

Abstract
Adenovirus (Ad) endosomal membrane penetration activates the NLRP3 inflammasome by releasing lysosomal cathepsin B (catB) into the cytoplasm. We therefore examined the extent to which inflammasome activation correlates with Ad colocalization with catB-enriched lysosomes. Inflammasome activation, is greater during infections with Ad5 possessing an Ad16 fiber (Ad5F16gfp), or Ad5gfp neutralized by human serum, than Ad5gfp alone. Enhanced IL-1β release by Ad5F16gfp is partially due to increased TLR9 signaling but also correlates with greater release of catB into the cytoplasm. This increased TLR9 signaling and catB release correlates with a greater localization of Ad5F16gfp to lysosomes prior to endosomal escape. Another nonenveloped virus, reovirus, requires catB to penetrate cell membranes. However, reovirus did not release catB into the cytoplasm despite significantly greater colocalization with lysosomes compared to Ad5gfp and efficient membrane penetration. Thus, not only lysosomal localization, but the mechanism of membrane penetration influences viral activation of the NLRP3 inflammasome.
Keywords: nonenveloped virus, inflammasome, lysosomes, cathepsin B, fiber
Introduction
Cells have evolved numerous strategies to sense invading viruses and initiate innate antiviral mechanisms which limit viral replication and recruit cells of the immune system to help clear infected cells. These strategies involve the recognition of virus associated molecules such as nucleic acids via Toll-like receptors (TLRs) and RIG-I-like Helicases (RLHs) (Kawai and Akira, 2008), or viral glycoproteins by TLRs (Boehme, Guerrero, and Compton, 2006; Compton et al., 2003; Kurt-Jones et al., 2004). It is also becoming increasingly apparent that cell-derived danger signals, produced in response to viral infection, serve as additional stimuli for innate antiviral responses. These danger signals have been found to include oxidized lipids and nuclear protein-derived “alarmins” such as HMGB1 or S100-family proteins (Bianchi, 2007; Wang et al., 2006). More recently, rupture of lysosomes and oxidative stress in response to adenovirus (Ad) cell entry was found to trigger the activation of the NOD-like receptor, NLRP3 (Barlan et al., 2010).
Adenoviruses are a family of nonenveloped dsDNA viruses commonly associated with acute upper respiratory, gastrointestinal and ocular infections. Over 50 human serotypes of Ad have been identified to date, which are currently split among seven subgroups based on hemagglutination properties and sequence homology (Benko and Harrach, 2003). Adenovirus infections are associated with acute inflammation which is known to be elicited by incoming virions (Cerullo et al., 2007; Di Paolo et al., 2009; Iacobelli-Martinez and Nemerow, 2007; Muruve, 2004; Muruve et al., 2008; Nociari et al., 2009; Nociari et al., 2007; Zhu, Huang, and Yang, 2007). To better understand the potential mechanisms through which Ad elicits a proinflammatory response, a description of the cell entry process is warranted.
To enter cells, Ads first bind to a primary attachment receptor via high affinity interactions between the virus fiber protein and the cell surface receptor (Howitt, Anderson, and Freimuth, 2003). For subgroup A, C, E and F, this receptor is the coxsackievirus and adenovirus receptor (Howitt, Anderson, and Freimuth, 2003). For subgroup B and D viruses, this receptor has been found to be CD46, sialic acid or CD80/CD86 (Sakurai, Kawabata, and Mizuguchi, 2007). Heparan sulfate proteoglycans can also mediate fiber-dependent infection by some subgroup B viruses (Tuve et al., 2008). After attachment, secondary engagement of alphav integrins by the RGD motif of the viral penton base protein induces a signal cascade which leads to clathrin mediated endocytosis of virions (Nemerow, 2000). Adenovirus engagement of αv integrins has been linked to proinflammatory signals such as NF-kappaB activation (Muruve, 2004). Although, it is difficult to rule out the contributions of additional downstream events in the entry process to this signaling event since preventing penton base interactions with integrins prevents subsequent entry steps.
Within endosomes, the virus begins a stepwise disassembly process (Greber et al., 1993) which correlates with acidification of the endosomal compartment by the vacuolar ATPase proton pump. This disassembly process is strictly required for virus escape from endosomes (Wiethoff et al., 2005). Disassembly in endosomes also contributes to recognition of the viral DNA by endosomally localized TLR9 leading to the production of TNF-α, IL-6 and type I interferons (Zhu, Huang, and Yang, 2007). One of the proteins released from the viral capsid during this disassembly process, protein VI, is membrane lytic and mediates the endosomal escape of partially disassembled virions by still poorly defined mechanisms (Wiethoff et al., 2005, Maier et al., 2010). Activation of p38 and ERK1/2 map kinases in a manner which is dependent upon Ad membrane penetration is another previously described proinflammatory signal elicited by Ad infection (Tibbles et al., 2002). Once in the cytoplasm, the virus moves along microtubules in a dynein-dependent manner towards the nucleus. Although the mechanisms remain unknown, evidence exists for the cytosolic detection of Ad leading to the activation of IRF3 and resulting in the production of interferon-β (Zhu, Huang, and Yang, 2007).
Most recently, recognition of Ad by the NLRP3 inflammasome was shown to mediate the release of IL-1β (Muruve et al., 2008). NLRP3 contains an N-terminal pyrin domain, a central NACHT domain, NACHT-associated domain and 7 C-terminal leucine rich repeats (Ye and Ting, 2008). Numerous stimuli can activate the NLRP3 inflammasome by induction of reactive oxygen species or release of lysosomal cathepsins into the cytoplasm (Hornung and Latz; Schroder, Zhou, and Tschopp, 2010; Tschopp and Schroder). We have shown that Ad5 recognition by TLR9 in human macrophages primes the inflammasome by inducing expression of NLRP3 and ASC (Barlan et al., 2010). Additionally, Ad5 protein VI-mediated rupture of lysosomal membranes during cell entry leads the cathepsin B- and ROS-dependent activation of the NLRP3 inflammasome. Adenovirus activation of the inflammasome leads to release of the cytokine IL-1β. Inflammasome activation by Ad5 also leads to NLRP3 and cathepsin B-dependent, but caspase-1-independent, necrotic cell death of macrophages resulting in the release of the proinflammatory molecule HMGB1. Thus, rupture of cathepsin-enriched lysosomes during Ad cell entry serves as a danger signal leading to release of proinflammatory mediators.
Since numerous studies have highlighted differences in intracellular trafficking of Ads from different subgroups with Subgroup C viruses thought to penetrate endosomal membranes at very early times post-endocytosis and subgroup B viruses localizing with late endosomes and lysosomes prior to endosomal membrane penetration (Miyazawa, Crystal, and Leopold, 2001; Miyazawa et al., 1999; Shayakhmetov et al., 2003), we sought to determine whether these differences in Ad endosomal sorting would influence NLRP3 inflammasome activation. Using Ad5 vectors possessing either Ad5 (Ad5gfp) or Ad16 knob domains (Ad5F16gfp), we find that although these viruses penetrate endosomal membranes with similar efficiency, greater localization of Ad5f16gfp to cathepsin-enriched lysosomes prior to endosomal escape leads to enhanced cytosolic release of cathepsins and augmented ROS production compared to Ad5gfp.
We also examined whether lysosomal rupture was a common danger signal recognized by cells during cell entry by other nonenveloped viruses. Reoviruses are a dsRNA nonenveloped virus which depends on lysosomal cathepsins to penetrate cell membranes and is therefore predicted to localize to cathepsin enriched lysosomes for productive infection. While we do observe lysosomal localization of reovirus T3D and T1L in macrophage-like THP-1 cells, release of cathepsin B into the cytoplasm is not observed. Furthermore, reovirus induced IL-1β release is also not observed, suggesting that not only the location, but the mechanism of nonenveloped virus endosomal escape dictates whether cathepsin-dependent inflammasome activation occurs during virus cell entry.
Results
We have previously demonstrated that Ad activation of the NLRP3 requires release of lysosomal cathepsins into the cytoplasm during viral penetration of endosomal membranes (Barlan et al., 2010). Previous studies have suggested that Ads possessing the Ad5 (subgroup C) or Ad3 (subgroup B) knob can both activate the inflammasome (Muruve et al., 2008). Several studies have suggested that Ads of subgroup B colocalize with late endosomal and lysosomal markers prior to endosomal escape to a greater extent than subgroup C viruses (Miyazawa, Crystal, and Leopold, 2001; Miyazawa et al., 1999; Shayakhmetov et al., 2003) suggesting that subgroup B viruses may induce greater release of lysosomal cathepsins upon endosomal rupture. Additionally, it was recently demonstrated that Ad5 neutralization by antibodies present in pooled human serum prior to infection of THP-1 cells augmented the extent of the Ad-induced proinflammatory response including greater activation of the NLRP3 inflammasome (Zaiss et al., 2009). Although, the mechanism of enhanced inflammasome activation was not described.
We first examined whether the extent of inflammasome activation was influenced by the route of Ad cell entry. PMA-differentiated THP-1 cells were infected with increasing MOIs of Ad5gfp, Ad5gfp incubated for 30 min in the presence of 10% heat inactivated, pooled human serum (Ad5gfp + HS), or Ad5F16gfp, a recombinant Ad5gfp vector in which the knob and shaft were replaced with that of Ad16 (Hsu et al., 2005), and IL-1β release into the supernatant was determined 6 hrs post infection. Compared to Ad5gfp, Ad5gfp + HS and Ad5f16gfp induced similar amounts of IL-1β at MOIs which were 30 and 100 fold lower, respectively when comparing the MOI at which IL-1β release above baseline is observed (Fig 1). To rule out whether the enhanced release of IL-1β observed for Ad5+HS and Ad5F16gfp was due to more efficient endosomal escape, we quantified Ad endosomal escape using a previously described assay which monitors the ability of Ads to facilitate the cytosolic translocation of a co-endocytosed, membrane impermeable, ribosomal toxin, saporin (Maier et al., 2010). Although the ability of Ad5gfp to deliver its genome to the nucleus is greatly inhibited in the presence of pooled human serum (data not shown and (Zaiss et al., 2009), other studies have shown that the anti-hexon antibodies present in human serum restrict Ad gene delivery at a step post-endosomal escape (Smith et al., 2008; Varghese et al., 2004). PMA-differentiated THP-1 cells were treated with increasing MOIs of Ad5gfp, Ad5+HS and Ad5F16gfp in the presence of saporin and saporin-dependent cytotoxicity was assessed 24 hrs post infection by MTT assay. While Ad5gfp + HS and Ad5F16gfp could more efficiently induce IL-1β release, each virus was similarly efficient in facilitating the saporin translocation into the cytoplasm as assessed by MTT assay (Fig 2A).
Figure 1. Efficiency of Ad-induced IL-1β release.

PMA-differentiated THP-1 cells were rested in SFM for 2 hrs before treatment with increasing MOIs of Ad5gfp(◆), Ad5gfp+ 10% human serum (■) or Ad5F16gfp(▲) for 6 hrs. Release of IL-1β into supernatant was determined by ELISA. Data represent the mean and standard error of 3 replicates.
Figure 2. Correlation of inflammasome activation with Ad membrane permeabilization, TLR9 signaling and cathepsin B release into the cytoplasm.

A) PMA-differentiated THP-1 cells were rested in SFM for 2 hrs before treatment with increasing MOIs of Ad5gfp(◆), Ad5gfp + 10% human serum(■), or Ad5F16gfp(▲) for 6 hrs in the presence of Saporin. Cells were then washed, incubated overnight and subjected to an MTT assay to assess cell viability. Data represent the mean and standard error of 3 replicates. B) PMA-differentiated THP-1tlr9KD cells stably expressing TLR9 specific shRNA were rested in SFM for 2 hrs before priming with 30ng/ml of TLR2 agonist Pam3CSK4 for 2 hrs, followed by treatment with increasing MOI of Ad5gfp(◆), Ad5gfp + 10% human serum(■), or Ad5F16gfp(▲) for 2 hr. Release of IL-1β was determined by ELISA. Data represent the mean and standard error of 3 replicates. Inset: western blot of cell lysates for TLR9 from THP-1cntrl and THP-1tlr9KD cells stably expressing control or tlr9 specific shRNA, respectively. C) Cells were not treated (NT), treated with 1000 GTU-equivalents of ts1, 100, 300 or 1000 GTU of Ad5gfp or 30, 100, or 300 GTU of Ad6F16gfp or Ad5gfp + 10% human serum for 1 hr. Cytosolic extracts from digitonin permeabilized cells were analyzed for cathepsin B by western blot. Extraction with 1% triton X-100 (TX100) was performed as a positive control.
We have previously shown that TLR9 recognition of the Ad5 genome primes the NLRP3 inflammasome in human cells by stimulating the expression of NLRP3, ASC and proIL-1β while Ad5-induced release of lysosomal cathepsin B into the cytoplasm activates the NLRP3 inflammasome (Barlan et al., 2010). Ad5F16gfp and Ad5gfp + HS were both previously shown to localize more strongly with lysosomes than Ad5gfp in epithelial cells and THP-1 cells, respectively. Lysosomes are enriched for TLR9 and cathepsin B. Thus greater signaling through TLR9 to prime the NLRP3 inflammasome and greater cytosolic release of cathepsin B which activates the NLRP3 inflammasome could both explain the greater release of IL-1β by Ad5F16gfp and Ad5gfp +HS from THP-1 cells. Ad5F16gfp was previously shown to induce greater secretion of interferon-α from human PBMCs than Ad5gfp in a TLR9-dependent manner (Iacobelli-Martinez and Nemerow, 2007). It was also recently shown that Ad5 + HS leads to an overall increase in the expression of several proinflammatory cytokines compared to Ad5 alone (Zaiss et al., 2009). Thus enhanced TLR9 signaling by Ad5F16gfp and Ad5gfp + HS likely contributes to greater priming of the NLRP3 inflammasome. However, since Ad5F16gfp and Ad5gfp + HS rupture endosomes with similar efficiency as Ad5gfp alone, we next examined whether the extent to which increased cathepsin B release into the cytoplasm contributes to greater inflammasome activation by Ad5F16gfp and Ad5gfp + HS. To determine the extent to which adenovirus lysosomal localization and cathepsin B release enhances NLRP3 activation, we used cells in which TLR9 was stably knocked down to reduce adenovirus priming of the inflammasome. We primed PMA-differentiated THP-1 cells stably expressing TLR9 shRNA, THP-1tlr9KD, with the TLR2 agonist, PAM3CSK4, for 2 hrs to induce expression of inflammasome components before infecting cells with increasing MOIs of Ad5gfp, Ad5gfp + HS or Ad5F16gfp for 2 hrs. Under these conditions, inflammasome priming via TLR2 effectively normalizes the priming process for all viral treatments. We observed that Ad5F16gfp and Ad5gfp + HS released similar amounts of IL-1β as Ad5gfp at only 3-fold lesser MOIs when comparing the MOI at which IL-1β is released above baseline (Fig 2B). The expression of TLR9 was reduced ~70% as assessed by western blot (Fig 2B, inset). These results suggest that when priming of the NLRP3 inflammasome is equivalent amongst the viral treatments, Ad5F16gfp and Ad5gfp + HS still more potently induce NLRP3-dependent IL-1β secretion compared to Ad5gfp.
The reported increased colocalization of Ad5+HS with lysosomal markers in THP-1 cells and Ad5F16gfp in epithelial cells seems to correlate with enhanced inflammasome activation in spite of the fact that Ad5gfp, Ad5+HS and Ad5F16gfp all escape from endosomes with similar efficiency. So we next examined whether the increased IL-1β release in response to Ad5+HS and Ad5F16gfp correlated with increased release of cathepsin B into the cytoplasm. PMA-differentiated THP-1 cells were infected with equivalent amounts of Ad5gfp, Ad5+HS or Ad5F16gfp for 2 hrs, cells were then washed extensively, the plasma membrane was selectively permeabilized with digitonin and the amount of cathepsin B in the cytosolic extract was examined by western blot. Control infections with equivalent amounts of the temperature sensitive mutant, ts1, which is unable to escape from endosomes when produced at the nonpermissive temperature (Cotten and Weber, 1995; Greber et al., 1996; Silvestry et al., 2009; Wiethoff et al., 2005), were also performed. Infection with all but ts1 facilitated release of lysosomal cathepsins into the cytoplasm, suggesting viral membrane penetration was required for this event (Fig 2C). However, Ad5+HS and Ad5F16gfp membrane penetration released substantially more cathepsin B into the cytoplasm compared to Ad5gfp. Taken together, data in Fig 2B and 2C suggest that greater release of lysosomal cathepsin B into the cytoplasm by Ad5F16gfp and Ad5gfp + HS contributes to their greater activation of the NLRP3 inflammasome
To determine whether the enhanced release of cathepsin B into the cytoplasm by Ad5F16gfp was due to greater colocalization of the virus with lysosomes, PMA-differentiated THP-1 cells were synchronously infected with either fluorescently labeled Ad5gfp, Ad5F16gfp or ts1 for various times. Cells were fixed and permeabilized before immunostaining for the lysosomal marker, LAMP1 and counterstaining the nuclei with DAPI. Fluorescence deconvolution images were collected (Fig 3A). Colocalization of virus with lysosomes was quantified by determining the number of cytoplasmic puncta positive for fluorescent virus (green) and LAMP1 (red) per cell. Ad5gfp demonstrated a minimal colocalization with LAMP1 particles, with maximal observation of an average of 0.2 dual positive puncta per cell observed at 1 hr post internalization (Fig 3B). This was 5-fold less then the average value of 1.1 puncta per cell for Ad6F16 at 1 hr post internalization (p=0.007). Lysosomal localization of ts1, which uses the same receptors and is endocytosed similarly to Ad5gfp, is similar at 20 minutes to that observed for Ad5F16gfp at 1 hr (p=0.335), in agreement with observations that Ad5gfp more efficiently escapes from endosomes prior to reaching lysosomes (Miyazawa, Crystal, and Leopold, 2001; Miyazawa et al., 1999; Shayakhmetov et al., 2003). This data suggest that there is a correlation between the extent of lysosomal colocalization of Ad and cathepsin B-dependent activation of the NLRP3 inflammasome.
Figure 3. Lysosomal localization of adenovirus.
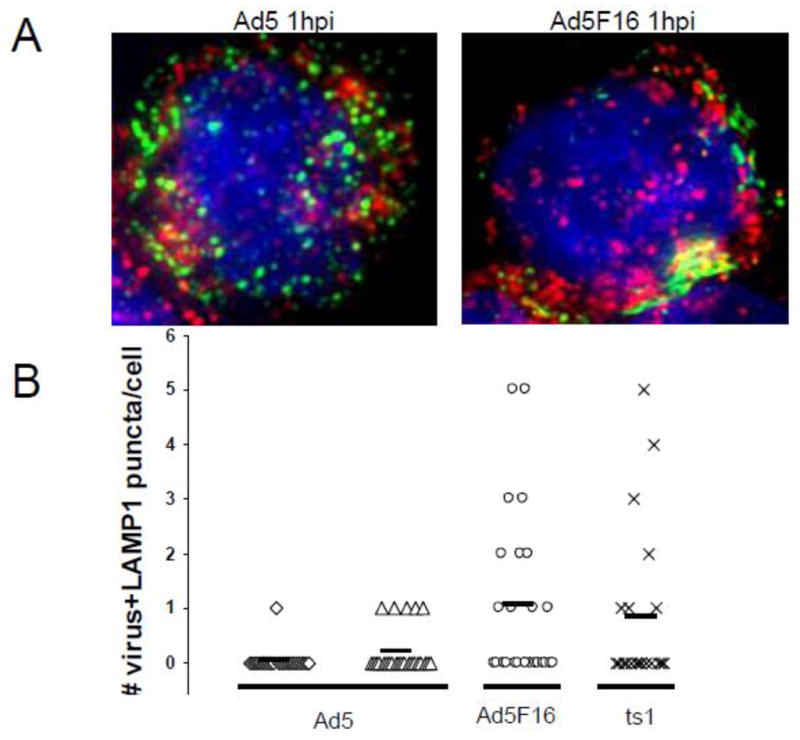
PMA-differentiated THP-1 cells were plated on fibronectin coated coverslips and infected w/Dylight 488 NHS-Ester labeled Ad5gfp, Ad5F16gfp, and ts1. Cells were then fixed and permeabilized with Triton X-100. Localization of A) Ad5gfp (left) or Ad5F16gfp (right) with lysosomes was analyzed by immunofluorescence with monoclonal anti-LAMP1 and secondary Alexa Fluor 568-conjugated Abs, followed by nuclear counterstaining w/Hoechst. The images show virus (green), LAMP-1 (red), and nucleus (blue). C) The number of puncta positive for both virus and LAMP-1 were counted for 25 infected cells per treatment: Ad5 at 20 min(◇) and 60 min(△) post infection (mpi), Ad5F16 at 60 mpi(○), and ts1 at 20 mpi(×). The solid horizontal bar is the average number of puncta units of colocalization per cell for each virus.
Numerous nonenveloped viruses gain access to the cytoplasm by penetrating endosomal membranes (Tsai, 2007). Thus release of cathepsin B into the cytoplasm as these viruses penetrate lysosomal membranes could be a common danger signal. Reoviruses are dsRNA viruses which require endocytosis to penetrate cell membranes and replicate in the cytoplasm (Danthi et al.). The Dearing and Lang strains of reovirus (T3D and T1L, respectively) require proteolytic processing of the capsids by cathepsins B and L to penetrate cell membranes (Ebert et al., 2002). Like Ads, cells respond to reovirus cell entry by activating proinflammatory transcription factors IRF-3 and NF-kappaB (Danthi et al., 2008; Hansberger et al., 2007; Holm et al., 2007). To determine whether reovirus releases cathepsin B into the cytoplasm during cell entry, PMA differentiated THP-1 cells were infected with Ad5F16gfp, reovirus T3D or T1L for 2 hrs and cathepsin B in the cytoplasm was monitored by western blot as described above. Compared to Ad5F15gfp, T3D induced no detectable release of cathepsin B release into the cytoplasm and T1L released marginal amounts (Fig 4A).
Figure 4. Cathepsin B release into the cytoplasm and lysosomal localization of reovirus.
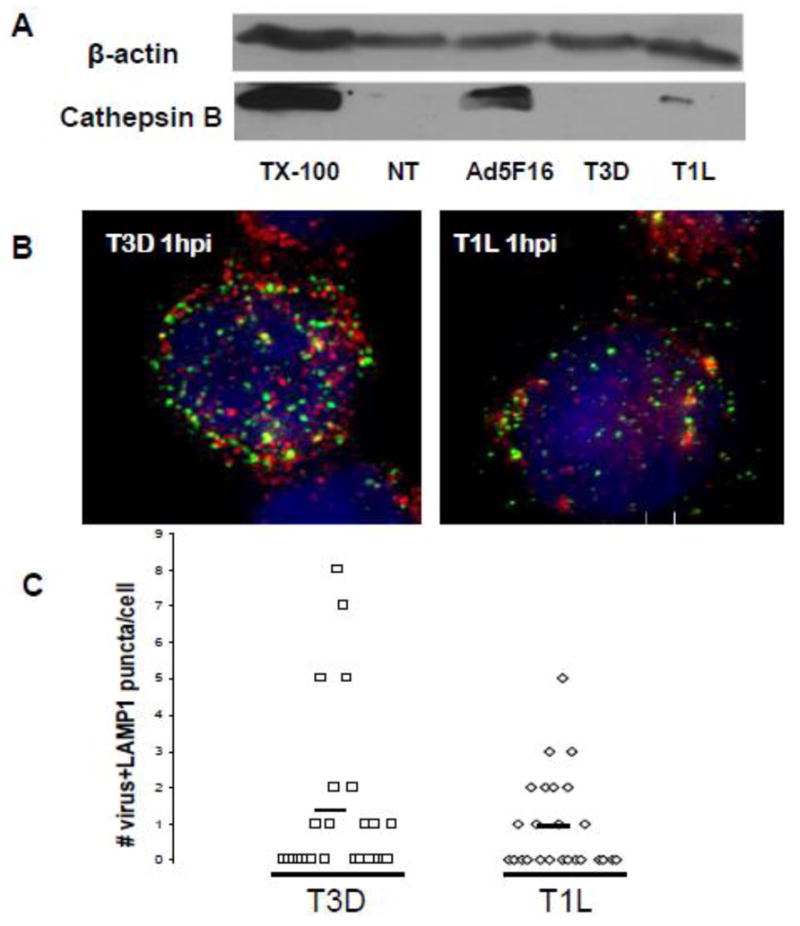
A) PMA-differentiated THP-1 cells were treated with 1000 PFU/cell of reovirus T3D or T1L or 300 GTU of Ad5F16gfp for 2 hrs. Cytosolic extracts from digitonin permeabilized cells were analyzed for cathepsin B by western blot. B) PMA-differentiated THP-1 cells were plated on fibronectin coated coverslips and then synchronously infected with 1000 PFU/cell of T3D (left) TIL (right) for 1 hr. Cells were then fixed, permeabilized and immunostained with polyclonal Abs to reovirus and mAb for LAMP-1 followed by secondary Alexa Fluor 488 and 568-conjugated Abs, respectively. Hoechst stain was used to counterstain nuclei. The images show virus (green), LAMP-1 (red), and nucleus (blue). C) The number of dually labeled puncta were counted for 25 infected cells for each virus type. Each symbol is one infected cell: T3D(□), T1L(◇). The solid horizontal bar is the average number of units of colocalization per cell for each virus.
Reovirus is not able to efficiently facilitate the cytosolic translocation of ribosomal toxins such as saporin in THP-1 cells (unpublished observation). So, to confirm that both T3D and T1L penetrated endosomal membranes, we assessed viral gene expression in PMA-differentiated THP-1 cells. Cells were infected with T3D or T1L viruses for 1hr. Excess virus was washed away and cells were either fixed or incubated for 8 hrs at 37 °C before cells were fixed. Cells were then permeabilized and immunostained with anti-μNS to quantify the percentage of expressing viral proteins. While no significant staining was observed for cells after 1 hr infection, 65% and 67% of cells stained intensely after 8 hrs for T3D and T1L viruses, respectively, which suggests that reovirus efficiently escaped from endosomes in these cells
Since reovirus reportedly requires cathepsin B to enter fibroblasts and epithelial cells, we next determined the efficiency with which reovirus localized to lysosomes in PMA-differentiated THP-1 cells. Cells were infected for 1 hrs with T3D and T1L strains, then fixed, permeabilized and immunostained with polyclonal anti-reovirus sera and anti-LAMP1 (Fig 4B). The number of puncta per cell staining for both reovirus and LAMP-1 were quantified as described above. Compared to Ad5gfp, T3D and T1L viruses more strongly colocalized with LAMP-1 positive vesicles (p=0.002 and 0.010, respectively) similar to that observed for Ad5F16gfp and in agreement with the requirement for lysosomal cathepsins to penetrate cell membranes (Fig 4C).
To determine whether reovirus activates the inflammasome during cell entry, PMA-differentiated THP-1 cells were primed with PAM3CSK4 for 2 hrs to ensure sufficient expression of NLRP3, ASC and proIL-1β before infecting with 1000 PFU/cell of reovirus T3D or T1L or 300 GTU/cell of Ad5gfp for 2 or 1 hrs, respectively. Compared to Ad5gfp, which induced release of significantly more IL-1β than PAM3CSK4 alone, reovirus did not induce IL-1β release above control although present at a higher MOI than Ad5gfp (Fig 5). Additional experiments examining reovirus-induced release of IL-1β from PAM3CSK4 primed cells at 4 and 6 hrs post infection yielded similar results (not shown). Thus, not only does the location of viral membrane penetration within the endolysosomal system influence NLRP3 activation, but the mechanism of viral membrane penetration may as well.
Figure 5. Reovirus endosomal escape is not sufficient for activation of the NLRP3 inflammasome in TLR primed cells.

PMA-differentiated THP-1 cells were rested in SFM for 2 hrs before prestimulation with 30ng/ml Pam3CSK4 for 2 hrs. After prestimulating cells, infected w/3000 PFU/Cell of T3D or T1L for 2hrs and 300 GTU/cell Ad5gfp for 2 hr. IL-1β release was determined by ELISA. Data represent the mean and standard error of 3 replicates.
Discussion
Detection of a virus by the host during cell entry represents the very earliest possible event leading to an innate and adaptive immune response. Several nonenveloped viruses including adenovirus (Di Paolo et al., 2009; Iacobelli-Martinez and Nemerow, 2007; Muruve et al., 2008; Nociari et al., 2009), reovirus (Holm et al., 2007), and parvovirus (Zhu, Huang, and Yang, 2009) are recognized by the host while entering cells although the exact details have been incompletely defined. Adenovirus cell entry triggers numerous different proinflammatory signaling pathways resulting in the activation of MAPK, NF-kappaB, and IRF3/IRF7 dependent expression of proinflammatory cytokines (Appledorn et al., 2008; Fejer et al., 2008; Muruve, 2004; Nociari et al., 2009; Nociari et al., 2007; Zhu, Huang, and Yang, 2007), MHC II and costimulatory molecules (Nociari et al., 2009; Nociari et al., 2007), as well as type I interferons, and subsequently, interferon stimulated genes (Granberg et al., 2005; Scibetta et al., 2005; Zhao et al., 2003). While a considerable amount of information is available regarding the downstream molecules activated during adenovirus infection and cell entry, much less is known regarding exactly which host cell and viral molecules are necessary to initiate these signaling cascades. A better understanding of the upstream events which regulate the downstream effector molecules will ultimately provide information which could ultimately be used for the design of more effective vaccines and antiviral agents.
We have previously demonstrated that Ad rupture of lysosomal membranes during cell entry activates the NLRP3 inflammasome. Activation of the NLRP3 inflammasome initiates several proinflammatory pathways which are important for antiviral immunity (Ichinohe et al., 2009) as well as the effectiveness of vaccine adjuvants in some circumstances (Eisenbarth et al., 2008; Li et al., 2008). Key mediators of inflammasome downstream signaling are IL-1β and IL-18 (Martinon, Mayor, and Tschopp, 2009). Both of these cytokines have been implicated in the proinflammatory response to replication defective adenovirus vectors administrated intratracheally (Berclaz et al., 2002) or intravenously to mice (Shayakhmetov et al., 2005). However, a more recent study found that neither IL-1β nor NLRP3 are involved in the proinflammatory response to i.v. administered Ad5 (Di Paolo et al., 2009). Instead, an interaction of Ad5 with β3 integrins on splenic marginal zone macrophages induces IL-1α dependent inflammation. Further study is required to determine whether NLRP3-dependent proinflammatory signals are involved in Ad-induced inflammation during infections of other organs such as the lung or heart.
Our current data suggest that the route of Ad endosomal trafficking during cell entry can greatly influence the proinflammatory response. Previous observations have noted that Ad5F16gfp more potently induces interferon-α release from plasmacytoid dendritic cells during infection of human PBMCs (Iacobelli-Martinez and Nemerow, 2007). Our current data suggest that enhanced TLR9-dependent signaling occurs in response to Ad5F16gfp infection when compared to infection by Ad5gfp. Additionally, targeting of Ad5 to lysosomes upon neutralization with pooled human serum has also been shown to greatly increase the release of proinflammatory cytokines (Zaiss et al., 2009). Our results demonstrate that targeting Ad virions to lysosomes increases IL-1β release. This increased release is partially due to the increased activation of TLR9 resulting in the upregulation of proIL-1β expression as well as the inflammasome components, ASC and NLRP3 (Barlan et al., 2010; Bauernfeind et al., 2009). In addition to increased activation of TLR9, targeting Ad virions to lysosomes enhances the release of cathepsin B into the cytoplasm as the virus ruptures endosomal membranes during cell entry. This increased release of cathepsin B by Ad5F16gfp and Ad5+HS correlates with enhanced IL-1β release from cells in which TLR9 is stably knocked down by shRNA.
Of the greater than 50 human adenovirus serotypes, Ad5 and members of subgroup C viruses are the most successful at infecting the human population, with >70% of the population thought possess preexisting immunity to these viruses (Ginsberg and Prince, 1994). In fact, members of subgroup B, such as Ad16, are more commonly associated with infections of immunocompromised individuals (Leen and Rooney, 2005). While much of this difference between subgroup C and other Ad subgroups is likely due to differences in the Ad early genes such as immune evasion genes encoded in the E3 transcriptional unit, subgroup C Ads may also possess a selective advantage over other subgroups due to their ability to more efficiently escape from endosomes prior to arrival at cathepsin and TLR9 enriched lysosomes and eliciting a weaker proinflammatory response.
Several intracellular bacterial pathogens are known to activate NLRP3 by rupturing lysosomes while gaining access to the cytoplasm during infection (Duncan et al., 2009; Willingham et al., 2007). Thus, it seems that release of cathepsin B into the cytoplasm has evolved as a danger-associated signal which triggers proinflammatory responses and necrotic cell death of infected macrophages (Hornung and Latz). Other nonenveloped viruses have been shown to pass through lysosomes during cell entry (Brabec, Blaas, and Fuchs, 2006; Marsh and Pelchen-Matthews, 2000; Miyazawa, Crystal, and Leopold, 2001; Qian et al., 2009; Suikkanen et al., 2003). Reoviruses, in particular, have evolved a requirement for lysosomal cathepsins to trigger viral capsid conformational changes which facilitate membrane penetration (Ebert et al., 2002). While reoviruses are though to disrupt endosomal membranes to allow passage of a ~70 nm diameter subvirion particle into the cytoplasm, their ability to facilitate the transport of coninternalized proteins and fluorescently labeled dextrans across cell membranes is much more size restricted than that of Ad virions (Agosto, Ivanovic, and Nibert, 2006; Ivanovic et al., 2008; Zhang et al., 2009). Cathepsin B is 28 kDa which is in the range of size selectivity reported for reovirus membrane pores. Since reovirus appears to colocalize with lysosomes to a similar extent as Ad5F16gfp in THP-1 cells and yet fails to release significant amounts of cathepsin B, it appears that differences in the mechanism of viral membrane penetration influence the induction of this danger-associated signal leading to inflammasome activation. Thus, future studies of the location and mechanism of nonenveloped virus cell entry may provide more information regarding the generality of cathepsin B-associated danger signals in innate immune responses to viral infection.
Proinflammatory responses to adenoviruses have greatly limited their utility as vectors for gene therapy despite the numerous qualities of these viruses which makes them excellent vectors for human gene therapies (Muruve, 2004; Raper et al., 2003). Our findings suggest that developing new Ad vectors which more efficiently avoid cathepsin and TLR9 enriched lysosomes during cell entry could greatly benefit the utility of these vectors for gene therapy. Additionally, temporary pharmacologic inhibition of lysosomal cysteine proteases such as cathepsin B, or TLR9 maturing asparagine endopeptidase (AEP), could potentially reduce the inflammatory response to Ad vectors. Conversely, studies with influenza virus suggest that NLRP3 activation aids in the development of a protective immune response (Ichinohe et al., 2009). In some cases, NLRP3 has been also been found to contribute to the humoral response elicited by alum-adjuvanted antigen (Eisenbarth et al., 2008; Li et al., 2008). Thus Ad vectors employed for genetic vaccination may benefit from being more efficiently targeted to cathepsin enriched lysosomes.
Methods
Viruses and reagents
An E1/E3-deleted adenovirus encoding egfp under the control of a cytomegalovirus promoter, Ad5gfp (Wiethoff et al., 2005), a temperature sensitive mutant, ts1 (Weber, 1976) or, Ad5F16gfp (kindly provided by Glen Nemerow (Hsu et al., 2005)) were propagated in HEK293 cells obtained from ATCC and twice purified by cesium chloride gradient centrifugation as previously described (Wiethoff et al., 2005). For these studies, the ts1 virus was propagated at 39.5 °C resulting in viral particles incapable of membrane penetration (Cotten and Weber, 1995; Greber et al., 1996; Wiethoff et al., 2005). Viral concentrations were determined by Bradford assay (Bio-Rad Laboratories, Inc.). 1 μg of protein corresponded to 4 × 10(9) viral particles. Viruses were titered by serial dilution on HeLa (ATCC) cells using flow cytometry to quantify egfp expression. The specific infectivity of viral preparations used in these studies ranged from 100–200 viral particles per GFP-transducing unit (GTU). Reovirus strains T3D and T1L were derived from lab stocks which were twice plaque purified. Virus was propagated on L929 cells and purified by cesium chloride density gradient centrifugation as previously described (Furlong, Nibert, and Fields, 1988). Virus was titered by plaque assay on L929 cells.
Cell lines and reagents
THP-1 cells were obtained from ATCC and were maintained in RPMI 1640 media, supplemented with 10% FBS, 100 IU/ml penicillin, 1 mg/ml streptomycin, 0.25 μg/ml amphotericin B, non-essential amino acids, 1 mM sodium pyruvate, 10 mM HEPES buffer and 2 mM glutamine from Mediatech and HyClone. THP-1 cells were differentiated by overnight stimulation with 100 nM phorbol-12-myristate-13-acetate (PMA; EMD Chemicals) to macrophage-like cells. HEK293 and HeLa cells were maintained in DMEM (Mediatech) containing the above additives used in the RPMI 1640 media. Murine L929 (L) cells were maintained in Joklik’s MEM (Lonza) supplemented to contain 5% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 ng/ml amphotericin B (Sigma-Aldrich). All other chemical reagents were from Fisher Chemicals.
Measurement of IL-1β secretion by ELISA
50,000 THP-1 cells per well were plated in a 96-well plate and differentiated with 100 nM PMA for 16 hrs. Cells were then rested in serum free media for 2 hrs. A subset of cells was then primed with 30ng/ml PAM3CSK4 (InvivoGen) for 2 hrs. Both primed and unprimed cells were then infected with various amounts of Ad5gfp (with 10% FBS or 10% pooled human AB serum (Sigma-Aldrich)), Ad5F16gfp, T3D and T1L. At various time points post infection, the supernatants were collected and secreted IL-1β was quantified with Human IL-1β ELISA Ready-SET-Go! kit obtained from eBioscience.
Saporin cell viability assay
Lyophilized saporin (Sigma-Aldrich) was reconstituted with water to 1mg/ml stock solution. Working concentration in complete media was 50ug/ml. 20,000 HeLa cells per well in a 96-well plate were infected with serial dilutions of Ad5gfp or Ad5F16gfp in the presence of saporin. 16 hrs post infection, 0.02% MTT (cat# M92050, RPI Corp.) was added to the media for 2 hrs incubation. Cells were then washed with PBS and dissolved with DMSO for 15min. Color intensity was measured by Biotech ELx800 plate reader using KC Junior software.
Measurement of Cathepsin B release into cytoplasm
PMA-differentiated THP-1 cells (2×10(6)/well) in 6 well plates were treated with media alone, 100, 300 or 1000 GTU/cell of Ad5gfp, 30, 100 or 300 GTU/cell of Ad5F16gfp Ad5gfp + 10% human serum for 1 hr. Cells were then permeabilized with ice cold 50 μg/ml digitonin (Calbiochem) in PBS pH 7.4 and 1 mM PMSF (MP Biomedicals) for 15 minutes at 4 °C which selectively permeabilizes the plasma membrane while leaving intracellular membranes intact (Ghavami et al., 2008). Cell extract using 0.1% triton X-100 was used as a positive control for cathepsin B release. These cytosolic extracts were subjected to SDS-PAGE, transferred to nitrocellulose membranes and immunoblotted for Cathepsin B (Santa Cruz cat# sc-13985) and Beta-actin (Sigma).
Immunofluorescence microscopy
Differentiated THP-1 cells were plated on coverslips coated with fibronectin (Sigma-Aldrich) and then infected with Ad5gfp, Ad5F16gfp, ts1, T1L, or T3D. Cells were then fixed with 4% paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.1% TritonX-100 (Sigma-Aldrich). Cells were immunostained with polyclonal reovirus Abs and LAMP-1 mAb from eBioscience. Secondary Alexa Fluor 488- and 568-conjugated Abs were used to visualize reovirus infection and lysosomes, respectively. Ad5gfp, Ad5F16gfp, ts1 were prelabeled with Dylight 488 NHS-Ester Fluorophores, according to manufacturer’s protocol prior to use. Labeling did not significantly affect viral infectivity. Hoechst stain from ImmunoChemistry Technologies was used to counterstain nuclei before coverslips were mounted on glass slides with ProLong Gold from Invitrogen. Z-stack images were acquired with a DeltaVision microscope (Applied Precision) using a CoolSnap HQ digital camera (Photometrics) with a 1.4-numerical aperture (NA) 100X objective lens, and deconvolved with SoftWorx deconvolution software (Applied Precision).
Quantification of lysosomal localization
Deconvolved images were analyzed for the localization of virus particles in relation to lysosomes with the “multichannel view” plugin of MacBiophotonics ImageJ. The number of units of colocalization of virus with LAMP-1 per cell were counted for 25 infected cells for each virus type. The significance of observed differences between the distributions of the number of dually positive puncta per cell was determined by a two-tailed t-test assuming equal variance using Microsoft Excel.
Acknowledgments
We would like to acknowledge financial assistance from the NIH (AI082430 to CMW, subcontract from HL054352 to Glen R. Nemerow) and the American Heart Association (09GRNT2261306 to CMW and 09DG2140019 to PD). We thank Glen Nemerow for providing the Ad5F16gfp virus.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agosto MA, Ivanovic T, Nibert ML. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc Natl Acad Sci U S A. 2006;103(44):16496–501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appledorn DM, Patial S, McBride A, Godbehere S, Van Rooijen N, Parameswaran N, Amalfitano A. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J Immunol. 2008;181(3):2134–44. doi: 10.4049/jimmunol.181.3.2134. [DOI] [PubMed] [Google Scholar]
- Barlan AU, Griffin TM, McGuire KA, Wiethoff CM. Adenovirus membrane penetration activates the NLRP3 inflammasome. Journal of Virology. 2010 doi: 10.1128/JVI.01265-10. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, Macdonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting Edge: NF-{kappa}B Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol. 2009 doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benko M, Harrach B. Molecular evolution of adenoviruses. Curr Top Microbiol Immunol. 2003;272:3–35. doi: 10.1007/978-3-662-05597-7_1. [DOI] [PubMed] [Google Scholar]
- Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100(12):4193–200. doi: 10.1182/blood-2002-04-1102. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol. 2006;177(10):7094–102. doi: 10.4049/jimmunol.177.10.7094. [DOI] [PubMed] [Google Scholar]
- Brabec M, Blaas D, Fuchs R. Wortmannin delays transfer of human rhinovirus serotype 2 to late endocytic compartments. Biochem Biophys Res Commun. 2006;348(2):741–9. doi: 10.1016/j.bbrc.2006.07.125. [DOI] [PubMed] [Google Scholar]
- Cerullo V, Seiler MP, Mane V, Brunetti-Pierri N, Clarke C, Bertin TK, Rodgers JR, Lee B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol Ther. 2007;15(2):378–85. doi: 10.1038/sj.mt.6300031. [DOI] [PubMed] [Google Scholar]
- Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77(8):4588–96. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten M, Weber JM. The adenovirus protease is required for virus entry into host cells. Virology. 1995;213(2):494–502. doi: 10.1006/viro.1995.0022. [DOI] [PubMed] [Google Scholar]
- Danthi P, Guglielmi KM, Kirchner E, Mainou B, Stehle T, Dermody TS. From Touchdown to Transcription: The Reovirus Cell Entry Pathway. Curr Top Microbiol Immunol. doi: 10.1007/82_2010_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol. 2008;82(1):161–72. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo NC, Miao EA, Iwakura Y, Murali-Krishna K, Aderem A, Flavell RA, Papayannopoulou T, Shayakhmetov DM. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31(1):110–21. doi: 10.1016/j.immuni.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182(10):6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Deussing J, Peters C, Dermody TS. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277(27):24609–17. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008 doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejer G, Drechsel L, Liese J, Schleicher U, Ruzsics Z, Imelli N, Greber UF, Keck S, Hildenbrand B, Krug A, Bogdan C, Freudenberg MA. Key role of splenic myeloid DCs in the IFN-alphabeta response to adenoviruses in vivo. PLoS Pathog. 2008;4(11):e1000208. doi: 10.1371/journal.ppat.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62(1):246–56. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S, Asoodeh A, Klonisch T, Halayko AJ, Kadkhoda K, Kroczak TJ, Gibson SB, Booy EP, Naderi-Manesh H, Los M. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med. 2008;12(3):1005–22. doi: 10.1111/j.1582-4934.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg HS, Prince GA. The molecular basis of adenovirus pathogenesis. Infect Agents Dis. 1994;3(1):1–8. [PubMed] [Google Scholar]
- Granberg F, Svensson C, Pettersson U, Zhao H. Modulation of host cell gene expression during onset of the late phase of an adenovirus infection is focused on growth inhibition and cell architecture. Virology. 2005;343(2):236–45. doi: 10.1016/j.virol.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Greber UF, Webster P, Weber J, Helenius A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996;15(8):1766–77. [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75(3):477–86. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. IkappaB kinase subunits alpha and gamma are required for activation of NF-kappaB and induction of apoptosis by mammalian reovirus. J Virol. 2007;81(3):1360–71. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm GH, Zurney J, Tumilasci V, Leveille S, Danthi P, Hiscott J, Sherry B, Dermody TS. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J Biol Chem. 2007;282(30):21953–61. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 40(3):620–3. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitt J, Anderson CW, Freimuth P. Adenovirus interaction with its cellular receptor CAR. Curr Top Microbiol Immunol. 2003;272:331–64. doi: 10.1007/978-3-662-05597-7_11. [DOI] [PubMed] [Google Scholar]
- Hsu C, Boysen M, Gritton LD, Frosst PD, Nemerow GR, Von Seggern DJ. In vitro dendritic cell infection by pseudotyped adenoviral vectors does not correlate with their in vivo immunogenicity. Virology. 2005;332(1):1–7. doi: 10.1016/j.virol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Iacobelli-Martinez M, Nemerow GR. Preferential activation of Toll-like receptor nine by CD46-utilizing adenoviruses. J Virol. 2007;81(3):1305–12. doi: 10.1128/JVI.01926-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008;27(8):1289–98. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A. 2004;101(5):1315–20. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Rooney CM. Adenovirus as an emerging pathogen in immunocompromised patients. Br J Haematol. 2005;128(2):135–44. doi: 10.1111/j.1365-2141.2004.05218.x. [DOI] [PubMed] [Google Scholar]
- Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181(1):17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier O, Galan DL, Wodrich H, Wiethoff CM. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology. 2010;402(1):11–9. doi: 10.1016/j.virol.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M, Pelchen-Matthews A. Endocytosis in viral replication. Traffic. 2000;1(7):525–32. doi: 10.1034/j.1600-0854.2000.010701.x. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Miyazawa N, Crystal RG, Leopold PL. Adenovirus serotype 7 retention in a late endosomal compartment prior to cytosol escape is modulated by fiber protein. J Virol. 2001;75(3):1387–400. doi: 10.1128/JVI.75.3.1387-1400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa N, Leopold PL, Hackett NR, Ferris B, Worgall S, Falck-Pedersen E, Crystal RG. Fiber swap between adenovirus subgroups B and C alters intracellular trafficking of adenovirus gene transfer vectors. J Virol. 1999;73(7):6056–65. doi: 10.1128/jvi.73.7.6056-6065.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004;15(12):1157–66. doi: 10.1089/hum.2004.15.1157. [DOI] [PubMed] [Google Scholar]
- Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452(7183):103–7. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- Nemerow GR. Cell receptors involved in adenovirus entry. Virology. 2000;274(1):1–4. doi: 10.1006/viro.2000.0468. [DOI] [PubMed] [Google Scholar]
- Nociari M, Ocheretina O, Murphy M, Falck-Pedersen E. Adenovirus induction of IRF3 occurs through a binary trigger targeting Jun N-terminal kinase and TBK1 kinase cascades and type I interferon autocrine signaling. J Virol. 2009;83(9):4081–91. doi: 10.1128/JVI.02591-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nociari M, Ocheretina O, Schoggins JW, Falck-Pedersen E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J Virol. 2007;81(8):4145–57. doi: 10.1128/JVI.02685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian M, Cai D, Verhey KJ, Tsai B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009;5(6):e1000465. doi: 10.1371/journal.ppat.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80(1–2):148–58. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Sakurai F, Kawabata K, Mizuguchi H. Adenovirus vectors composed of subgroup B adenoviruses. Curr Gene Ther. 2007;7(4):229–38. doi: 10.2174/156652307781369137. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- Scibetta AG, Copier J, Barrett A, Chaplin T, Taylor-Papadimitriou J. Gene expression changes induced by a recombinant E1-/E3- adenovirus type 5 vector in human mammary epithelial cells. Intervirology. 2005;48(6):350–61. doi: 10.1159/000086062. [DOI] [PubMed] [Google Scholar]
- Shayakhmetov DM, Li ZY, Ni S, Lieber A. Interference with the IL-1-signaling pathway improves the toxicity profile of systemically applied adenovirus vectors. J Immunol. 2005;174(11):7310–9. doi: 10.4049/jimmunol.174.11.7310. [DOI] [PubMed] [Google Scholar]
- Shayakhmetov DM, Li ZY, Ternovoi V, Gaggar A, Gharwan H, Lieber A. The interaction between the fiber knob domain and the cellular attachment receptor determines the intracellular trafficking route of adenoviruses. J Virol. 2003;77(6):3712–23. doi: 10.1128/JVI.77.6.3712-3723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestry M, Lindert S, Smith JG, Maier O, Wiethoff CM, Nemerow GR, Stewart PL. Cryoelectron microscopy structure of the adenovirus type 2 temperature sensitive mutant 1 reveals insight into the cell entry defect. J Virol. 2009 doi: 10.1128/JVI.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JG, Cassany A, Gerace L, Ralston R, Nemerow GR. Neutralizing antibody blocks adenovirus infection by arresting microtubule-dependent cytoplasmic transport. J Virol. 2008;82(13):6492–500. doi: 10.1128/JVI.00557-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suikkanen S, Antila M, Jaatinen A, Vihinen-Ranta M, Vuento M. Release of canine parvovirus from endocytic vesicles. Virology. 2003;316(2):267–80. doi: 10.1016/j.virol.2003.08.031. [DOI] [PubMed] [Google Scholar]
- Tibbles LA, Spurrell JC, Bowen GP, Liu Q, Lam M, Zaiss AK, Robbins SM, Hollenberg MD, Wickham TJ, Muruve DA. Activation of p38 and ERK signaling during adenovirus vector cell entry lead to expression of the C-X-C chemokine IP-10. J Virol. 2002;76(4):1559–68. doi: 10.1128/JVI.76.4.1559-1568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B. Penetration of nonenveloped viruses into the cytoplasm. Annu Rev Cell Dev Biol. 2007;23:23–43. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 10(3):210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- Tuve S, Wang H, Jacobs JD, Yumul RC, Smith DF, Lieber A. Role of cellular heparan sulfate proteoglycans in infection of human adenovirus serotype 3 and 35. PLoS Pathog. 2008;4(10):e1000189. doi: 10.1371/journal.ppat.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese R, Mikyas Y, Stewart PL, Ralston R. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J Virol. 2004;78(22):12320–32. doi: 10.1128/JVI.78.22.12320-12332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ward MF, Fan XG, Sama AE, Li W. Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol. 2006;19(1):3–9. doi: 10.1089/vim.2006.19.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J. Genetic analysis of adenovirus type 2 III. Temperature sensitivity of processing viral proteins. J Virol. 1976;17(2):462–71. doi: 10.1128/jvi.17.2.462-471.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol. 2005;79(4):1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Bergstralh DT, O’Connor W, Morrison AC, Taxman DJ, Duncan JA, Barnoy S, Venkatesan MM, Flavell RA, Deshmukh M, Hoffman HM, Ting JP. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2(3):147–59. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr Opin Immunol. 2008;20(1):3–9. doi: 10.1016/j.coi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zaiss AK, Vilaysane A, Cotter MJ, Clark SA, Meijndert HC, Colarusso P, Yates RM, Petrilli V, Tschopp J, Muruve DA. Antiviral antibodies target adenovirus to phagolysosomes and amplify the innate immune response. J Immunol. 2009;182(11):7058–68. doi: 10.4049/jimmunol.0804269. [DOI] [PubMed] [Google Scholar]
- Zhang L, Agosto MA, Ivanovic T, King DS, Nibert ML, Harrison SC. Requirements for the formation of membrane pores by the reovirus myristoylated micro1N peptide. J Virol. 2009;83(14):7004–14. doi: 10.1128/JVI.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Granberg F, Elfineh L, Pettersson U, Svensson C. Strategic attack on host cell gene expression during adenovirus infection. J Virol. 2003;77(20):11006–15. doi: 10.1128/JVI.77.20.11006-11015.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol. 2007;81(7):3170–80. doi: 10.1128/JVI.02192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huang X, Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest. 2009;119(8):2388–98. doi: 10.1172/JCI37607. [DOI] [PMC free article] [PubMed] [Google Scholar]