

Abstract
Pandemic influenza viral infections have been associated with viral pneumonia. Chimeric influenza viruses with the hemagglutinin segment of the 1918, 1957, 1968 or 2009 pandemic influenza viruses in the context of a seasonal H1N1 influenza genome were constructed to analyze the role of hemagglutinin (HA) in pathogenesis and cell tropism in a mouse model. We also explored whether there was an association between the ability of lung surfactant protein D (SP-D) to bind to the HA and the ability of the corresponding chimeric virus to infect bronchiolar and alveolar epithelial cells of the lower respiratory tract. Viruses expressing the hemagglutinin of pandemic viruses were associated with significant pathology in the lower respiratory tract, including acute inflammation, and showed low binding activity for SP-D. In contrast, the virus expressing the HA of a seasonal influenza strain induced only mild disease with little lung pathology in infected mice and exhibited strong in vitro binding to SP-D.
Introduction
Influenza A virus (IAV) in humans primarily causes an acute respiratory infection with inflammation of the upper respiratory tree and trachea. While in most cases pneumonic involvement is not clinically prominent, virus can be recovered from both the upper and lower respiratory tract. Under certain still poorly understood circumstances, infection can lead to the development of viral pneumonia, characterized by injury to the gas exchange tissue, often with severe clinical consequences (Taubenberger and Morens, 2008). Severe disease is more often associated with the emergence of novel influenza viruses that cause pandemics (Simonsen et al., 1998; Wright, Neumann, and Kawaoka, 2007). In the last 100 years, influenza pandemics occurred in 1918, 1957, 1968, and 2009, and were associated with varying levels of morbidity and excess mortality (Morens, Taubenberger, and Fauci, 2009). Postmortem examinations from patients dying during these pandemics have revealed striking similarities in the spectrum of pathological changes in the lower respiratory tract (Gill et al., 2010; Kuiken and Taubenberger, 2008; Morens, Taubenberger, and Fauci, 2008; Mulder and Hers, 1972; Taubenberger and Morens, 2008). One key pathological feature in fatal cases, also prominent in postmortem examination of lung sections from victims of the 2009 H1N1 pandemic, was severe lower respiratory tract pathological changes associated with widespread infection of bronchiolar and alveolar epithelial cells and alveolar macrophages (Gill et al., 2010).
IAV hemagglutinin (HA) and neuraminidase (NA) are viral surface glycoproteins that mediate receptor binding and virion release from the host cell, respectively. HA recognizes sialic acid bound to underlying sugars on host cell glycoproteins. IAV adapted to birds have an HA receptor binding specificity for SA-α2,3-galactose, while HA from IAV adapted to humans typically have higher specificity for SA-α2,6-galactose (Matrosovich et al., 2000; Matrosovich et al., 1997; Rogers and Paulson, 1983). This binding preference is believed to be an important determinant of IAV cell tropism within a host. Lectin histochemistry studies have demonstrated that the respiratory tract of humans bears both α2-6 and α2-3 linked sialic acids, but that α2-3 sialic acids are more prevalent in the lower respiratory tract (Nicholls et al., 2007; Shinya et al., 2006). Although pandemic viruses display preference for α2-6 linked sialic acids, autopsy reports indicate that, they still can cause severe alveolitis, a consequence of infection of the lower respiratory tract. Moreover, in a recent study, Qi, et al. demonstrated that IAV expressing the 1918 HA possessing either an SA-α2,6-galactose specificity, an SA-α2,3-galactose specificity, or a mixed SA-α2,6-galactose/SA-α2,3-galactose specificity were lethal to mice, with similar pathology and cellular tropism (Qi et al., 2009). This suggests that IAV respiratory tract tropism both in humans and mice must be influenced by other features of HA proteins than their SA binding specificity.
It has been reported that mannose-containing glycans on the viral HA and NA proteins can be recognized by components of the innate immune system, such as, for instance, C-type lectins of the collectin family, including surface surfactant protein D (SP-D) (Crouch, 2000; Hartshorn, 2010; Hartshorn et al., 1994; Hartshorn et al., 2008; Hartshorn et al., 1997; Hartshorn et al., 2007; Hartshorn et al., 2000; LeVine et al., 2001; Reading et al., 1997). SP-D is constitutively expressed by alveolar type II pneumocytes and non-ciliated bronchiolar (Clara) cells in the lower respiratory tract where it is considered to represent a counterpart of IgA, thereby playing a key role in the innate immune system (Crouch, 2000; Litvack et al., 2010). SP-D, together with other proteins, is believed to establish a first line of defense against both bacterial and viral pathogens by binding to carbohydrate moieties on the pathogen surface (Hartshorn, 2010). Most interestingly, SP-D has previously been shown to bind to carbohydrate residues on some influenza A viruses, thereby inhibiting their HA binding activity, causing viral aggregation (Hartshorn et al., 2008) and eventually leading to pathogen inactivation through activation of neutrophil and macrophage activity (Crouch, 2000; Hartshorn et al., 1994). Furthermore, SP-D may enhance viral clearance by binding to the carbohydrate side chains of IAV, blocking access of the HA receptor binding domain to cell surface receptors, and thus, interfering with virus internalization by host cells (Hartshorn et al., 2008). The ability of SP-D to bind the glycan component of HA protein has been shown to require high mannose type II glycans.
Previous analysis of the glycosylation patterns of the HA proteins from representative viruses of the 1957 H2N2 pandemic (A/Singapore/1/1957), the 1968 H3N2 pandemic (A/Aichi/1/1968), and a close relative of the pandemic 1918 influenza virus, (A/Swine/Iowa/1931) observed an absence of type II glycans (Hartshorn et al., 2008; Schwarz and Klenk, 1981; Vigerust et al., 2007). While the exact glycosylation pattern of the 1918 and 2009 pandemic IAV HA proteins has yet to be determined, the deduced potential glycosylation pattern based on amino acid sequence predicts a lack of type II glycans (Reid et al., 1999; Stevens et al., 2004; Wei et al., 2010), and we hypothesized that the 2009 pandemic HA and the HA proteins of other pandemic IAV would have low binding activity with SP-D that would likely be associated with an enhanced ability of these viruses to invade deeper into the lower lung and infect alveolar lining cells.
In the present study we tested this hypothesis and show that the HA proteins from pandemic influenza viruses show low SP-D binding in vitro and that chimeric viruses expressing the 1918, 1957, 1968 or 2009 pandemic virus HA induced more severe disease in mice as compared to seasonal IAV with increased SP-D binding activity. Additionally, we show enhanced alveolar and bronchiolar epithelial cell tropism is associated with severe lower lung pathology and more significant activation of host inflammatory responses. Our results demonstrate that low SP-D binding is a common feature of the HA proteins from the past four influenza pandemics, and that this is also associated with increased morbidity, mortality, lung pathology and inflammatory responses in mice infected with an otherwise isogenic virus.
Materials and Methods
Virus preparation and growth
The control IAV, A/New York/312/2001 (H1N1) [NY312] was produced via reverse genetics as previously described (Qi et al., 2009). The HA gene segments of A/SouthCarolina/1/1918 (H1N1) (Reid et al., 1999), A/Japan/305+/1957 (H2N2) (virus kindly supplied by Sue Epstein at the FDA), A/Hong Kong/1/68 (H3N2), A/Mexico/4108/09 (H1N1) (virus kindly supplied by Heinz Feldmann at the NIH) were subcloned into the rescue plasmid pHH21 (kindly supplied by Yoshihiro Kawaoka at the University of Wisconsin at Madison). Isogenic “1:7” chimeric influenza viruses containing different HA gene segments from the viruses described above on the NY312 backbone were generated as described (Qi et al., 2009). Rescued viruses were both grown and titers determined by plaque assay in MDCK cells as previously described (Qi et al., 2009). All rescued viruses had their HA genes confirmed by comparison with the sequences deposited in GenBank. Similarly, isogenic “2:6” chimeric viruses using cognate HA and NA gene segments from the viruses described above on the NY312 backbone were also generated and sequence verified.
Ethics statement
All experimental animal work was performed in an enhanced ABSL3 laboratory at the National Institutes of Heath (NIH) using animal safety protocols approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee.
Mouse infection studies
Groups of 8-10 week old female BALB/c mice (Jackson Labs, Bar Harbor, ME) were lightly anesthetized in a chamber with isoflurane supplemented with O2 (1.5 L/min) and mice were intranasally inoculated with 2×105 PFU of influenza A virus under enhanced animal BSL3 conditions. Viruses were diluted in sterile DMEM where appropriate. Body weights were measured daily and mice were humanely euthanized if they lost more than 25% of their initial body weight. Lungs from 3 animals were collected at 1, 2, 3, 4, and 5 dpi for viral titration and cytokine assays for each virus and time point. Lungs collected for pathology (from two mice for each virus at 1-5 dpi) were inflated with 10% neutral buffered formalin at time of isolation to prevent atelectasis. Lung viral titers were determined by plaque assay (described above) in 10% (w/v) lung suspensions prepared by homogenization in sterile 1× L15 media.
Pathology and immunohistochemistry
Mice were humanely euthanized with CO2 and lungs were removed and inflated using 10% formalin. Following fixation for at least 24 h in 10% formalin, lungs were dehydrated, embedded in paraffin, and 5 μm sections on positively charged slides were either stained with hematoxylin and eosin for histopathologic examination (American HistoLabs, Gaithersburg, MD) or were processed for immunohistochemistry as previously described (Memoli et al., 2009; Qi et al., 2009). Briefly, tissue sections were dewaxed, rehydrated, and blocked with methanol/hydrogen peroxide followed by a heat induced epitope retrieval (HIER) procedure using citrate buffer, pH 6.0. The samples were then rinsed in PBS, serum blocked, and either incubated with goat anti-Influenza A (Abcam, Inc., Cambridge, MA; #ab20841) and stained with horse anti-goat-peroxidase (Vector Laboratories, Burlingame, CA; #PI-9500) or Vector ImmPRESS anti-rabbit-IgG-peroxidase (Vector Labs #MP7401) respectively. The samples were reacted with diaminobenzidine (DAB), counterstained with hematoxylin, rinsed, and coverslipped. Lung sections were given a histopathology severity score as follows: 0: no significant pathological changes; 1+: mild pathological changes; 2+: moderate pathological changes; 3+ moderately severe pathological changes; 4+ severe pathological changes. Pathological changes included in the severity scale were degree and extent of bronchiolitis, alveolitis, inflammatory infiltrates, pulmonary edema and hemorrhage.
Determination of cytokine and chemokine protein levels
A mouse multiplex Ab bead kit (Bio-Plex Pro Mouse Cytokine 23-plex Assay; Bio-Rad Laboratories, Hercules, CA) was used to measure chemokine and cytokine secretion from infected mice lung tissue. The method allows simultaneous evaluation of murine cytokines, chemokines, and growth factors. The multiplexed assay was performed according to the manufacturer's instructions. To generate a standard curve, 2-fold serial dilutions of appropriate standards were prepared in L-15 medium. Standards and supernatant samples were pipetted at 50 μl/well and mixed with 50 μl of the bead mixture. After a 30 min incubation, wells were washed three times with washing buffer using a vacuum manifold. Detection antibodies were added for 30 min, the wells were washed three times, PE-conjugated secondary antibody was added for 10 min, the wells were washed three times, and samples were analyzed using the Bio-Plex suspension array system (Bio-Rad). One hundred beads were counted for each analyte per well and values (pg/g) were calculated.
Determination of SP-D binding by hemagglutination inhibition (HAI) assay
Recombinant human SP-D protein (112.5μg/ml) was prepared as previously described (Brown-Augsburger et al., 1996; Doss et al., 2009). SP-D protein (at 2 μg/ml) was serially diluted in D-PBS (containing Ca2+ and Mg2+) (Quality Biological, Inc., Gaithersburg, MD) in 96 well plates with V-shaped bottoms (Costar). After adding an equal volume (25 μl) of virus at a final concentration of 4 HA units/well, the virus and SP-D mixture was incubated for 15 min at room temperature followed by addition of 50 μl of a 0.5% turkey erythrocyte suspension (Lampire Biological Laboratories, Inc., Pipersville, PA). The minimum concentration of protein required to inhibit fully the hemagglutinating activity of the viral suspension was determined by noting the highest dilution of protein that still inhibited hemagglutination. Inhibition of HA activity in a given well was demonstrated by presence of an erythrocyte pellet. If no inhibition of HA activity was observed at the highest protein concentration used then the value was expressed as greater than the maximal protein concentration tested.
Determination of SP-D binding by plaque reduction neutralization (PRN) assay
MDCK cells were seeded into 24 well plates and grown to confluency. SP-D protein (240 ng/ml) was sequentially diluted two-fold in D-PBS (up to 15 ng/ml) and each dilution was mixed with an equal volume of virus corresponding to 20-30 plaques per well. Following pre-incubation at room temperature for 45-60 minutes, the mixed samples (100 μl) were layered onto the MDCK cells. Each sample was tested in triplicate wells. Virus alone without SP-D protein was included as a control. After incubation at 37 C for 1 h, samples were aspirated, wells washed with PBS and cells were overlayed with 2% agar (w/v) in 2× D-MEM. Plaques were visualized using crystal violet 48 h later using standard protocols (Cottey, Rowe, and Bender, 2003).
Results
Rescue of chimeric viruses containing the HA gene of pandemic IAVs
Using reverse genetics, a series of isogenic chimeric 1:7 influenza viruses that contained the same seven genomic segments derived from A/New York/312/2001 (H1N1) [NY312], but different HA segments were constructed. The latter encoded the HA gene of the 1918, 1957, 1968 or 2009 pandemic IAV, termed 1918-HA, 1957-HA, 1968-HA, or 2009-HA, respectively. The parental NY312 virus was also rescued. All rescued 1:7 chimeric viruses replicated to titers of 106-107 PFU/ml in cell culture (Table 1). To address if the HA/NA mismatch of the HA gene from H2 (1957) and H3 (1968) viruses on the NY312 H1N1 background could influence replication of the chimeric viruses, we also tested the 1918-HA, the 1957-HA, and the 1968-HA chimeric viruses in the context of their cognate NA by rescuing 2:6 viruses containing six NY312 gene segments and the HA and NA gene segments of each of the pandemic viruses and measuring growth in MDCK cells in vitro and their pathogenicity in vivo. These studies demonstrated that the properties of the 1:7 HA and 2:6 HA+NA expressing chimeric viruses tested in this study were very similar (Table1). Similarly, the properties of the 2009-HA virus were similar to the wild-type A/Mexico/4108/09 (H1N1) virus (data not shown).
Table 1. Properties of viruses tested.
| Virus | Subtype | MDCK titer (PFU/ml log10) |
Surfactant D protein (SP-D) binding assays | MLD50 (PFU/ml log10) |
|
|---|---|---|---|---|---|
| HAI+ | IC50† | ||||
| 1918-HA:NY312 | H1N1 | 7.2 | >2 μg | >120 ng | 5.25 |
| 1957-HA:NY312 | H2N1 | 6.7 | >2 μg | >120 ng | 5.54 |
| 1968-HA:NY312 | H3N1 | 6.5 | >2 μg | >120 ng | 5.41 |
| 2009-HA:NY312 | H1N1 | 6.1 | >2 μg | >120ng | >5.6 |
| NY312 | H1N1 | 7.0 | 62.5 ng | 7 ng | >5.7 |
| 1918-HA,NA:NY312 | H1N1 | 6.8 | >2 μg | >120 ng | 3.8 |
| 1957-HA,NA:NY312 | H2N2 | 6.5 | >2 μg | >120 ng | 5.04 |
| 1968-HA,NA:NY312 | H3N2 | 6.6 | >2 μg | >120 ng | 5.45 |
Mass of human SP-D protein required to inhibit agglutination of turkey RBCs
Determined by plaque reduction assay.
Morbidity and mortality of pandemic HA expressing isogenic viruses in mice
To test whether the isogenic chimeric viruses expressing seasonal or pandemic HA could cause differential disease and pathology in mice, groups of five 8-10 week old BALB/c mice were intranasally inoculated with each virus (Table 1) at a dose of 2×105 PFU. As shown in Figure 1A, mice infected with the NY312 parental virus showed only very little weight loss during the first three days post infection. Whereas there was an average weight loss of about 10% in these mice by day 4 p.i., all mice eventually recovered and none of the animals died. In contrast, mice inoculated with the four pandemic HA expressing chimeric viruses all exhibited rapid and significant weight loss beginning from day one post-infection (dpi) reaching a nadir at 3 to 4 dpi (Figure 1A). Even though peak weight loss in mice infected with 1957-HA and 1968-HA was similar to the peak weight loss observed with mice infected with NY312, by 4 dpi, mice infected with 1957-HA, 1968-HA each exhibited 20% mortality, which was statistically significant compared to NY312 (n = 20; P = 0.002). Similarily, the 2009-HA virus infected mice displayed peak weight loss comparable to NY312, but exhibited 10% mortality, which did not reach statistical significance (n = 20; P = 0.273) (Figure 1B). Interestingly, mice infected with 1918-HA virus continued to lose weight until 6 dpi with an overall mortality rate of 60% by 7 dpi (n = 25; P < 0.001). To characterize further the virulence of the different viruses, the mouse 50% lethal dose (MLD50) of each of the pandemic HA expressing viruses was determined where possible (Table 1). However, we were unable to obtain a stock of 2009-HA with sufficiently high titer to calculate a mouse LD50 for this virus (MLD50 >5.6). The calculated MLD50 for the 1918-HA, 1957-HA, 1968-HA viruses ranged between 5.25 and 5.54 log10 PFU using the method of Reed and Muench (Reed and Muench, 1938).
Figure 1. Weight loss and survival of mice infected with wild-type influenza viruses and chimeric influenza viruses expressing the HA genes of different influenza viruses.
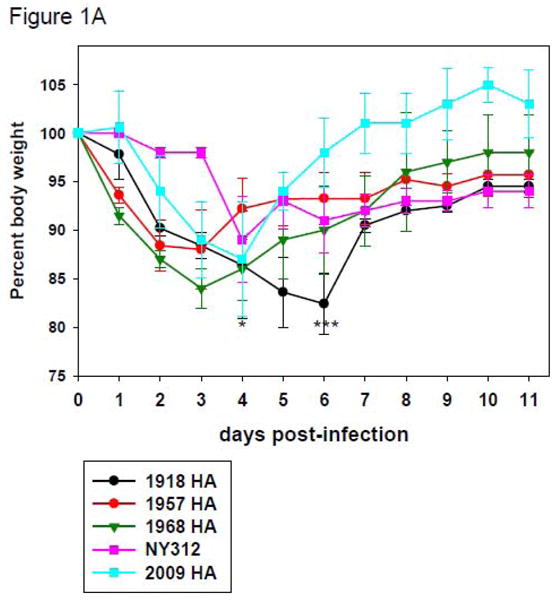
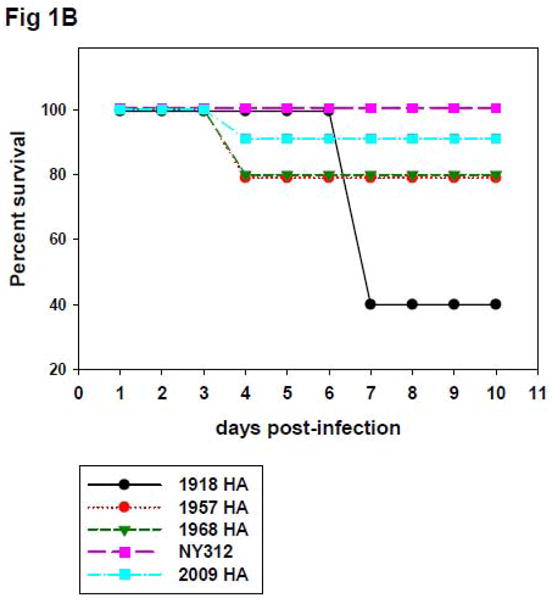
(A) Representative weight loss of mice infected with rescued influenza viruses. Eight-to-ten-week-old female BALB/c mice were intranasally inoculated with 2 × 105 PFU of the indicated viruses and weighed daily (n=5 mice per virus per group). *Note that by day 4 post infection, one animal each infected with 1957-HA, 1968-HA, and 2009-HA had died. ***Note that by day 6 post infection, 3 animals infected with 1918-HA had died. (B) Kaplan-Meier survival curve of 8-to-10-week-old female BALB/c mice intranasally inoculated with 2 × 105 PFU of the indicated viruses (n=20 mice per virus group, except 1918-HA virus which had n=25).
Evaluation of mouse lung pathology
To evaluate mouse lung pathology, 2 mice each infected with the various viruses were sacrificed at 1, 2, 3 and 4 dpi and lung sections were subjected to histopathological examination and immunohistochemistry (IHC) for IAV antigen. Pathological changes were scored on a severity scale from 0 (no significant changes) to 4+ (severe). In lungs of mice infected with the 1918-HA, 1957-HA, 1968-HA and 2009-HA viruses, viral antigen was found as early as 1 dpi throughout the entire lung (data not shown) and until the last time point analyzed (4 dpi, Figures 2 and 3). Intriguingly, however, in lungs of mice infected with the seasonal NY312 virus, detection of viral antigen was restricted to the large airways and the bronchiolar epithelium throughout the observation period (Figures 2 and 3, and data not shown). Mice inoculated with the 1918-HA virus developed prominent histopathologic changes at 3 and 4 dpi. Lung sections showed a multifocal moderate-to-severe bronchiolitis, including focal necrotizing changes, and multifocal alveolitis, with a neutrophil-predominant inflammatory infiltrate (pathological score of 3+) (Figure 2 panel A and Figure 3 panel A). Influenza viral antigen was detected in the bronchiolar epithelium (Figure 2 panel B), alveolar macrophages, and alveolar lining cells (Figure 3 panels B and C). Mice inoculated with the 1957-HA (Fig. 2 panel C and Figure 3 panel D), 1968-HA (Figure 2 panel E and Figure 3 panel G), and 2009-HA (Figure 2 panel G and Figure 3 panel J) viruses showed similar histopathologic changes beginning at three dpi. Lung sections showed a multifocal mild-to-moderate bronchiolitis and alveolitis pattern (pathological score of 2+ in each case). The inflammatory infiltrates were predominantly lymphocytic and histiocytic with very few granulocytes. Influenza viral antigen was detected in the bronchiolar epithelium (Figure 2 panels D, F and H), alveolar macrophages, and alveolar lining cells (Figure 3 panels E, F, H, I, K and L). In comparison, the parental NY312 (Figure 2 panels I and K and Figure 3 panel M) virus showed little-to-no pathologic changes at 4 dpi (pathological score of 0). Influenza viral antigen was detected in the bronchiolar epithelium and tracheobronchial airway epithelium of the NY312 inoculated mice (Figure 2 panels J and L), in rare alveolar macrophages (Figure 3 panel N), but not in alveolar lining cells (Figure 3 panels N and O). The histopathologic changes outlined above for the 1:7 chimeric viruses were very similar to those observed with the 2:6 chimeric viruses (data not shown). The 2:6 1918-HA+NA virus induced bronchiolitis and alveolitis, with a neutrophil-predominant inflammatory infiltrate (score of 3+) (Qi et al., 2009). The 1957 and 1968 chimeric 2:6 pandemic HA+NA viruses also induced histopathologic changes (score of 2+) similar to the cognate 1:7 HA viruses described above.
Figure 2. Lung pathology induced with different influenza A viruses.
Pathology and immunohistochemistry of influenza A virus-infected mouse lung tissue. Photomicrographs of hematoxylin-and-eosin-stained tissue sections (A, C, E, G, I, K) and immunohistochemically-stained sections (B, D, F, H, J, L) to detect influenza viral antigen from mice infected with isogenic viruses expressing different HAs at 4 dpi. Viral antigen is stained red-brown on a hematoxylin-stained background. The 1918- HA virus (A, B) induced a moderate-severe alveolitis and bronchiolitis with viral antigen in both bronchiolar epithelial and alveolar lining cells. The 1957- HA virus (C, D), the 1968 - HA virus (E, F), and the 2009- HA virus (G, H) induced similar pathologic changes with mild-moderate alveolitis and bronchiolitis with viral antigen in both bronchiolar and alveolar lining cells. The NY312 virus (I, J, K, H) induced no pathologic changes but did show viral antigen staining in tracheobronchial and bronchiolar epithelial cells and occasional alveolar macrophages [Original magnifications 100×].
Figure 3. Pathology and immunohistochemistry of influenza A virus-infected mouse lung tissue.

Photomicrographs of hematoxylin-and-eosin-stained tissue sections and immunohistochemically-stained sections to detect influenza viral antigen from mice infected with isogenic viruses expressing different HAs at 4 dpi. Viral antigen is stained red-brown on a hematoxylin-stained background. (A-C) 1918-HA virus induced an alveolitis with a mixed cellular inflammatory infiltrate including numerous neutrophils (arrowheads), lymphocytes, and macrophages (arrows). Viral antigen was detected in alveolar macrophages (B, arrows), and alveolar lining cells (C, arrows). Viruses expressing the 1957 (D-F), 1968 (G-I), or 2009 (J-L) pandemic HAs induced similar pulmonary pathologic changes, consisting of an alveolitis with a mixed cellular inflammatory infiltrate including numerous macrophages (arrows). Viral antigen was detected in alveolar macrophages (E, H, K, arrows), and alveolar lining cells (F, I, L, arrows). The NY312 (P-R) virus induced little appreciable pulmonary pathologic changes, with no alveolitis (M, P). The lungs of NY312 virus-infected mice showed occasional viral antigen positive alveolar macrophages (N, arrows), but no alveolar lining cell staining (O) [Original magnifications 1000×].
Determination of cytokine and chemokine expression in lungs of infected mice
The severity of influenza induced disease in experimental animals correlates well with the ability of virulent virus strains to induce a robust expression of proinflammatory cytokines (Kobasa et al., 2004; Peiris et al., 2009). Moreover, there is a strong association between cytokine levels and the severity of clinical symptoms in infected people (Deng et al., 2008; Yuen and Wong, 2005). Furthermore, the pathological studies outlined above demonstrated a select tropism of viruses with pandemic IAV HA for bronchiolar epithelial and type II alveolar lining cells that was associated with more severe lower lung pathology. Because infected type II alveolar cells have also been shown to play an important role in activation of innate immune responses through increased expression of chemokines and cytokines (Wang et al., 2009), we compared levels of cytokines/chemokines in lungs of mice infected with the seasonal and pandemic HA expressing viruses at 1 to 5 dpi (Figure 4 and data not shown). Notably, among the various chemokines and cytokines analyzed, only expression levels of chemokines CCL2 (MCP-1) and colony-stimulating-factor-3 (CSF3) displayed apparent differences between mice infected with NY312 versus mice infected with chimeric pandemic HA viruses (Figure 4). Employing 2-way ANOVA statistical analysis, we found a significant difference in CCL2 and CSF3 expression levels between mice infected with 1918-HA and NY312 viruses (P = <0.001), 1968-HA and NY312 viruses (P = <0.001), and 2009-HA and NY312 viruses (P = <0.001) over the time course of the experiment. Moreover, there also was a statistically significant difference in CSF3 expression levels between 1957-HA and NY312 infected mice (P = <0.001) while the difference in CCL2 expression levels between 1957-HA and NY312 infected mice did not reach statistical significance (P = 0.062). Intriguingly, except for day 1 dpi, CCL2 and CSF3 expression levels were significantly higher in 1918-HA infected lungs at all other time points as compared to the other viruses. Indeed, 2-way ANOVA analysis also revealed statistically significant differences in CSF3 and CCL2 expression levels between mice infected with 1918-HA and 1968-HA, 1957-HA and 2009-HA, respectively (P = < 0.001). Of note, CCL2 and CSF3 expression levels in NY312 infected lungs also differed significantly from lungs of mock-infected mice (P = < 0.001). Mice infected with 1918-HA virus also showed higher expression levels of the chemokine CXCL1 (KC) and chemokines IL-6 and TNFα were observed when compared to NY312 (2-way ANOVA; data not shown).
Figure 4. Expression of CCL2 (MCP-1) and CSF3 (G-CSF) in lungs of mice infected with chimeric influenza viruses expressing the HA genes of different influenza viruses.
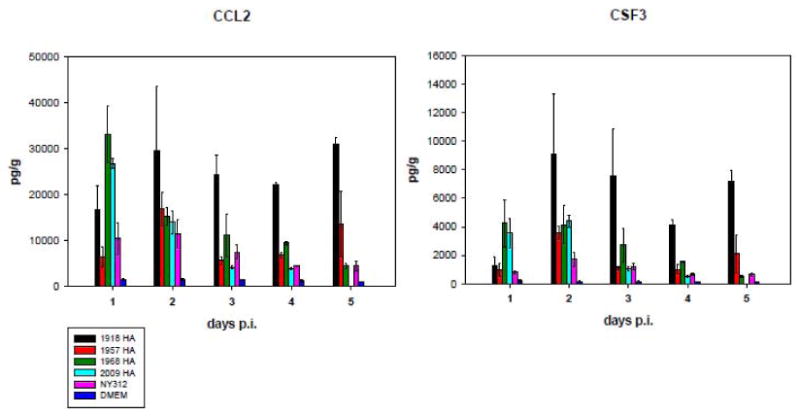
Cytokine and chemokine protein concentrations were determined from mouse lung homogenates as described in the methods from infected animals at indicated days post infection (note that no samples were available for 2009-HA at day 5 post infection) A) CCL2 (MCP-1) B) CSF3 (G-CSF). Mean protein concentrations are shown in pg/g ±SD. See key for different viruses.
Determination of viral replication in mouse lung
To further gain insight into the pathogenesis of these different viruses, and to determine whether differential CCL2 and CSF3 expression levels as described above correlated with the extent of viral replication, titers in lungs of mice at 1, 3, 4 and 6 dpi were determined (Figure 5). Virus replication was detected at 1, 3 and 4 dpi, but not 6 dpi. 2-way ANOVA revealed that virus titers in lungs of mice infected with the 1918-HA virus were significantly different over the entire time course of the experiment as compared to lungs of mice infected with 1957-HA, 2009-HA and NY312 viruses (P = 0.008, P = 0.001 and P = 0.011, respectively). Nevertheless, a per day post hoc statistical analysis using t-test only revealed statistically significant differences between 1918-HA and 2009-HA infected mice at days 3 and 4 dpi (P = 0.024 and P = 0.037, respectively). A comparison of virus titers between 1918-HA and 1968-HA infected mice revealed that statistical significance was narrowly missed (P = 0.065). The only other significant differences detected by 2-Way ANOVA analysis were between mice infected with 1957-HA and 2009-HA (P = 0.038) and between 2009-HA and NY312 infected mice (P = 0.01) indicating that viral titers in lungs infected with 2009-HA were lower when compared to NY312 infected lungs. However, a post hoc analysis using t-test failed to demonstrate significant differences in virus titer between 2009-HA and NY312 infected lungs at individual time points (data not shown). In general, virus titers between the various viruses were small, with only 1918-HA growing to slightly higher titer in mouse lung compared to NY312 as assessed over the entire time course of the experiment.
Figure 5. Lung titers in mice infected with wild-type influenza viruses and chimeric influenza viruses expressing the HA genes of different influenza viruses.

Virus titers in the lungs of infected BALB/c mice (n=3) at days 1, 3, 4, and 6 post infection, plotted as PFU per gram (Log10) of lung tissue as determined by plaque assay. N.D. = Not done. B.D. = below the limit of detection of the assay.
In vitro binding of SP-D to HA proteins from pandemic viruses
In order to evaluate the ability of these chimeric viruses to bind to SP-D protein in vitro, we carried out hemagglutination inhibition assays (HAI) as well as plaque reduction neutralization assays (PRN) using different concentrations of recombinant human SP-D protein (Table 1 and Figure 6). Human SP-D protein was used because of its high homology to mouse SP-D and since both native rodent and human SP-D exhibit similar antiviral activities against several strains of IAV (Hartshorn et al., 1996). These assays demonstrated that while only 62.5 ng/ml of SP-D were sufficient to inhibit agglutination of turkey red blood cells by NY312 in the HAI assay, none of the pandemic HA expressing viruses (1918-HA, 1957-HA, 1968-HA or 2009-HA) was inhibited from agglutinating turkey erythrocytes even in the presence of 2 μg/ml of SP-D, the highest amount of SP-D tested (Table 1).
Figure 6. SP-D neutralization assay with wild-type influenza viruses and chimeric influenza viruses expressing the HA genes of different influenza viruses.
Chimeric IAV expressing the HA of pandemic or seasonal IAV (see Table 1) were tested for their ability to bind to SP-D using a SP-D neutralization and plaque reduction assay as described in the Material and Methods section. Data are presented as mean ± SD. Two-tailed P values were calculated with an unpaired t test. (P = 0.002)
Neutralization of viruses by SP-D
Similarly, PRN assays showed that only 7 ng/ml of SP-D protein was sufficient to cause 50% inhibition (IC50) in the ability of the NY312 virus to form plaques in MDCK cells. In contrast, incubation with the highest concentration of SP-D tested (120 ng/ml) caused no significant reduction in the number of plaques of any of the pandemic HA expressing viruses (Figure 6). Taken together, these data support a hypothesis that low SP-D binding activity of pandemic HA proteins may contribute to infection of alveolar lining cells, increased activation of inflammatory responses and more severe lung pathology.
Discussion
Influenza A viruses (IAV) have the ability to infect a wide variety of warm-blooded animal hosts including wild and domestic avian and mammalian species, as well as humans. In humans, IAV are among the most common causes of acute respiratory infections, and among the most significant because of their high associated morbidity and mortality (Thompson et al., 2003). In the past 100 years there were pandemics in 1918, 1957, 1968, 1977, and 2009 (Taubenberger and Morens, 2009). In 1918, the worst pandemic in recorded history caused approximately 546,000 excess deaths (675,000 total deaths) in the United States (Taubenberger and Morens, 2006) and killed up to 50 million people worldwide (Johnson and Mueller, 2002). The 1957 and 1968 pandemics caused approximately 70,000 and 34,000 excess deaths in the United States, respectively (Noble, 1982). While it is too early to assess the full impact of the 2009 pandemic, in its first year, there have been approximately 12,500 deaths in the United States of which the vast majority have been in individuals under age 65 (CDC, 2010).
IAV infection is initiated by viral attachment mediated by its HA proteins binding to SA-bearing cell surface receptors (Matrosovich, Stech, and Klenk, 2009). HA glycoproteins bind to certain SA isomers on host glycoproteins, strongly supporting a role in determining host range and likely cell tropism (Matrosovich et al., 2000; Nicholls et al., 2007; Rogers and Paulson, 1983; Salomon and Webster, 2009; Taubenberger, 2006). However, several recent studies have demonstrated that enhanced binding to α2-6 SA is not in itself adequate for host switch of an avian-adapted IAV to humans, nor is it a requirement for human infections as IAV bearing the 1918 HA with different receptor binding domain (RBD) configurations (α2-3, α2-6, and blended α2-3/α2-6 SA specificities) were all shown to be virulent in mice (Qi et al., 2009) and ferrets (Tumpey et al., 2007). Notably though, viral constructs with the 1918 HA having only α2-3 SA specificity did not transmit between ferrets (Tumpey et al., 2007). Moreover, mice infected with avian viruses with the HA RBD mutated to exclusively bind α2-6 SA do not show infection of lower respiratory tract epithelial cells or increased virulence (Qi et al., 2009; Stevens et al., 2006). Thus, a number of fundamental questions about the significance of HA RBD specificity as it relates to cellular tropism in the respiratory tract remain unanswered.
Autopsy studies have demonstrated that all IAV pandemics since 1918 have been associated with viral pneumonia and viral replication in the lower respiratory tract (Gill et al., 2010; Taubenberger and Morens, 2008). In this study we explored the hypothesis that SP-D binding activity would be associated with the ability of pandemic IAV to replicate in the lower respiratory tract. SP-D has previously been shown to bind to high mannose type II glycans on some IAV (Crouch, 2000; Hartshorn et al., 1994; Hartshorn et al., 2008). The data presented here suggest that the HA proteins of pandemic IAV show significantly lower SP-D binding compared to the HA from seasonal IAVs, which was associated with the ability of these viruses to invade deeper into the lower lung and infect alveolar lining cells. These findings correlate with the in vivo data such that chimeric viruses expressing the 1918, 1957, 1968 and 2009 pandemic virus HA gene segments induced more severe disease in mice and enhanced alveolar and bronchiolar epithelial cell tropism as compared to seasonal IAV.
Pandemic HAs, derived from zoonotic sources, generally have fewer potential N-linked glycosylation sites, which are observed in later seasonal IAV strains presumably as a consequence of serial antigenic drift pressure (Bush et al., 1999; Hensley et al., 2009; Kash et al., 2010; Wei et al., 2010). More work is needed to correlate differential glycosylation patterns of the globular head of HA with pathogenicity and antigenicity. Clearly, the relationship of HA glycosylation patterns and their pathophysiologic significance is poorly understood. For example, how does the HA glycosylation pattern of a particular IAV differ in different hosts or particular cell types? How uniform are the glycans placed at particular N-X-S/T consensus sites within one virus population or between different IAV HAs sharing the same site? All available pandemic HAs have low SP-D binding activity in vitro and are associated with enhanced lower respiratory tract tropism in mice, but it is not yet clear whether other HAs from divergent avian or mammalian IAV that escape SP-D binding would also cause a similar phenomenon. Indeed, previous reports suggested that pandemic 1968 H3N2 IAV that was shown to evade SP-D binding induced more severe disease in mice as compared to IAV that were able to bind to SP-D (Reading et al., 1997; Reading et al., 2009; Vigerust et al., 2007). Moreover, the latter viruses were more virulent in SP-D-/- mice as compared to wild-type mice, demonstrating an important role of SP-D in IAV pathogenesis (Vigerust et al., 2007).
Elevated expression of inflammatory cytokines and chemokines has been shown to be associated with immunopathogenic responses to highly virulent influenza viruses, such as 1918 and H5N1 HPAI (Kash et al., 2006; Kobasa et al., 2007). Mice infected with chimeric viruses expressing pandemic HA genes showed marked elevation of CCL2 and CSF3 compared to the parental NY312 virus when measured over the whole time course from 1 to 5 dpi. We also observed a statistically significant increased viral replication of the 1918-HA virus compared to the 1957-HA, 2009-HA and NY312 viruses that might partially account for the high CCL2 and CSF3 expression in lungs of 1918-HA infected mice compared to all other viruses. Overall, however, there was only a weak correlation between cytokine expression and virus titers. Therefore, the observed pathological changes among the different viruses were unlikely to be simply related to the extent of viral replication in the lungs.
However, the fact that CCL2 and CSF3 expression levels were higher in mice infected with pandemic HA chimeric viruses compared to NY312 infected mice, and the fact that the major site of replication in NY312 infected lungs was restricted to the epithelium of the large airways compared to the bronchiolar and type II alveolar lining cells in lungs infected with pandemic HA chimeric viruses suggests that cell tropism, and not whole organ viral load, can influence inflammatory responses characterized by different chemokine and cytokine expression profiles and infiltrating immune cell populations. This hypothesis is supported by the prevalence of lymphocytes, macrophages and, in the case of 1918-HA, neutrophils, in the alveolar and interstitial spaces in the lungs of mice infected with these viruses. Interestingly, we observed differences in magnitude, duration and types of inflammatory mediators expressed during infection with the different pandemic HA expressing viruses (Figure 4 and data not shown). Infection with virus expressing 1957, 1968 or 2009 pandemic virus HA resulted in significant infiltration of macrophages and lymphocytes (in agreement with elevated CCL2 expression), whereas in 1918-HA infected mice, we additionally observed a significant infiltration of neutrophils (Figure 3, panel A). While the reason for this phenomenon is unknown, we observed a correlation between increased presence of neutrophils and sustained expression of neutrophil-related chemokines CSF3 and CXCL1 (KC) in lungs of 1918-HA infected mice (Figure 4 and data not shown). Collectively, our results suggest that evasion of SP-D binding was associated with infection of bronchiolar and alveolar epithelial cells, elevated expression of inflammatory cytokines and chemokines and the increased infiltration of innate immune cells into the lungs of infected mice. Finally, viruses expressing the 1918 HA were the most virulent and caused the most severe lung pathology suggesting that the 1918 HA protein contains virulence properties in addition to the evasion of SP-D binding.
In future work it will be important to determine the composition of the glycans of the different HAs examined here to help address these questions. In a previous study, Qi et al. demonstrated that a chimeric NY312 virus expressing an avian-derived H1 HA did not induce lung pathology even with an α2-6 SA RBD specificity (Qi et al., 2009). It will also be important to assess SP-D binding and mouse virulence patterns in avian and swine IAV.
While chimeric viruses expressing the HA of all the available pandemic IAV all displayed minimal SP-D binding and induced lower respiratory tract pathology, the 1918 HA was associated with more severe disease and a unique inflammatory cell infiltrate dominated by neutrophils. Thus, while the extent of SP-D binding is a likely component, it is not the only determinant of pathogenicity of chimeric pandemic HA viruses and it is clear that individual HA proteins can affect pathogenicity via multiple, if still poorly defined, mechanisms. Importantly, as in other recent studies, lung titers alone did not correlate well with histopathology or outcome (Jagger et al., 2010; Qi et al., 2009). For example, whereas there was an overall statistical significant difference between virus titers of lungs infected with the 1918-HA virus versus the parental NY312 virus, post hoc analyses did not reveal significant differences at a particular day, indicating that both viruses replicated to similar titers in mouse lungs. Nevertheless, the 1918 HA virus induced severe and often lethal disease. Moreover, the 1918 HA virus showed a unique inflammatory infiltrate, higher morbidity and mortality, and the longer duration of disease and the inflammatory response compared to the other pandemic HA viruses examined, but the mechanisms for this are not yet clear. In sum these data reinforce the importance of immunopathology in determining the outcome of IAV infection and expand our understanding of influenza pathogenicity beyond the direct cytopathic effects of viral replication. This study suggests that reduced SP-D binding could be associated with the ability of IAV to invade the lower respiratory tract and to induce severe pathology in otherwise healthy individuals. Finally, these findings suggest new avenues for developing new classes of antiviral drugs that interfere with the ability of IAV to cause lower respiratory tract pathology
Acknowledgments
We thank Christian Sauder (CBER/FDA, Bethesda) for helpful discussions. We are grateful for the help provided by the staff of the NIH building 33 animal facility (NIAID/CMB). This work was supported by the Intramural Research Program of the NIH and the NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brown-Augsburger P, Chang D, Rust K, Crouch EC. Biosynthesis of surfactant protein D. Contributions of conserved NH2-terminal cysteine residues and collagen helix formation to assembly and secretion. J Biol Chem. 1996;271(31):18912–9. doi: 10.1074/jbc.271.31.18912. [DOI] [PubMed] [Google Scholar]
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the evolution of human influenza A. Science. 1999;286(5446):1921–5. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- CDC . CDC Estimates of 2009 H1N1 Influenza Cases, Hospitalizations and Deaths in the United States, April 2009 – February 13, 2010. Vol. 2010. CDC; Atlanta: 2010. [Google Scholar]
- Cottey R, Rowe CA, Bender BS. Influenza Virus. In: Coico R, editor. Current Protocols in Immunology. John Wiley and Sons; Hoboken, NJ: 2003. pp. 19.11.6–19.11.9.pp. 19.11.19–19.11.20. [DOI] [PubMed] [Google Scholar]
- Crouch EC. Surfactant protein-D and pulmonary host defense. Respir Res. 2000;1(2):93–108. doi: 10.1186/rr19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng R, Lu M, Korteweg C, Gao Z, McNutt MA, Ye J, Zhang T, Gu J. Distinctly different expression of cytokines and chemokines in the lungs of two H5N1 avian influenza patients. J Pathol. 2008;216(3):328–36. doi: 10.1002/path.2417. [DOI] [PubMed] [Google Scholar]
- Doss M, White MR, Tecle T, Gantz D, Crouch EC, Jung G, Ruchala P, Waring AJ, Lehrer RI, Hartshorn KL. Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J Immunol. 2009;182(12):7878–87. doi: 10.4049/jimmunol.0804049. [DOI] [PubMed] [Google Scholar]
- Gill JR, Sheng ZM, Ely SF, Guinee DG, Beasley MB, Suh J, Deshpande C, Mollura DJ, Morens DM, Bray M, Travis WD, Taubenberger JK. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch Pathol Lab Med. 2010;134(2):235–43. doi: 10.5858/134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K, Chang D, Rust K, White M, Heuser J, Crouch E. Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am J Physiol. 1996;271(5 Pt 1):L753–62. doi: 10.1152/ajplung.1996.271.5.L753. [DOI] [PubMed] [Google Scholar]
- Hartshorn KL. Role of surfactant protein A and D (SP-A and SP-D) in human antiviral host defense. Front Biosci (Schol Ed) 2010;2:527–46. doi: 10.2741/s83. [DOI] [PubMed] [Google Scholar]
- Hartshorn KL, Crouch EC, White MR, Eggleton P, Tauber AI, Chang D, Sastry K. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J Clin Invest. 1994;94(1):311–9. doi: 10.1172/JCI117323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn KL, Webby R, White MR, Tecle T, Pan C, Boucher S, Moreland RJ, Crouch EC, Scheule RK. Role of viral hemagglutinin glycosylation in anti-influenza activities of recombinant surfactant protein D. Respir Res. 2008;9:65. doi: 10.1186/1465-9921-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn KL, White MR, Shepherd V, Reid K, Jensenius JC, Crouch EC. Mechanisms of anti-influenza activity of surfactant proteins A and D: comparison with serum collectins. Am J Physiol. 1997;273(6 Pt 1):L1156–66. doi: 10.1152/ajplung.1997.273.6.L1156. [DOI] [PubMed] [Google Scholar]
- Hartshorn KL, White MR, Tecle T, Tornoe I, Sorensen GL, Crouch EC, Holmskov U. Reduced influenza viral neutralizing activity of natural human trimers of surfactant protein D. Respir Res. 2007;8:9. doi: 10.1186/1465-9921-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn KL, White MR, Voelker DR, Coburn J, Zaner K, Crouch EC. Mechanism of binding of surfactant protein D to influenza A viruses: importance of binding to haemagglutinin to antiviral activity. Biochem J. 2000;351(Pt 2):449–58. [PMC free article] [PubMed] [Google Scholar]
- Hensley SE, Das SR, Bailey AL, Schmidt LM, Hickman HD, Jayaraman A, Viswanathan K, Raman R, Sasisekharan R, Bennink JR, Yewdell JW. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science. 2009;326(5953):734–6. doi: 10.1126/science.1178258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagger BW, Memoli MJ, Sheng ZM, Qi L, Hrabal RJ, Allen GL, Dugan VG, Wang R, Digard P, Kash JC, Taubenberger JK. The PB2 E627K Mutation Attenuates Viruses Containing the 2009 H1N1 Influenza Pandemic Polymerase. mBio. 2010;1(1):pii: e00067–10. doi: 10.1128/mBio.00067-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76(1):105–15. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- Kash JC, Qi L, Dugan VG, Jagger BW, Hrabal RJ, Memoli MJ, Morens DM, Taubenberger JK. Prior infection with classical swine H1N1 influenza viruses is associated with protective immunty to the 2009 pandemic H1N1 virus. Influenza Other Respi Viruses. 2010 doi: 10.1111/j.1750-2659.2010.00132.x. E-published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443(7111):578–81. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445(7125):319–23. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- Kobasa D, Takada A, Shinya K, Hatta M, Halfmann P, Theriault S, Suzuki H, Nishimura H, Mitamura K, Sugaya N, Usui T, Murata T, Maeda Y, Watanabe S, Suresh M, Suzuki T, Suzuki Y, Feldmann H, Kawaoka Y. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature. 2004;431(7009):703–7. doi: 10.1038/nature02951. [DOI] [PubMed] [Google Scholar]
- Kuiken T, Taubenberger JK. The pathology of human influenza revisited. Vaccine. 2008;26(Suppl. 4):D59–D66. doi: 10.1016/j.vaccine.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol. 2001;167(10):5868–73. doi: 10.4049/jimmunol.167.10.5868. [DOI] [PubMed] [Google Scholar]
- Litvack ML, Djiadeu P, Sri Renganathan SD, Sy S, Post M, Palaniyar N. Natural IgM and innate immune collectin SP-D bind to late apoptotic cells and enhance their clearance by alveolar macrophages in vivo. Mol Immunol. 2010;48(1-3):37–47. doi: 10.1016/j.molimm.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Matrosovich M, Stech J, Klenk HD. Influenza receptors, polymerase and host range. Rev Sci Tech. 2009;28(1):203–17. doi: 10.20506/rst.28.1.1870. [DOI] [PubMed] [Google Scholar]
- Matrosovich M, Tuzikov A, Bovin N, Gambaryan A, Klimov A, Castrucci MR, Donatelli I, Kawaoka Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J Virol. 2000;74(18):8502–12. doi: 10.1128/jvi.74.18.8502-8512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrosovich MN, Gambaryan AS, Teneberg S, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Karlsson KA. Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor-binding site. Virology. 1997;233(1):224–34. doi: 10.1006/viro.1997.8580. [DOI] [PubMed] [Google Scholar]
- Memoli MJ, Tumpey TM, Jagger BW, Dugan VG, Sheng ZM, Qi L, Kash JC, Taubenberger JK. An early ‘classical’ swine H1N1 influenza virus shows similar pathogenicity to the 1918 pandemic virus in ferrets and mice. Virology. 2009;393(2):338–45. doi: 10.1016/j.virol.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198(7):962–70. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, Fauci AS. The Persistent Legacy of the 1918 Influenza Virus. N Engl J Med. 2009;361(3):225–229. doi: 10.1056/NEJMp0904819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J, Hers JFP. Influenza. Wolters-Noordhoff Publishing; Groningen: 1972. [Google Scholar]
- Nicholls JM, Bourne AJ, Chen H, Guan Y, Peiris JS. Sialic acid receptor detection in the human respiratory tract: evidence for widespread distribution of potential binding sites for human and avian influenza viruses. Respir Res. 2007;8:73. doi: 10.1186/1465-9921-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble GR. Epidemiological and clinical aspects of influenza. In: Beare AS, editor. Basic and Applied Influenza Research. CRC Press; Boca Raton: 1982. pp. 11–50. [Google Scholar]
- Peiris JS, Cheung CY, Leung CY, Nicholls JM. Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol. 2009;30(12):574–84. doi: 10.1016/j.it.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Kash JC, Dugan VG, Wang R, Jin G, Cunningham RE, Taubenberger JK. Role of sialic acid binding specificity of the 1918 influenza virus hemagglutinin protein in virulence and pathogenesis for mice. J Virol. 2009;83(8):3754–61. doi: 10.1128/JVI.02596-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading PC, Morey LS, Crouch EC, Anders EM. Collectin-mediated antiviral host defense of the lung: evidence from influenza virus infection of mice. J Virol. 1997;71(11):8204–12. doi: 10.1128/jvi.71.11.8204-8212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading PC, Pickett DL, Tate MD, Whitney PG, Job ER, Brooks AG. Loss of a single N-linked glycan from the hemagglutinin of influenza virus is associated with resistance to collectins and increased virulence in mice. Respir Res. 2009;10:117. doi: 10.1186/1465-9921-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. American Journal of Hygiene. 1938;27:493–497. [Google Scholar]
- Reid AH, Fanning TG, Hultin JV, Taubenberger JK. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci U S A. 1999;96(4):1651–6. doi: 10.1073/pnas.96.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GN, Paulson JC. Receptor determinants of human and animal influenza virus isolates: differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology. 1983;127(2):361–73. doi: 10.1016/0042-6822(83)90150-2. [DOI] [PubMed] [Google Scholar]
- Salomon R, Webster RG. The influenza virus enigma. Cell. 2009;136(3):402–10. doi: 10.1016/j.cell.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz RT, Klenk HD. Carbohydrates of influenza virus. IV. Strain-dependent variations. Virology. 1981;113(2):584–93. doi: 10.1016/0042-6822(81)90186-0. [DOI] [PubMed] [Google Scholar]
- Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440(7083):435–6. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- Simonsen L, Clarke MJ, Schonberger LB, Arden NH, Cox NJ, Fukuda K. Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J Infect Dis. 1998;178(1):53–60. doi: 10.1086/515616. [DOI] [PubMed] [Google Scholar]
- Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol. 2006;355(5):1143–55. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303(5665):1866–70. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- Taubenberger JK. Influenza hemagglutinin attachment to target cells: ‘birds do it, we do it…’. Future Virol. 2006;1(4):415–418. doi: 10.2217/17460794.1.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM. 1918 Influenza: the mother of all pandemics. Emerg Infect Dis. 2006;12(1):15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM. Pandemic influenza - including a risk assessment of H5N1. Rev Sci Tech. 2009;28(1):187–202. doi: 10.20506/rst.28.1.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289(2):179–86. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- Tumpey TM, Maines TR, Van Hoeven N, Glaser L, Solorzano A, Pappas C, Cox NJ, Swayne DE, Palese P, Katz JM, Garcia-Sastre A. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science. 2007;315(5812):655–9. doi: 10.1126/science.1136212. [DOI] [PubMed] [Google Scholar]
- Vigerust DJ, Ulett KB, Boyd KL, Madsen J, Hawgood S, McCullers JA. N-linked glycosylation attenuates H3N2 influenza viruses. J Virol. 2007;81(16):8593–600. doi: 10.1128/JVI.00769-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol. 2009;182(3):1296–304. doi: 10.4049/jimmunol.182.3.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CJ, Boyington JC, Dai K, Houser KV, Pearce MB, Kong WP, Yang ZY, Tumpey TM, Nabel GM. Cross-Neutralization of 1918 and 2009 Influenza Viruses: Role of Glycans in Viral Evolution and Vaccine Design. Science Translational Medicine. 2010 doi: 10.1126/scitranslmed.3000799. E-published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PF, Neumann G, Kawaoka Y. Orthomyxoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1691–1740. [Google Scholar]
- Yuen KY, Wong SS. Human infection by avian influenza A H5N1. Hong Kong Med J. 2005;11(3):189–99. [PubMed] [Google Scholar]