 8.Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res. 2004;72:41–51. doi: 10.1016/j.schres.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Niekawa N, Sakuraba Y, Uto H, Kumazawa Y, Matsuda O. Relationship between financial competence and cognitive function in patients with schizophrenia. Psychiatry Clin Neurosci. 2007;61:455–461. doi: 10.1111/j.1440-1819.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 10.Gold JM. Cognitive deficits as treatment targets in schizophrenia. Schizophr Res. 2004;72:21–28. doi: 10.1016/j.schres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Buchanan RW, Davis M, Goff D, Green MF, Keefe RSE, Leon AC, Nuechterlein KH, Laughren T, Levin R, Stover E, Fenton W, Marder SR. A summary of the FDA-NIMH-MATRICS Workshop on Clinical Trial Design for Neurocognitive Drugs for Schizophrenia. Schizophr Bull. 2005;31:5–19. doi: 10.1093/schbul/sbi020. [DOI] [PubMed] [Google Scholar]
- 12.Harvey PD. Pharmacological cognitive enhancement in schizophrenia. Neuropsychology Review. 2009;19:324–335. doi: 10.1007/s11065-009-9103-4. [DOI] [PubMed] [Google Scholar]
- 13.Fisher M, Holland C, Merzenich MM, Vinogradov S. Using neuroplasticity-based auditory training to improve verbal memory in schizophrenia. Am J Psychiatry. 2009;166:805–811. doi: 10.1176/appi.ajp.2009.08050757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tulving E. Episodic and semantic memory. In: Tulving E,E, Donaldson W, editors. Organization of memory. Academic Press; NY: 1972. [Google Scholar]
- 15.Warrington EK. Neuropsychological evidence for multiple memory systems. Ciba Foundation Symposium. 1979;69:153–166. doi: 10.1002/9780470720523.ch9. [DOI] [PubMed] [Google Scholar]
- 16.Schachter DL, Tulving E. What are the memory systems of 1994? In: Schachter DL, Tulving E, editors. Memory Systems 1994. The MIT Press; Cambridge, MA: 1994. [Google Scholar]
- 17.Schachter DL, Wagner AD, Buckner RL. Memory systems of 1999. In: Tulving EE, Craik FIM, editors. The Oxford Handbook of Memory. Oxford University Press; NY: 2000. [Google Scholar]
- 18.Baddeley A. Working memory. Oxford University Press; Oxford: 1986. [Google Scholar]
- 19.Baddeley A. Working memory: the interface between memory and cognition. In: Daniel L, Schachter DL, Tulving E, editors. Memory Systems. The MIT Press; Cambridge, MA: 1994. [Google Scholar]
- 20.Squire LR, Zola-Morgan S. The medial temporal lobe memory system. Science. 1991;253:1380–1386. doi: 10.1126/science.1896849. [DOI] [PubMed] [Google Scholar]
- 21.Aleman A, Hijman R, de Haan EHF, Kahn RS. Memory impairment in schizophrenia: a meta-analysis. Am J Psychiatry. 1999;156:1358–1366. doi: 10.1176/ajp.156.9.1358. [DOI] [PubMed] [Google Scholar]
- 22.Cirillo MA, Seidman LJ. Verbal declarative memory dysfunction in schizophrenia: from clinical assessment to genetics and brain mechanisms. Neuropsychol Rev. 2003;13:43–77. doi: 10.1023/a:1023870821631. [DOI] [PubMed] [Google Scholar]
- 23.Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology. 1998;12:426–445. doi: 10.1037//0894-4105.12.3.426. [DOI] [PubMed] [Google Scholar]
- 24.Nuechterlein KH, Barch DM, Gold JM, Goldberg TE, Green MF, Heaton RK. Identification of separable cognitive factors in schizophrenia. Schizophr Res. 2004;72:29–39. doi: 10.1016/j.schres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26:119–136. doi: 10.1093/oxfordjournals.schbul.a033430. [DOI] [PubMed] [Google Scholar]
- 26.Cannon M, Caspi A, Moffitt TE, Harrington HL, Taylor A, Murray RM, Poulton R. Evidence for early-childhood, pan-developmental impairment specific to schizophreniform disorder: results from a longitudinal birth cohort. Arch Gen Psychiatry. 2002;59:449–456. doi: 10.1001/archpsyc.59.5.449. [DOI] [PubMed] [Google Scholar]
- 27.Crow TJ, Done DJ, Sacker A. Childhood precursors of psychosis as clues to its evolutionary origins. Eur Arch Psychiatry Clin Neurosci. 1995;245:61–69. doi: 10.1007/BF02190732. [DOI] [PubMed] [Google Scholar]
- 28.Docherty NM, Gottesman II. A twin study of communication disturbances in schizophrenia. J Nerv Ment Dis. 2000;188:395–401. doi: 10.1097/00005053-200007000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Docherty NM, Gordinier SW, Hall MJ, Cutting LP. Communication disturbances in relatives beyond the age of risk for schizophrenia and their associations with symptoms in patients. Schizophr Bull. 1999;25:851–862. doi: 10.1093/oxfordjournals.schbul.a033424. [DOI] [PubMed] [Google Scholar]
- 30.Docherty NM, Gordinier SW, Hall MJ, Dombrowski ME. Referential communication disturbances in the speech of nonschizophrenic siblings of schizophrenia patients. J Abnorm Psychol. 2004;113:399–405. doi: 10.1037/0021-843X.113.3.399. [DOI] [PubMed] [Google Scholar]
- 31.Maher BA. A tentative theory of schizophrenic utterance. Prog Exp Pers Res. 1983;12:1–52. [PubMed] [Google Scholar]
- 32.Minzenberg MJ, Ober BA, Vinogradov S. Semantic priming in schizophrenia: a review and synthesis. J Int Neuropsychol Soc. 2002;8:699–720. doi: 10.1017/s1355617702801357. [DOI] [PubMed] [Google Scholar]
- 33.Doughty OJ, Done DJ. Is semantic memory impaired in schizophrenia? A systematic review and meta-analysis of 91 studies. Cogn Neuropsychiatry. 2009;14:473–509. doi: 10.1080/13546800903073291. [DOI] [PubMed] [Google Scholar]
- 34.Pomarol-Clotet E, Oh TMSS, Laws KR, McKenna PJ. Semantic priming in schizophrenia: systematic review and meta-analysis. The British Journal of Psychiatry. 2008;192:92–97. doi: 10.1192/bjp.bp.106.032102. [DOI] [PubMed] [Google Scholar]
- 35.Kutas M, Hillyard SA. Reading senseless sentences: brain potentials reflect semantic incongruity. Science. 1980;207:203–205. doi: 10.1126/science.7350657. [DOI] [PubMed] [Google Scholar]
- 36.Kutas M, Federmeier KD. Electrophysiology reveals semantic memory use in language comprehension. Trends Cognitive Sciences. 2000;4:463–470. doi: 10.1016/s1364-6613(00)01560-6. [DOI] [PubMed] [Google Scholar]
- 37.Kuperberg GR, Kreher DA, Ditman T. What can event-related potentials tell us about language, and perhaps even thought, in schizophrenia? Int J Psychophysiol. 2010 doi: 10.1016/j.ijpsycho.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathalon DH, Roach BJ, Ford JM. Automatic semantic priming abnormalities in schizophrenia. Int J Psychophysiol. 2010;75:157–166. doi: 10.1016/j.ijpsycho.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kreher DA, Goff D, Kuperberg GR. Why all the confusion? Experimental task explains discrepant semantic priming effects in schizophrenia under “automatic” conditions: evidence from event-related potentials. Schizophr Res. 2009;111:174–181. doi: 10.1016/j.schres.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Condray R, Steinhauer SR, Cohen JD, van Kammen DP, Kasparek A. Modulation of language processing in schizophrenia: effects of context and haloperidol on the event-related potential. Biol Psychiatry. 1999;45:1336–1355. doi: 10.1016/s0006-3223(98)00317-5. [DOI] [PubMed] [Google Scholar]
- 41.Condray R, Siegle GJ, Cohen JD, van Kammen DP, Steinhauer SR. Automatic activation of the semantic network in schizophrenia: Evidence from event-related potentials. Biol Psychiatry. 2003;54:1134–1148. doi: 10.1016/s0006-3223(03)00699-1. [DOI] [PubMed] [Google Scholar]
- 42.Condray R, Siegle GJ, Keshavan MS, Steinhauer SR. Effects of word frequency on semantic memory in schizophrenia: electrophysiological evidence for a deficit in linguistic access. Int J Psychophysiology. 2010;75:141–156. doi: 10.1016/j.ijpsycho.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Condray R, Yao JK, Steinhauer SR, van Kammen DP, Reddy RD, Morrow LA. Semantic memory in schizophrenia: association with cell membrane essential fatty acids. Schizophr Res. 2008;106:13–28. doi: 10.1016/j.schres.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saykin AJ, Shtasel DL, Gur RE, Kester DB, Mozley LH, Stafiniak P, Gur RC. Neuropsychological deficits in neuroleptic naive patients with first-episode schizophrenia. Arch Gen Psychiatry. 1994;51:124–131. doi: 10.1001/archpsyc.1994.03950020048005. [DOI] [PubMed] [Google Scholar]
- 45.Saykin AJ, Gur RC, Gur RE, Mozley PD, Mozley LH, Resnick SM, Kester DB, Stafiniak P. Neuropsychological function in schizophrenia: selective impairment in memory and learning. Arch Gen Psychiatry. 1991;48:618–624. doi: 10.1001/archpsyc.1991.01810310036007. [DOI] [PubMed] [Google Scholar]
- 46.Faraone SV, Seidman LJ, Kremen WS, Pepple JR, Lyons MJ, Tsuang MT. Neuropsychological functioning among the nonpsychotic relatives of schizophrenic patients: a diagnostic efficiency analysis. J Abnorm Psychol. 1995;104:286–304. doi: 10.1037//0021-843x.104.2.286. [DOI] [PubMed] [Google Scholar]
- 47.Faraone SV, Seidman LJ, Kremen WS, Toomey R, Pepple JR, Tsuang MT. Neuropsychological functioning among the nonpsychotic relatives of schizophrenic patients: a 4-year follow-up study. J Abnorm Psychol. 1999;108:176–181. doi: 10.1037//0021-843x.108.1.176. [DOI] [PubMed] [Google Scholar]
- 48.Gur RE, Calkins ME, Gur RC, Horan WP, Nuechterlein KH, Seidman LJ, Stone WS. The consortium on the genetics of schizophrenia: neurocognitive endophenotypes. Schizophr Bul. 2007;33:49–68. doi: 10.1093/schbul/sbl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kremen WS, Seidman LJ, Pepple JR, Lyons MJ, Tsuang MT, Faraone SV. Neuropsychological risk indicators for schizophrenia: a review of family studies. Schizophr Bull. 1994;20:103–119. doi: 10.1093/schbul/20.1.103. [DOI] [PubMed] [Google Scholar]
- 50.Kremen WS, Faraone SV, Seidman LJ, Pepple JR, Tsuang MT. Neuropsychological risk indicators for schizophrenia: a preliminary study of female relatives of schizophrenic and bipolar probands. Psychiatry Res. 1998;79:227–240. doi: 10.1016/s0165-1781(98)00042-0. [DOI] [PubMed] [Google Scholar]
- 51.Snitz BE, MacDonald AW, III, Carter CS. Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull. 2006;32:179–194. doi: 10.1093/schbul/sbi048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson JL, Watson JR, Steinhauer S.t R., Goldstein G, Pogue-Geile MF. Indicators of genetic liability to schizophrenia: a sibling study of neuropsychological performance. Schizophr Bull. 2005;31:85–96. doi: 10.1093/schbul/sbi009. [DOI] [PubMed] [Google Scholar]
- 53.Cannon TD, Huttunen MO, Lonnqvist J, Tuulio-Henriksson A, Pirkola T, Glahn D, Finkelstein J, Hietanen M, Kaprio J, Koskenvuo M. The inheritance of neuropsychological dysfunction in twins discordant for schizophrenia. Am J Hum Genet. 2000;67:369–382. doi: 10.1086/303006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toulopoulou T, Picchioni M, Rijsdijk F, Hua-Hall M, Ettinger U, Sham P, Murray R. Substantial genetic overlap between neurocognition and schizophrenia: genetic modeling in twin samples. Arch Gen Psychiatry. 2007;64:1348–1355. doi: 10.1001/archpsyc.64.12.1348. [DOI] [PubMed] [Google Scholar]
- 55.Greenwood TA, Braff DL, Light GA, Cadenhead KS, Calkins ME, Dobie DJ, Freedman R, Green MF, Gur RE, Gur RC, Mintz J, Nuechterlein KH, Olincy A, Radant AD, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Turetsky BI, Schork NJ. Initial heritability analyses of endophenotypic measures for schizophrenia: the consortium on the genetics of schizophrenia. Arch Gen Psychiatry. 2007;64:1242–1250. doi: 10.1001/archpsyc.64.11.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tuulio-Henriksson A, Haukka J, Partonen T, Varilo T, Paunio T, Ekelund J, Cannon TD, Meyer JM, Lönnqvist J. Heritability and number of quantitative trait loci of neurocognitive functions in families with schizophrenia. Am J Med Genet Part B: Neuropsych Genet. 2002;114:483–490. doi: 10.1002/ajmg.10480. [DOI] [PubMed] [Google Scholar]
- 57.Flint J, Munafo MR. The endophenotype concept in psychiatric genetics. Psychological Med. 2007;37:163–180. doi: 10.1017/S0033291706008750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thaker GK. Neurophysiological endophenotypes across bipolar and schizophrenia psychosis. Schizophr Bull. 2008;34:760–773. doi: 10.1093/schbul/sbn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: Version III – the final common pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Javitt DC. Glutamate and schizophrenia: Phencyclidine, N-Methyl-D-Aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- 61.Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of γ-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- 62.Pettegrew JW, Keshavan MS, Panchalingam K, Strychor S, Kaplan DB, Tretta MG, Allen M. Alterations in brain high-energy phosphate and membrane phospholipid metabolism in first-episode, drug-naive schizophrenics: a pilot study of the dorsal prefrontal cortex by in vivo phosphorus 31 nuclear magnetic resonance spectroscopy. Archives of General Psychiatry. 1991;48:563–568. doi: 10.1001/archpsyc.1991.01810300075011. [DOI] [PubMed] [Google Scholar]
- 63.Keshavan MS, Mallinger AG, Pettegrew JW, Dippold C. Erythrocyte membrane phospholipids in psychotic patients. Psychiatry Res. 1993;49:89–95. doi: 10.1016/0165-1781(93)90032-c. [DOI] [PubMed] [Google Scholar]
- 64.Horrobin DF. The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res. 1998;30:193–208. doi: 10.1016/s0920-9964(97)00151-5. [DOI] [PubMed] [Google Scholar]
- 65.Skosnik PD, Yao JK. From phospholipid and fatty acid defects to altered neurotransmission: is arachidonic acid a nexus in the pathophysiology of schizophrenia? Prostaglandins Leukot Essent Fatty Acids. 2003;69:367–384. doi: 10.1016/j.plefa.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 66.Mahadik SP, Yao JK. Phospholipids in schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, Stroup TS, Perkins DO, editors. Textbook of Schizophrenia. The American Psychiatric Publishing, Inc.; Washington, DC: 2006. [Google Scholar]
- 67.Chalon S. Omega-3 fatty acids and monoamine neurotransmission. Prostaglandins Leukot Essent Fatty Acids. 2006;75:259–269. doi: 10.1016/j.plefa.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 68.du Bois TM, Deng C, Huang X-F. Membrane phospholipid composition, alterations in neurotransmitter systems and schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry. 2005;29:878–888. doi: 10.1016/j.pnpbp.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 69.Ohara K. The n-3 polyunsaturated fatty acid/dopamine hypothesis of schizophrenia. Prog Neuro-psychopharmacol Biol Psychiatry. 2007;31:469–474. doi: 10.1016/j.pnpbp.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 70.Zimmer L, Delpal S, Guilloteau D, Aïoun J, Durand G, Chalon S. Chronic n-3 polyunsaturated fatty acid deficiency alters dopamine vesicle density in the rat frontal cortex. Neurosci Letters. 2000;284:25–28. doi: 10.1016/s0304-3940(00)00950-2. [DOI] [PubMed] [Google Scholar]
- 71.Fedorova I, Hussein N, Baumann MH, Martino CD, Salem N., Jr An n-3 fatty acid deficiency impairs rat spatial learning in the Barnes Maze. Behavioral Neurosci. 2009;123:196–205. doi: 10.1037/a0013801. [DOI] [PubMed] [Google Scholar]
- 72.Bhattacharjee AK, Chang L, Lee H-J, Bazinet RP, Seemann R, Rapoport SI. D2 but not D1 dopamine receptor stimulation augments brain signaling involving arachidonic acid in unanesthetized rats. Psychopharmacology. 2005;180:735–742. doi: 10.1007/s00213-005-2208-4. [DOI] [PubMed] [Google Scholar]
- 73.Cohen JD, Servan-Schreiber D. Context, cortex, and dopamine: a connectionist approach to behavior and biology in schizophrenia. Psychological Rev. 1992;99:45–77. doi: 10.1037/0033-295x.99.1.45. [DOI] [PubMed] [Google Scholar]
- 74.Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- 75.Daniel DG, Weinberger DR, Jones DW, Zigun JR, Coppola R, Handel S, Bigelow LB, Goldberg TE, Berman KF, Kleinman JE. The effect of amphetamine on regional cerebral blood flow during cognitive activation in schizophrenia. J Neurosci. 1991;11:1907–1917. doi: 10.1523/JNEUROSCI.11-07-01907.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophr Res. 2005;77:43–58. doi: 10.1016/j.schres.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 77.Kischka U, Kammer TH, Maier S, Weisbrod M, Thimm M, Spitzer M. Dopaminergic modulation of semantic network activation. Neuropsychologia. 1996;34:1107–1113. doi: 10.1016/0028-3932(96)00024-3. [DOI] [PubMed] [Google Scholar]
- 78.Knecht S, Breitenstein C, Bushuven S, Wailke S, Kamping S, Flöel A, Zwitserlood P, Ringelstein EB. Levodopa: faster and better word learning in normal humans. Ann Neurol. 2004;56:20–26. doi: 10.1002/ana.20125. [DOI] [PubMed] [Google Scholar]
- 79.Roesch-Ely D, Weiland S, Scheffel H, Schwaninger M, Hundemer H-P, Kolter T, Weisbrod M. Dopaminergic modulation of semantic priming in healthy volunteers. Biol Psychiatry. 2006;60:604–611. doi: 10.1016/j.biopsych.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 80.Arnsten AFT, Cai JX, Murphy BL, Goldman-Rakic PS. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology. 1994;116:143–151. doi: 10.1007/BF02245056. [DOI] [PubMed] [Google Scholar]
- 81.Castner SA, Goldman-Rakic PS. Enhancement of working memory in aged monkeys by a sensitizing regimen of dopamine D1 receptor stimulation. J Neurosci. 2004;24:1446–1450. doi: 10.1523/JNEUROSCI.3987-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Costa A, Peppe A, Dell’Agnello G, Caltagirone C, Carlesimo GA. Dopamine and cognitive functioning in de novo subjects with Parkinson’s disease: effects of pramipexole and pergolide on working memory. Neuropsychologia. 2009;47:1374–1381. doi: 10.1016/j.neuropsychologia.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 83.Takahashi H, Kato M, Takano H, Arakawa R, Okumura M, Otsuka T, Kodaka F, Hayashi M, Okubo Y, Ito H, Suhara T. Differential contributions of prefrontal and hippocampal dopamine D1 and D2 receptors in human cognitive functions. J Neurosci. 2008;28:12032–12038. doi: 10.1523/JNEUROSCI.3446-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Connolly PM, Maxwell C, Liang Y, Kahn JB, Kanes SJ, Abel T, Gur RE, Turetsky BI, Siegel SJ. The effects of ketamine vary among inbred mouse strains and mimic schizophrenia for the P80, but not P20 or N40 auditory components. Neurochem Res. 2004;29:1179–1188. doi: 10.1023/b:nere.0000023605.68408.fb. [DOI] [PubMed] [Google Scholar]
- 85.Ehrlichman RS, Gandal MJ, Maxwell CR, Lazarewicz MT, Finkel LH, Contreras D, Turetsky BI, Siegel SJ. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience. 2009;158:705–712. doi: 10.1016/j.neuroscience.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 86.Turetsky BI, Fein G. Alpha2-noradrenergic effects on ERP and behavioral indices of auditory information processing. Psychophysiology. 2002;39:147–157. doi: 10.1017/S0048577202991298. [DOI] [PubMed] [Google Scholar]
- 87.Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- 88.Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann J, Van Heertum RL, Gorman JM, Laruelle M. From the cover: Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. PNAS. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Howes OD, Montgomery AJ, Asselin M-C, Murray RM, Valli I, Tabraham P, Bramon-Bosch E, Valmaggia L, Johns L, Broome M, McGuire PK, Grasby PM. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66:13–20. doi: 10.1001/archgenpsychiatry.2008.514. [DOI] [PubMed] [Google Scholar]
- 90.Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang D-R, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. doi: 10.1001/archgenpsychiatry.2010.10. [DOI] [PubMed] [Google Scholar]
- 91.Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, Moore H, Kandel ER. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron. 2006;49:603–615. doi: 10.1016/j.neuron.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 92.Bach M-E, Simpson EH, Kahn L, Marshall JJ, Kandel ER, Kellendonk C. Transient and selective overexpression of D2 receptors in the striatum causes persistent deficits in conditional associative learning. PNAS. 2008;105:16027–16032. doi: 10.1073/pnas.0807746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benjamins JA, Hajra AK, Agranoff BW. Lipids. In: Siegel GJ, Wayne AR, Brady ST, Price DL, editors. Basic neurochemistry, molecular, cellular, and medical aspects. 7th Ed Elsevier; NY: 2006. [Google Scholar]
- 94.Fenton WS, Hibbeln J, Knable M. Essential fatty acids, lipid membrane abnormalities, and the diagnosis and treatment of schizophrenia. Biol Psychiatry. 2000;47:8–21. doi: 10.1016/s0006-3223(99)00092-x. [DOI] [PubMed] [Google Scholar]
- 95.Fedorova I, Salem N., Jr Omega-3 fatty acids and rodent behavior. Prostaglandins Leukot Essent Fatty Acids. 2006;75:271–289. doi: 10.1016/j.plefa.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 96.Innis SM. Essential fatty acids in growth and development. Prog Lipid Res. 1991;30:39–103. doi: 10.1016/0163-7827(91)90006-q. [DOI] [PubMed] [Google Scholar]
- 97.Hoffman DR, Boettcher JA, Diersen-Schade DA. Toward optimizing vision and cognition in term infants by dietary docosahexaenoic and arachidonic acid supplementation: a review of randomoized controlled trials. Prostaglandins Leukot Essent Fatty Acids. 2009;81:151–158. doi: 10.1016/j.plefa.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 98.Ryan AS, Astwood JD, Gautier S, Kuratko CN, Nelson EB, Salem N., Jr. Effects of long-chain polyunsaturated fatty acid supplementation on neurodevelopment in childhood: a review of human studies. Prostaglandins Leukot Essent Fatty Acids. 2010;82:305–314. doi: 10.1016/j.plefa.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 99.Simmer K, Patole S, Rao SC. Longchain polyunsaturated fatty acid supplementation in infants born at term. Cochrane Database of Systematic Reviews 1. 2008 doi: 10.1002/14651858.CD000376.pub2. Art.No.: CD000376. [DOI] [PubMed] [Google Scholar]
- 100.Freeman MP, Hibbeln JR, Wisner KL, Davis JM, Mischoulon D, Peet M, Keck PE, Marangell LB, Richardson AJ, Lake J, Stoll AL. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67:1954–1967. doi: 10.4088/jcp.v67n1217. [DOI] [PubMed] [Google Scholar]
- 101.McNamara RK, Carlson SE. Role of omega-3 fatty acids in brain development and function: potential implications for the pathogenesis and prevention of psychopathology. Prostaglandins Leukot Essent Fatty Acids. 2006;75:329–349. doi: 10.1016/j.plefa.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 102.Peet M. Omega-3 polyunsaturated fatty acids in the treatment of schizophrenia. Isr J Psychiatry Relat Sci. 2008;45:19–25. [PubMed] [Google Scholar]
- 103.Richardson AJ. Omega-3 fatty acids in ADHD and related neurodevelopmental disorders. International review of psychiatry. 2006;18:155–172. doi: 10.1080/09540260600583031. [DOI] [PubMed] [Google Scholar]
- 104.Holguin S, Huang Y, Liu J, Wurtman R. Chronic administration of DHA and UMP improves the impaired memory of environmentally impoverished rats. Behav Brain Res. 2008;191:11–16. doi: 10.1016/j.bbr.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petursdottir AL, Farr SA, Morley JE, Banks WA, Skuladottir GV. Effect of dietary n-3 polyunsaturated fatty acids on brain lipid fatty acid composition, learning ability, and memory of senescence-accelerated mouse. J Gerontol A Biol Sci Med Sci. 2008;63A:1153–1160. doi: 10.1093/gerona/63.11.1153. [DOI] [PubMed] [Google Scholar]
- 106.Eilander A, Hundscheid DC, Osendarp SJ, Transler C, Zock PL. Effects of n-3 long chain polyunsaturated fatty acid supplementation on visual and cognitive development throughout childhood: A review of human studies. Prostaglandins Leukot Essent Fatty Acids. 2007;76:189–203. doi: 10.1016/j.plefa.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 107.Schuchardt JP, Huss M, Stauss-Grabo M, Hahn A. Significance of long-chain polyunsaturated fatty acids (PUFAs) for the development and behaviour of children. Eur J Pediatr. 2010;169:149–164. doi: 10.1007/s00431-009-1035-8. [DOI] [PubMed] [Google Scholar]
- 108.Colombo J, Kannass KN, Shaddy DJ, Kundurthi S, Maikranz JM, Anderson CJ, Blaga OM, Carlson SE. Maternal DHA and the development of attention in infancy and toddlerhood. Child Dev. 2004;75:1254–1267. doi: 10.1111/j.1467-8624.2004.00737.x. [DOI] [PubMed] [Google Scholar]
- 109.Dunstan JA, Simmer K, Dixon G, Prescott SL. Cognitive assessment of children at age 2 years after maternal fish oil supplementation in pregnancy: a randomised controlled trial. Arch Dis Child Fetal Neonatal Ed. 2008;93:F45–F50. doi: 10.1136/adc.2006.099085. [DOI] [PubMed] [Google Scholar]
- 110.Jacobson L, Jacobson SW, Muckle JG, Kaplan-Estrin M, Ayotte P, Dewailly E. Beneficial effects of a polyunsaturated fatty acid on infant development: evidence from the Inuit of Arctic Quebec. J Pediatrics. 2008;152:356–364. doi: 10.1016/j.jpeds.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 111.Beydoun MA, Kaufman JS, Satia JA, Rosamond W, Folsom AR. Plasma n–3 fatty acids and the risk of cognitive decline in older adults: the Atherosclerosis Risk in Communities Study. Am J Clin Nutr. 2007;85:1103–1111. doi: 10.1093/ajcn/85.4.1103. [DOI] [PubMed] [Google Scholar]
- 112.Dullemeijer C, Durga J, Brouwer IA, van de Rest O, Kok FJ, Brummer R-JM, van Boxtel MPJ, Verhoef P. n–3 Fatty acid proportions in plasma and cognitive performance in older adults. Am J Clin Nutr. 2007;86:1479–1485. doi: 10.1093/ajcn/86.5.1479. [DOI] [PubMed] [Google Scholar]
- 113.van de Rest O, Geleijnse JM, Kok FJ, van Staveren WA, Dullemeijer C, OldeRikkert MGM, Beekman ATF, de Groot CPGM. Effect of fish oil on cognitive performance in older subjects: A randomized, controlled trial. Neurology. 2008;71:430–438. doi: 10.1212/01.wnl.0000324268.45138.86. [DOI] [PubMed] [Google Scholar]
- 114.van de Rest O, Spiro A, III, Krall-Kaye E, Geleijnse JM, de Groot LCPGM, Tucker KL. Intakes of (n-3) fatty acids and fatty fish are not associated with cognitive performance and 6-year cognitive change in men participating in the Veterans Affairs Normative Aging Study. J Nutr. 2009;139:2329–2336. doi: 10.3945/jn.109.113647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Amminger GP, Schäfer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan SM, Mackinnon A, McGorry PD, Berger GE. Long-chain ω-3 fatty acids for indicated prevention of psychotic disorders: A randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67:146–154. doi: 10.1001/archgenpsychiatry.2009.192. [DOI] [PubMed] [Google Scholar]
- 116.Berger GE, Proffitt T-M, McConchie M, Yuen H, Wood SJ, Amminger GP, Brewer W, McGorry PD. Ethyl-eicosapentaenoic acid in first-episode psychosis: A randomized, placebo-controlled trial. J Clin Psychiatry. 2007;68:1867–1873. doi: 10.4088/jcp.v68n1206. [DOI] [PubMed] [Google Scholar]
- 117.Fenton WS, Dickerson F, Boronow J, Hibbeln JR, Knable M. A placebo-controlled trial of omega-3 fatty acid (ethyl eicosapentaenoic acid) supplementation for residual symptoms and cognitive impairment in schizophrenia. Am J Psychiatry. 2001;158:2071–2074. doi: 10.1176/appi.ajp.158.12.2071. [DOI] [PubMed] [Google Scholar]
- 118.Peet M, Horrobin DF. A dose-ranging exploratory study of the effects of ethyl-eicosapentaenoate in patients with persistent schizophrenic symptoms. J Psychiatric Resea. 2002;36:7–18. doi: 10.1016/s0022-3956(01)00048-6. [DOI] [PubMed] [Google Scholar]
- 119.Emsley R, Myburgh C, Oosthuizen P, van Rensburg SJ. Randomized, placebo-controlled study of ethyl-eicosapentaenoic acid as supplemental treatment in schizophrenia. Am J Psychiatry. 2002;159:1596–1598. doi: 10.1176/appi.ajp.159.9.1596. [DOI] [PubMed] [Google Scholar]
- 120.Whalley LJ, Deary IJ, Starr JM, Wahle KW, Rance KA, Bourne VJ, Fox HC. n-3 fatty acid erythrocyte membrane content, APOE ε4, and cognitive variation: an observational follow-up study in late adulthood. Am J Clin Nutr. 2008;87:449–454. doi: 10.1093/ajcn/87.2.449. [DOI] [PubMed] [Google Scholar]
- 121.Arendash GW, Jensen MT, Salem N, Jr, Hussein N, Cracchiolo J, Dickson A, Leighty R, Potter H. A diet high in omega-3 fatty acids does not improve or protect cognitive performance in Alzheimer’s transgenic mice. Neuroscience. 2007;149:286–302. doi: 10.1016/j.neuroscience.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 122.Bang HO, Dyerberg J, Sinclair HM. The composition of the Eskimo food in north western Greenland. Am J Clin Nutr. 1980;33:2657–2661. doi: 10.1093/ajcn/33.12.2657. [DOI] [PubMed] [Google Scholar]
- 123.Cyhlarova E, Bell JG, Dick JR, Mackinlay EE, Stein JF, Richardson AJ. Membrane fatty acids, reading and spelling in dyslexic and non-dyslexic adults. European Neuropsychopharmacology. 2007;17:116–121. doi: 10.1016/j.euroneuro.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 124.Ishikura Y, Ikeda G, Akimoto K, Hata M, Kusumoto A, Kidokoro A, Kontani M, Kawashima H, Kiso Y, Koga Y. Arachidonic acid supplementation decreases P300 latency and increases P300 amplitude of event-related potentials in healthy elderly men. Neuropsychobiology. 2009;60:73–79. doi: 10.1159/000236447. [DOI] [PubMed] [Google Scholar]
- 125.Horrobin DF, Jenkins K, Bennett CN, Christie WW. Eicosapentaenoic acid and arachidonic acid: collaboration and not antagonism is the key to biological understanding. Prostaglandins Leukot Essent Fatty Acids. 2002;66:83–90. doi: 10.1054/plef.2001.0338. [DOI] [PubMed] [Google Scholar]
- 126.Bazan NG. The onset of brain injury and neurodegeneration triggers the synthesis of docosanoid neuroprotective signaling. Cell Mol Neurobiol. 2006;26:901–913. doi: 10.1007/s10571-006-9064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Akhondzadeh S, Tabatabaee M, Amini H, Abhari SAA, Abbasi SH, Behnam B. Celecoxib as adjunctive therapy in schizophrenia: A double-blind, randomized and placebo-controlled trial. Schizophr Res. 2007;90:179–185. doi: 10.1016/j.schres.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 128.Yao JK. Abnormalities of fatty acid metabolism in red cells, platelets and brain in schizophrenia. In: Peet M, Glen I, Horrobin DF, editors. Phospholipid spectrum disorders in psychiatry and neurology. 2nd edition Marius Press; Lancashire, UK: 2003. [Google Scholar]
- 129.Peet M. Membrane fatty acid deficit in schizophrenia and mood disorders. In: Reddy R, Yao JK, editors. Fatty acids and oxidative stress in neuropsychiatric disorders. Nova Science Publishers, Inc.; NY: 2007. [Google Scholar]
- 130.Conklin SM, Runyan C, Leonard S, Reddy RD, Muldoon MF, Yao JK. Age-related changes of n-3 and n-6 polyunsaturated fatty acids in the anterior cingulate cortex of individuals with major depressive disorder. Prostaglandins Leukot Essent Fatty Acids. 2010;82:111–119. doi: 10.1016/j.plefa.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Horrobin DF. Schizophrenia as a prostaglandin deficiency disease. Lancet. 1977;309:936–937. doi: 10.1016/s0140-6736(77)92228-0. [DOI] [PubMed] [Google Scholar]
- 132.Eaton WW, Hayward C, Ram R. Schizophrenia and rheumatoid arthritis: a review. Schizophr Res. 1992;6:181–192. doi: 10.1016/0920-9964(92)90001-l. [DOI] [PubMed] [Google Scholar]
- 133.Oken RJ, Schulzer M. At Issue: Schizophrenia and rheumatoid arthritis: the negative association revisited. Schizophr Bull. 1999;25:625–638. doi: 10.1093/oxfordjournals.schbul.a033407. [DOI] [PubMed] [Google Scholar]
- 134.Vinogradov S, Gottesman II, Moises HW, Nicol S. Negative association between schizophrenia and rheumatoid arthritis. Schizophr Bull. 1991;17:669–678. doi: 10.1093/schbul/17.4.669. [DOI] [PubMed] [Google Scholar]
- 135.Davis GC, Buchsbaum MS, van Kammen DP, Bunney WE., Jr Analgesia to pain stimuli in schizophrenics and its reversal by naltrexone. Psychiatry Res. 1979;1:61–69. doi: 10.1016/0165-1781(79)90029-5. [DOI] [PubMed] [Google Scholar]
- 136.Wanibuchi F, Usuda S. Synergistic effects between D-1 and D-2 dopamine antagonists on catalepsy in rats. Psychopharmacology (Berl) 1990;102:339–342. doi: 10.1007/BF02244101. [DOI] [PubMed] [Google Scholar]
- 137.Ono N, Abiru T, Sugiyama K, Kamiya H. Influences of cyclooxygenase inhibitors on the cataleptic behavior induced by haloperidol in mice. Prostaglandins Leukotrienes Essent Fatty Acids. 1992;46:59–63. doi: 10.1016/0952-3278(92)90060-v. [DOI] [PubMed] [Google Scholar]
- 138.Barcelos RCS, Benvegnú DM, Boufleur N, Reckziegel P, Müller LG, Pase C, Emanuelli T, Bürger ME. Effects of ω-3 essential fatty acids (ω-3 EFAs) on motor disorders and memory dysfunction typical neuroleptic-induced: behavioral and biochemical parameter. Neurotox Res. 2010;17:228–237. doi: 10.1007/s12640-009-9095-0. [DOI] [PubMed] [Google Scholar]
- 139.Drtil J. Analgesics-antipyretics and etiology and pathogenesis of psychosis. Activitas Nervosa Superior. 1971;13:237–238. [PubMed] [Google Scholar]
- 140.Hoppmann RA, Peden JG, Ober SK. Central nervous system side effects of nonsteroidal anti-inflammatory drugs: aseptic meningitis, psychosis, and cognitive dysfunction. Arch Intern Med. 1991;151:1309–1313. [PubMed] [Google Scholar]
- 141.Sussman N, Magid S. Psychiatric manifestations of nonsteroidal anti-inflammatory drugs. Primary Psychiatry. 2000;7:26–30. [Google Scholar]
- 142.Braff DL, Freedman R, Schork NJ, Gottesman II. Deconstructing schizophrenia: an overview of the use of endophenotypes in order to understand a complex disorder. Schizophr Bull. 2007;33:21–32. doi: 10.1093/schbul/sbl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Keshavan MS, Tandon R, Boutros NN, Nasrallah HA. Schizophrenia, “Just the facts”: what we know in 2008 Part 3: neurobiology. Schizophr Res. 2008;106:89–107. doi: 10.1016/j.schres.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 144.Yao JK, van Kammen DP, Welker JA. Red blood cell membrane dynamics in schizophrenia. II. Fatty acid composition. Schizophr Res. 1994;13:217–226. doi: 10.1016/0920-9964(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 145.Mahadik SP, Evans DR. Is Schizophrenia a metabolic brain disorder? Membrane phospholipid dysregulation and its therapeutic implications. Psychiatry Clinics North America. 2003;26:85–102. doi: 10.1016/s0193-953x(02)00033-3. [DOI] [PubMed] [Google Scholar]
- 146.Yao JK, van Kammen DP. Membrane phospholipids and cytokine interaction in schizophrenia. Int Rev Neurobiology. 2004;59:297–326. doi: 10.1016/S0074-7742(04)59012-8. [DOI] [PubMed] [Google Scholar]
- 147.Chalon S, Vancassel S, Zimmer L, Guilloteau D, Durand G. Polyunsaturated fatty acids and cerebral function: focus on monoaminergic neurotransmission. Lipids. 2001;36:937–944. doi: 10.1007/s11745-001-0804-7. [DOI] [PubMed] [Google Scholar]
- 148.Delion S, Chalon S, Hérault J, Guilloteau D, Besnard J-C, Durand G. Chronic dietary-linolenic acid deficiency alters dopaminergic and serotoninergic neurotransmission in rats. J Nutr. 1994;124:2466–2476. doi: 10.1093/jn/124.12.466. [DOI] [PubMed] [Google Scholar]
- 149.Besnard C, Durand G. α-linolenic acid dietary deficiency alters age-related changes of dopaminergic and serotoninergic neurotransmission in the rat frontal cortex. J Neurochem. 1996;66:1582–1591. doi: 10.1046/j.1471-4159.1996.66041582.x. [DOI] [PubMed] [Google Scholar]
- 150.Zimmer L, Hembert S, Durand G, Breton P, Guilloteau D, Besnard J-C, Chalon S. Chronic n-3 polyunsaturated fatty acid diet-deficiency acts on dopamine metabolism in the rat frontal cortex: a microdialysis study. Neurosci Letters. 1998;240:177–181. doi: 10.1016/s0304-3940(97)00938-5. [DOI] [PubMed] [Google Scholar]
- 151.Chalon S, Delion-Vancassel S, Belzung C, Guilloteau D, Leguisquet A-M, Besnard J-C, Durand G. Dietary fish oil affects monoaminergic neurotransmission and behavior in rats. J Nutr. 1998;128:2512–2519. doi: 10.1093/jn/128.12.2512. [DOI] [PubMed] [Google Scholar]
- 152.Ng K-F, Innis SM. Behavioral responses are altered in piglets with decreased frontal cortex docosahexaenoic acid. J Nutr. 2003;133:3222–3227. doi: 10.1093/jn/133.10.3222. [DOI] [PubMed] [Google Scholar]
- 153.van Kammen DP, Kelley ME, Gurklis JA, Gilbertson MW, Yao JK, Condray R, Peters JL. Predicting duration of clinical stability following haloperidol withdrawal in schizophrenic patients. Neuropsychopharmacology. 1996;14:275–283. doi: 10.1016/0893-133X(95)00135-Z. [DOI] [PubMed] [Google Scholar]