

Abstract
The amygdala, perhaps more than any other brain region, has been implicated in numerous neuropsychiatric and neurodevelopmental disorders. It is part of a system initially evolved to detect dangers in the environment and modulate subsequent responses, which can profoundly influence human behavior. If its threshold is set too low, normally benign aspects of the environment are perceived as dangers, interactions are limited, and anxiety may arise. If set too high, risk taking increases and inappropriate sociality may occur. Given that many neurodevelopmental disorders involve too little or too much anxiety or too little of too much social interaction, it is not surprising that the amygdala has been implicated in many of them. In this chapter, we begin by providing a brief overview of the phylogeny, ontogeny, and function of the amygdala and then appraise data from neurodevelopmental disorders which suggest amygdala dysregulation. We focus on neurodevelopmental disorders where there is evidence of amygdala dysregulation from postmortem studies, structural MRI analyses or functional MRI. However, the results are often disparate and it is not totally clear whether this is due to inherent heterogeneity or differences in methodology. Nonetheless, the amygdala is a common site for neuropathology in neurodevelopmental disorders and is therefore a potential target for therapeutics to alleviate associated symptoms.
Keywords: Autism, schizophrenia, bipolar, fragile X, anxiety, Williams syndrome
Introduction
The amygdala is an almond-shaped brain region located in the anterior portion of the temporal lobe (Fig. 1). Occupying a volume of approximately 2.25 cm3 on each side, it makes up barely 0.3% of the volume of the human brain. Yet, perhaps more than any other brain region, it has been implicated in a plethora of neuropsychiatric disorders. The amygdala is, in fact, a complex region made up of at least 13 nuclei and cortical areas in the nonhuman primate and human brains (Amaral, Price, Pitkänen, & Carmichael, 1992) (Fig. 2). The fact that the amygdala appears to interact with several functional systems of the brain (Johnston, 1923) has led some (Swanson & Petrovich, 1998) to question whether it should be considered an integrated entity at all. However, our own neuroanatomical data (Pitkänen & Amaral, 1998; Pitkänen, Kelly, & Amaral, 2002) indicate that the internal components of what we prefer to call the amydgaloid complex are linked together by powerful intrinsic connections. Moreover, others have demonstrated that the various nuclei of the amygdala have undergone a more coherent evolution than other brain regions suggesting a functional integrity (Barton, Aggleton, & Grenyer, 2003). Thus, despite its complexity and its interaction with systems that mediate functions ranging from olfaction, to sexual behavior, to detecting environmental dangers, there is every reason to believe that its components act in a coordinated fashion just as the various parts of the hippocampal formation act together to carry out episodic memory processing.
Figure 1.
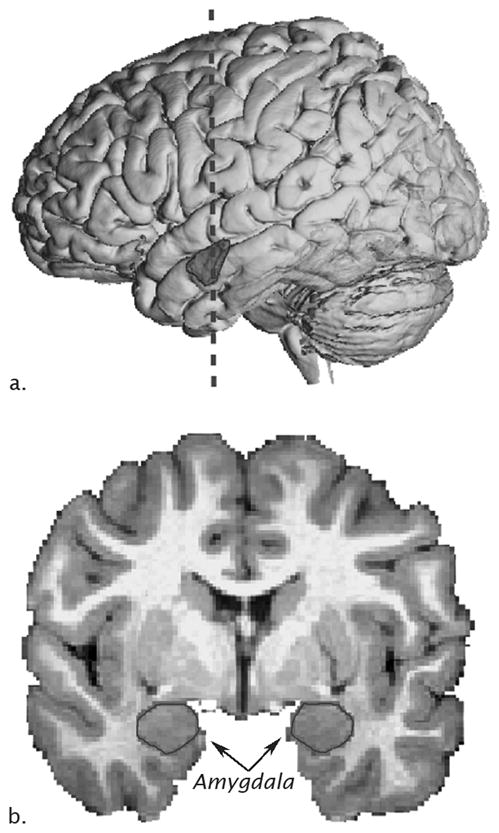
Neuroanatomy of the human amygdala on MRI. A) Lateral view of a three-dimensional reconstruction of an MRI of a human brain with amygdala outlined (dashed line represents location of coronal slide in panel B and B) MRI coronal image with amygdala outlined.
Figure 2.

Neuroanatomy of the human amygdala postmortem. A) Coronal section of human brain tissue (box around amygdala) and B) Nissl-stained section of amygdala nuclei.
The evolution of the amygdala has not been entirely worked out. This is due to the fact that not all components of the amygdala are present in all classes of animals. There is substantial evidence that a homolog of the amygdala is found in amphibia (Laberge, Muhlenbrock-Lenter, Grunwald, & Roth, 2006). Here the portions of the amygdala associated with the olfactory and autonomic systems are apparent but nuclei that would constitute the “deep” nuclei of the amygdala are not. This would suggest that a prototype of the amygdala was established some 250 million years prior to the emergence of mammals. Even within the mammalian brain, there has been substantial evolution of the structure of the amygdaloid complex. In general, as one progresses from basal insectivores to primates, there is a diminution of the olfactory regions of the amygdala (particularly those involved with the vomeronasal organ and pheromone detection) and an increase in the size of the deep nuclei, such as the basal and lateral nuclei (Stephan, Frahm, & Baron, 1987). In fact, quantitative analyses have shown that there is a much larger lateral nucleus in the human amygdala than would be expected for a nonhuman primate of comparable size (Barger, Stefanacci, & Semendeferi, 2007). Undoubtedly, this reflects the fact that the lateral nucleus is in receipt of input from the neocortex and that there has been a very large expansion of the neocortex in the human brain. But, this also makes the point that the amygdaloid complex cannot be thought of as a “primitive” or vestigial brain region since it is privy to the highest levels of sensory processing that has been afforded by the expansion of association cortex in the human brain.
In this chapter, we provide a brief overview of the development of the nonhuman primate and human amygdaloid complex. We then review data indicating the roles of the amygdala in primate development and function. We then provide an overview of data indicating that the structure and/or function of the human amygdala is compromised in a variety of neurodevelopmental disorders (Table 1). We conclude with some discussion concerning why pathology of the amygdala is such a common component of developmental neuropsychiatric disorders.
Table 1.
MRI reports of amygdala volume differences in neurodevelopmental disorders by mean age of subject population.
| Age group | Autism | Pediatric Anxiety | Pediatric Bipolar | Schizophrenia | William’s Syndrome | Fragile X |
|---|---|---|---|---|---|---|
| Young children | ↑ Mosconi ‘09 ↑ Schumann ‘09 ↑ Sparks ‘02 ↑ Schumann ‘04 |
unknown | unknown | unknown | unknown | ↓ Hazlett ‘09 — Kates ‘97 — Hoeft ‘08 |
| Adolescents | — Schumann ‘04 — Palmen ‘05 ↓ Nacewicz ‘06 |
↑ De Bellis ‘00 ↓ Milham ‘05 |
— Frazier ‘05 ↓ Blumberg ‘03 ↓ Blumberg ‘05 ↓ Chang ‘05 — Chen ‘04 ↓ DelBello ‘04 |
↑ Welch ‘09a —Giedd ‘99b ↑ Levitt ‘01b — Frazier ‘08 — Jacobsen ‘98b ↑ Velakoulis ‘10c |
— Campbell ‘09 ↑ Martens ‘09 |
↓ Gothelf ‘08 |
| Adults | ↓ Aylward ‘99 ↑ Howard ‘00 — Hazendar ‘00 ↓ Pierce ‘01 |
*** | *** | ↓ Joyal ‘03c ↓ Witthaus ‘09c — Gibbs ‘08c ↓ Exner ‘04 — Velakoulis ‘10 — Tanskanen ‘05 |
↑ Reiss ‘04 — Chiang ‘07 — Meyer Lindenberg‘04 |
↓ Moore ‘04d — Hessl ‘07d |
Only the childhood-onset or pediatric form of the disorder was included in this review, with the exception of Schizophrenia.
high-risk with psychotic and/or schizotypal symptoms,
early-onset or childhood schizophrenia,
following first-episode psychoses,
permutation carrier
Structural Development of the Primate Amygdala
Few studies have been carried out on the structural development of the human amygdala; however investigation of the nonhuman primate amygdala has provided preliminary information. There is evidence, for example, that the amygdaloid complex begins to develop early in gestation, derived from the ganglionic eminence, a thickening of the telencephalic and diencephalic proliferative zone, which protrudes into the ventricular cavity (Kordower, Piecinski, & Rakic, 1992; Muller & O’Rahilly, 2006). Neurogenesis of the macaque monkey amygdala occurs relatively early - in the first trimester of fetal development (Kordower, et al., 1992). The first amygdaloid neurons are generated around embryonic day (ED) 33 in the ~165 day macaque gestation making them among the earliest postmitotic neurons in the telencephalon, beginning first in the superficial nuclei followed by the deep nuclei. Amygdala neurogenesis peaks around ED40 and ceases by ED56.
In the human around the fifth gestational month, there are cell dense columns in the inferior portion of the amygdala within the proliferative zone that serve as migrational routes for neurons (Nikolic & Kostovic, 1986; Ulfig, Setzer, & Bohl, 1998). These columns are thought to be formed by radial glia that provide a scaffold for migrating neurons (Rakic, 1995). Humphrey (1968) noted that the superficial, more medial, nuclei are identifiable in the human embryo earlier than the deep nuclei (lateral, basal, accessory basal). The superficial nuclei and central nucleus show evidence of synaptogenesis in the fifth month, potentially due to early connections with the brainstem, whereas the deep nuclei do not show evidence of synaptogenesis until the seventh month possibly due to the strong interconnectivity with association areas in the neocortex that also develop later (Ulfig, Setzer, & Bohl, 1999, 2003). In the sixth and seventh month of fetal development, the columns lose contact with the ganglionic eminence and cellular differentiation and reorganization takes place (Setzer & Ulfig, 1999). In the eighth month of human gestation, migration has essentially ended and the cytoarchitecture closely resembles that of the mature amygdala (Humphrey, 1972; Setzer & Ulfig, 1999; Ulfig, et al., 1998, 2003).
During the early postnatal period, the macaque monkey amygdalocortical connections already closely resemble connections in the mature animals (Amaral, 2000). Likewise, adult-like projections from inferior temporal areas to both amygdala and orbitofrontal areas have been observed in one-week-old macaques (Webster, Bachevalier, & Ungerleider, 1994; Webster, Ungerleider, & Bachevalier, 1991). Though little is known regarding development of specific neurochemical systems, it appears that the distribution of opiate receptors within the amygdala is comparable to adult patterns as early as one week of age (Bachevalier, Ungerleider, O’Neill, & Friedman, 1986) and that the pattern of serotonergic innervation of the amygdala resembles the adult pattern within the first postnatal month (Prather & Amaral, 2000). While these neurochemical systems appear to develop relatively early in the primate amygdala, other systems may undergo a more delayed maturational process. For example, androgen receptors that are identified immunohistochemically in the hypothalamus as early as the end of the first trimester in the male rhesus monkey are not observed in the amygdala until the postnatal period (Choate, Slayden, & Resko, 1998).
Taken together, these neuroanatomical studies indicate that the structure of the amygdala is well-developed at the time of birth. Interestingly, human MRI studies indicate that the typically-developing amygdala continues to undergo substantial growth throughout development even into adolescence. The amygdala continues to increase in volume even at a time when the neocortex is decreasing in size. Three independent cross-sectional MRI studies have found that the amygdala increases in size from five years of age to adulthood by ~40% in typically developing males (Giedd, 1997; Giedd, Snell, & Lange, 1996; Ostby, et al., 2009; Schumann, et al., 2004). The underlying neurobiology that contributes to the protracted amygdala growth trajectory remains to be determined. Clearly much more work is needed to provide a comprehensive assessment of both prenatal and postnatal neuroanatomical development of the amygdala. There is evidence (discussed below) that in some neurodevelopmental disorders, such as autism, the amygdala undergoes an aberrant growth trajectory throughout development relative to typically developing children.
Functional Development of the Primate Amygdala
In order to evaluate the role of the amygdala in neurodevelopmental disorders, it is necessary to understand the functional role of the amygdala in typical development. A widely held view is that the amygdala is essential for learning the emotional significance of a stimulus in the environment. This is particularly true for negative emotions such as fear and anger. Both human and animal studies demonstrate amygdala involvement in the acquisition, storage, and expression of conditioned fear learning (LeDoux, 2007; Phelps & LeDoux, 2005). As reviewed elsewhere in this issue, the amygdala, through its primary role of monitoring the environment for potential danger and adjusting levels of vigilance, also plays a modulatory role in social behavior. There is extensive literature from Ralph Adolphs and colleagues on the contribution of the amygdala to a network of brain structures involved in social cognition in adults, such as recognizing emotion in faces, judging the trustworthiness of a person, and generating a sense of personal space (Adolphs, 2009, 2010; Kennedy, Glascher, Tyszka, & Adolphs, 2009).
Although it is clear that the amygdala serves as an important structure for emotional learning and response, the precise function of the amygdala during early development is not well established. Children need to learn early in development to identify potential dangers by monitoring the responses of their caregivers and eventually making these determinations on their own. The amygdala is particularly recruited during the initial period of learning when associations are ambiguous (LaBar, Gatenby, Gore, LeDoux, & Phelps, 1998), which is often the case early in development. Advances in functional neuroimaging techniques have allowed initial studies of the functional development of the human amygdala. These studies are still limited to late childhood due to methodological limitations. Some of these limitations can be circumvented by studying non-human primates and they thus provide a useful tool for understanding the early functional development of the amygdala. Below we discuss relevant functional imaging studies of the intact, typically-developing human amygdala and lesion studies in the human and non-human primate in the context of development.
Amygdala lesions in humans early in life produce impairments in identifying the emotion in facial expressions, particularly those depicting fear (Adolphs, Tranel, Damasio, & Damasio, 1994). However a lesion suffered in adulthood produces a less severe impairment (Hamann & Adolphs, 1999). These studies indicate that the amygdala may be critical for learning to judge emotions and determining the safety of a situation; this role may be diminished later in development once those associations have been made. Functional imaging studies in children and adolescents also support the notion that the amygdala is recruited early in development to form relationships of stimuli that may or may not be dangerous (Monk, et al., 2003). As in adults, children and adolescents recruit the amygdala to interpret the emotion in facial expressions, albeit with a different pattern of activity (Baird, et al., 1999; Killgore, Oki, & Yurgelun-Todd, 2001; Thomas, Drevets, Whalen, et al., 2001). Thomas and colleagues (Thomas, Drevets, Whalen, et al., 2001) found that in children, the amygdala is more active in response to neutral facial expressions than to those depicting fear. This suggests that the amygdala plays a dynamic role in learning to interpret emotions when the stimuli may be ambiguous in early development. Additional evidence that the amygdala may play an enhanced role in early development comes from studies of theory of mind, the ability to attribute mental states to others, in children and adults with amygdala lesions. Shaw and colleagues (Shaw, et al., 2004) found that subjects who acquired damage either congenitally or early in development displayed deficits in understanding the emotional states and attributing the beliefs of others. Subjects who acquired damage to the amygdala in adulthood, by contrast, were not impaired on theory of mind tasks.
Nonhuman primate research has benefited from the ability to directly record from neurons in awake, behaving animals. Electrophysiological recordings from adult macaques indicate that neurons responding to faces, body movements, eye gaze etc. are found throughout the amygdala (Brothers & Ring, 1993; Brothers, Ring, & Kling, 1990; Leonard, Rolls, Wilson, & Baylis, 1985; Rolls, 1984). What is not known is whether these response properties are already established at birth or if social experience plays a role in developing these highly selective response properties. Unfortunately, little is known regarding how and when the amygdala becomes specialized for processing facial information because of the many challenges of conducting this type of research in infant monkeys (Rodman, 1994).
At present, lesion research in nonhuman primates provides the most commonly utilized approach to evaluating the developmental role of the primate amygdala. The logic for these studies is that if a brain region plays an obligatory role in a particular function, then the loss of that brain region should substantially alter the expression of the function. The first studies of neonatal amygdala damage in nonhuman primates reported few changes in behavior (Kling & Green, 1967) or changes in fear-related behaviors (Thompson, 1968; Thompson, 1981; Thompson, Bergland, & Towfighi, 1977; Thompson, Schwartzbaum, & Harlow, 1969). More recently, Bachevalier and colleagues (Bachevalier, 1994) re-evaluated the effects of medial temporal lobe damage in neonatal rhesus monkeys. These studies included a group of six neonates that received bilateral aspiration lesions of the amygdala, resulting in damage to the amygdala, piriform cortex, rostral portion of entorhinal cortex and inferior temporal cortical area TE (Bachevalier, Alvarado, & Malkova, 1999). When placed in social pairs at two and six months of age, the neonatal amygdala-lesioned infants showed less overall activity, exploration of the testing environment and initiation of social behavior as compared to age-matched controls. Although this finding was consistent with a proposed role of the amygdala in social development, it was possible that the behavioral changes in these monkeys may have been influenced by unintended collateral damage of neural tissue surrounding the amygdala and/or by the restricted rearing practices that were employed.
To address this question, we conducted a series of experiments to evaluate the effects of neurotoxic amygdala damage in maternally and socially-reared rhesus monkeys (Bauman, Lavenex, Mason, Capitanio, & Amaral, 2004a, 2004b; Prather, et al., 2001). We found that macaque monkeys that are reared by their mothers in a social environment and receive discrete, amygdala lesions at two weeks of age do not demonstrate profound impairments in social development within the first year of life. The amygdala-lesioned monkeys were able to produce and respond to a variety of species-typical social signals and did not differ from control monkeys in the amount of their social interactions (Bauman, et al., 2004a, 2004b). The amygdala-lesioned monkeys did, however, show abnormal fear responses to both social and nonsocial stimuli (e.g., heightened fear of conspecifics and absence of fear to normally aversive objects) (Bauman, et al., 2004b; Prather, et al., 2001). Thus, neonatal lesions of the macaque amygdala resulted in a sparing of species-typical social behavior, while profoundly impacting fear processing abilities. The collective results from the studies described above indicate that the amygdala is not essential for the early development of fundamental components of social behavior (i.e., the ability to produce and respond to species typical social behaviors and engage in social interactions with conspecifics). The current research does, however, suggest that the amygdala has a significant modulatory role on social behavior, especially in potentially threatening contexts. For humans and other group-living primates, the ability to rapidly and accurately evaluate social signals for signs of impending danger is an essential social skill that must be acquired early in development. If was assume that this ability is dependent upon a properly functioning amygdala, then it is reasonable to speculate that amygdala pathology may have a profound influence on aspects of social behavior. For further reading on the various theoretical perspectives of amygdala function, the reader is referred to the book, The Human Amygdala Ed. Whalen and Phelps (2009) which provides several excellent reviews.
The link of amygdala dysfunction to neurodevelopmental disorders
Given this short overview on the structure, development and function of the primate and human amygdaloid complex, we now turn to evidence that the amygdala is pathological in a variety of neurodevelopmental disorders. Interestingly, in autism where data are most extensive, it is the trajectory of amygdala development rather than the ultimate size that seems to be most perturbed. As we will review, it is not clear what leads to the altered developmental growth curve i.e. what are the neurobiological substrates? At this point, it is equally likely that this is accounted for by phenomena as diverse as abnormal development of connections or abnormal proliferation of glial cells. We will review data from structural magnetic resonance imaging (MRI) studies across several disorders. These were selected based on two criteria: 1) that the disorder was clearly developmental in origin and 2) that there is replicated evidence that the amygdala is pathological or dysfunctional. Though the results are often disparate and it is not totally clear whether this is due to the inherent heterogeneity in the conditions or differences is methodology. Nonetheless, the data highlight the point that the amygdala is very commonly associated with several neurodevelopmental disorders.
Alterations in Amygdala Development - Neurodevelopmental Disorders
Autism
Autism is a behaviorally-defined severe disorder of neural development affecting one in 110 children and more likely to occur in males than females at a ratio of 4:1 (Rice, 2006). The etiology(ies) of autism remains unknown and the neuropathology has not yet been clearly established. In toddlers, some of the first signs of autism are unusual affective behavior, reduced social interest, and poor eye contact (Dawson, Meltzoff, Osterling, & Rinaldi, 1998; Werner, Dawson, Osterling, & Dinno, 2000). By three years of age, a child may be given a diagnosis of autism if they display three core behavioral features: (i) impairments in reciprocal social interactions; (ii) abnormal development and use of language; and (iii) repetitive and ritualized behaviors and a narrow range of interests (American Psychiatric Association, 1994). In addition to the core features of autism, there are common co-morbid neurological disorders (Amaral, Schumann, & Nordahl, 2008), such as epilepsy and anxiety (Bishop, 2007; Pitkanen, Tuunanen, Kalviainen, Partanen, & Salmenpera, 1998).
The amygdala has long been a site of intense interest in the search for neuropathology in the brains of people with autism, given its role in modulating anxiety levels and social behavior. However, the heterogeneity and unknown etiologies of autism, as well as the varying methods employed across laboratories on subjects of different ages, has made it difficult to find consistent results across studies of amygdala volume. A recent pattern has emerged that the age of the subject population studied is a critical factor in determining outcome (Schumann, et al., 2004). Four studies of toddlers and young children with autism have found the amygdala to be enlarged by ~15% relative to age-matched controls (Mosconi, et al., 2009; Schumann, Barnes, Lord, & Courchesne, 2009; Schumann, et al., 2004; Sparks, et al., 2002). However, studies of older adolescents, adults, or a wide age range of subjects, have found either no difference in (Haznedar, et al., 2000) or even smaller (Aylward, et al., 1999; Nacewicz, et al., 2006; Pierce, Müller, Ambrose, Allen, & Courchesne, 2001) amygdala volumes in individuals with autism. In addition, the amygdala continues to grow in size throughout adolescence in typically-developing male children (Giedd, 1997; Giedd, Snell, Lange, & al., 1996), but this growth pattern does not seem to take place in male children with autism (Schumann, et al., 2004). Recent evidence from children early in development suggests considerable heterogeneity in amygdala growth among children with autism and it is possible that subgroups may emerge as larger populations are studied (Nordahl, Scholz, Simon, Rogers, & Amaral, 2009). Therefore, on average, the amygdala appears to be initially larger than normal in children with autism, but does not undergo the same preadolescent increase in volume that takes place in typically-developing children. This hypothesis parallels the general theory of early brain overgrowth (Courchesne, et al., 2001; Courchesne, et al., 2007), although the aberrant growth trajectory of the amygdala may extend to a later age (Schumann, et al., 2004).
Sparks and colleagues were the first to suggest that the amygdala may be enlarged in young children with autism at 36–56 months of age (Sparks, et al., 2002). In this same cohort of children, Munson et al. (Munson, et al., 2006) found that the amygdala enlargement in the children with autism was associated with more severe social and communication impairments (based on the ADI and Vineland) and poorer outcome at 6 years of age. In a recent longitudinal study of children at 2 and 4 years of age, bilateral amygdala enlargement was also observed in children with autism compared to a control group that consisted of typical and developmentally-delayed children (Mosconi, et al., 2009).
Schumann, Courchesne and colleagues (Schumann, et al., 2009) recently carried out a prospective study to measure amygdala volume in toddlers at risk for autism at the age of first clinical detection compared to age-matched typically-developing children. The children underwent an MRI around 2.5 years of age and returned at 4 years of age for clinical diagnosis and testing. They found further evidence that the amygdala is enlarged in young children with autism and that the overgrowth begins before 3 years of age at about the time symptoms become clinically evident. As found in the Sparks (Sparks, et al., 2002) and Mosconi (Mosconi, et al., 2009) studies, the amygdala was disproportionately enlarged relative to total cerebral volume at 3 years of age. This study differed from previous studies on amygdala volume in that one of the primary goals was to characterize the neuropathological and behavioral profiles of males and females with autism independently. Strikingly, amygdala enlargement in females with autism was found to be more pronounced, compared to age- and gender-matched typically-developing counterparts, than in males with autism. In male, but not female, toddlers who later received a diagnosis of autism, the degree of amygdala enlargement at 3 years of age was also associated with the severity of the child’s social and communication impairments at final clinical evaluation at ~4 years of age. In contrast, in adolescents, Nacewicz and colleagues (Nacewicz, et al., 2006) found that a smaller amygdala volume was associated with gaze avoidance and more severe behavioral impairments as measured with the ADI-R. This relationship is the opposite pattern of that observed in younger children in both the Munson et al (Munson, et al., 2006) and Schumann et al. (Schumann, et al., 2009) studies. One might speculate that individuals with autism that demonstrate early overgrowth and more severe behavioral impairments may ultimately demonstrate reduced amygdala size later in adulthood; this notion awaits validation from a longitudinal study on brain growth throughout development in people with autism.
No study to date has evaluated younger postmortem cases with autism to determine what cellular properties account for the early overgrowth in volume. But, there is some evidence that a decreased number of neurons may be contributing to the reduction in amygdala volume later in adulthood. We carried out a neuropathological study of the brain in individuals with autism using unbiased stereological methods to measure the number and size of neurons in the entire amygdaloid complex and in individual nuclei in nine male postmortem cases of autism, without seizure disorder, compared to ten typically-developing age-matched male controls ranging in age from 10–44 years at death (Schumann and Amaral, 2006). We found that the autism group had significantly fewer neurons in the total amygdala and in the lateral nucleus than the controls. Whereas the average number of neurons in the control amygdala was about 12.2 million, the average in the amygdala in the autism cases was about 10.8 million or about 85% of the total in the control brains.
Taken together with the findings from previous magnetic resonance imaging and postmortem studies, the autistic amygdala shows multiple signs of pathology. In general, it appears to undergo an abnormal pattern of postnatal development that includes precocious enlargement. In the adult, the overall volume of the amygdala may not be different in individuals with autism (although it may be smaller) but may contain fewer neurons. It will be important to determine whether decreased neuronal numbers is unique to the amygdala or occurs in other brain regions as well.
How might pathology in the amygdala relate to specific behavioral abnormalities in autism? An emerging hypothesis is that the amygdala plays a role in mediating or directing visual attention (Adolphs, et al., 2005; Grelotti, Gauthier, & Schultz, 2002; Pierce, Haist, Sedaghat, & Courchesne, 2004; Schultz, 2005). Dalton and colleagues (Dalton, et al., 2005) carried out a series of studies utilizing functional imaging and eye-tracking technology simultaneously while showing subjects familiar and unfamiliar faces. They found that the amount of time persons with autism spent looking at the eye region of the face was strongly positively correlated with amygdala activation, but not in typically-developing control subjects. Two possible theories may explain these findings. The first is that the normal function of the amygdala is to direct attention, based on motivation, to attend to the eye region of the face and that individuals with autism are simply less motivated to look at the eyes. Pierce and colleagues (2001) initially found reduced amygdala activation when individuals with autism viewed faces depicting fear. However, when autistic subjects viewed familiar faces, they were able to activate the amygdala appropriately in response to familiar and unfamiliar faces (Pierce, et al., 2004), suggesting that the familiar faces may have enhanced motivation or attention to all of the stimuli. A second emerging theory is that individuals with autism perceive social interactions as threatening, and therefore avoid socialization as a means of alleviating the fear that it triggers. Given the amygdala’s role in fear and anxiety, one would predict heightened amygdala activation during eye contact in persons with autism, as found in the Dalton et al., 2005 study. This may be due to a heightened emotional, or even fearful, response when autistic individuals look at another person’s eyes, regardless of whether they are familiar or a stranger. As mentioned earlier, Nacewicz et al. (Nacewicz, et al., 2006) found that individuals with autism (8–25 years of age) who had a smaller amygdala also spent the least amount of time fixating on the eye region of the face. Spezio et al. (Spezio, Adolphs, Hurley, & Piven, 2007) confirmed that individuals with autism show less fixation on the eyes and mouth, but also a greater tendency to saccade away from the eyes when information was present in those regions. Recently, Kleinhans and colleagues (Kleinhans, et al., 2009) found evidence of reduced amygdala habituation to faces in their autism spectrum disorder group and the degree of reduction was related to the severity of their social impairments. This study lends further support to the theory that the amygdala is hyper-aroused in people with autism in response to socially relevant stimuli. These studies provide insight into the aberrant manner in which people with autism view faces, which is, in turn, likely related to their heightened anxiety due to abnormal amygdala activation.
Anxiety/Social Phobia
Pediatric anxiety disorders, including social phobia, separation anxiety and generalized anxiety disorder (GAD) represent the most common forms of childhood psychopathology (Klein & Pine, 2002). Behavioral inhibition is an early identifiable temperament characterized by a tendency to withdraw in novel social situations (Kagan, Reznick, Snidman, Gibbons, & Johnson, 1988) that may predispose individuals to develop anxiety disorders (Kagan, Snidman, McManis, & Woodward, 2001; Perez-Edgar & Fox, 2005). Given the association between behavioral inhibition and the subsequent emergence of anxiety disorders, we will discuss amygdala pathology associated with both of these conditions.
The neurobiology of adult onset anxiety disorders is complex, and may not accurately reflect the pathophysiology of pediatric anxiety disorders (Charney & Deutch, 1996). Consequently, we will focus our discussion on studies from pediatric populations with anxiety. Using volumetric MRI analysis, De Bellis et al. demonstrated that children and adolescents with GAD (mean age 12.7 years) had larger right and total amygdala volume as compared to age-matched controls (De Bellis, et al., 2000). A similar pattern of amygdala enlargement has recently been reported in adolescents (average age 14.7 years) who showed characteristics of behavioral inhibition earlier in childhood (Hill, Tessner, Wang, Carter, & McDermott, 2010). However, not all studies have reported increases in amygdala volume in individuals with pediatric anxiety or behavioral inhibition. For example, a voxel-based morphometry analysis that included children with separation anxiety, social phobia or GAD (mean age 12.9 years) reported a significant grey matter volume reduction within the left amygdala of children with a pediatric anxiety disorder compared to healthy controls (Milham, et al., 2005). The authors highlighted the fact that pediatric anxiety disorders are heterogeneous conditions, and although changes in amygdala structure may be present, the changes depend on the specific diagnoses of the participants.
Research in animal models has established the amygdala to be a central component of fear processing in the brain and these findings have shaped our understanding of human disorders of fear (LeDoux, 2000). In human populations with anxiety, hyperactivation of the amygdala is thought to underlie fear responses to innocuous stimuli incorrectly perceived as threatening (Pine, 2007). Indeed, a number of studies have found that adult anxiety disorders are associated with atypical amygdala activation (Kilts, et al., 2006; Phan, Fitzgerald, Nathan, & Tancer, 2006; Rauch, et al., 2000; Stein, Goldin, Sareen, Zorrilla, & Brown, 2002; Straube, Mentzel, & Miltner, 2006; Yoon, Fitzgerald, Angstadt, McCarron, & Phan, 2007). However, as described above, the developing amygdala appears not to respond to threatening stimuli in the same way as in the adult (Monk, et al., 2003; Thomas, Drevets, Whalen, et al., 2001). Thus, in order to understand the neurobiology of pediatric anxiety, it is essential to evaluate younger populations. Using functional imaging, Thomas et al. reported that anxious children show heightened amygdala activity in response to fearful faces compared with healthy children (Thomas, Drevets, Dahl, et al., 2001). McClure and colleagues further evaluated the issue of amygdala hyperactivation in pediatric generalized anxiety by controlling for attention states of the participants (McClure, Monk, et al., 2007). They found that anxious adolescents displayed right amygdala hyperactivity to fearful faces compared to happy faces, but only when attention was directed to an evaluation of fear (i.e., rating “how afraid are you?”), but not during other attention states (i.e., rating “how wide is the nose?”). The finding of hyperactivation of the amygdala during internal representation of fear was recently replicated in a study comparing depressive versus anxious adolescents (Beesdo, et al., 2009). Reports of amygdala hyperactivation are also found in studies attempting to replicate more naturalistic social settings. Compared to controls, anxious adolescents are more likely to perceive unfamiliar peers as less likely to want to interact with them and show hyperactivation of the amygdala when viewing seemingly innocuous stimuli such as smiling peers (Guyer, et al., 2008).
Amygdala hyperactivation has also been reported in experimental paradigms that do not require conscious perception of the stimuli. Monk and colleagues utilized “masked” emotional (angry, happy) or neutral faces presented for 17 milliseconds and found that anxious adolescents showed greater right amygdala activation relative to controls (Monk, et al., 2008). A functional connectivity analysis revealed that the heightened amygdala response in the GAD individuals occurred in the absence of compensatory activation in ventrolateral prefrontal cortex observed in control participants. Another fMRI study comparing anxious and healthy adolescents utilized a similar paradigm, but presented the images for 500 milliseconds (Monk, et al., 2006). In this scenario, the anxious adolescents did not differ from controls in patterns of amygdala activation, but they did show hyperactivity in the ventrolateral prefrontal cortex. During the 500 millisecond presentation, the prefrontal cortex activity was negatively correlated with severity of anxiety. In contrast, during the 17 millisecond presentation paradigm, amygdala activity was positively correlated with severity of anxiety. Taken together, the authors suggest that in pediatric anxiety, amygdala activation reflects processes associated with the actual anxiety, while activation of the prefrontal cortex reflects modulation of the amygdala and the emotional response (Pine, Guyer, & Leibenluft, 2008).
A similar pattern of amygdala hyperactivation has been reported in studies of children identified as behaviorally inhibited. Schwartz et al. found greater amygdala activation to emotionally neutral faces in young adults who had been identified as having high levels of behavioral inhibition early in development (Schwartz, Wright, Shin, Kagan, & Rauch, 2003). Likewise, behaviorally inhibited adolescents show exaggerated amygdala activity when asked to rate subjective fear states (Perez-Edgar, et al., 2007), though the behaviorally inhibited individuals show this response across all emotional stimuli, while adolescents with GAD showed heightened amygdala activity in response to fearful and angry faces (McClure, Monk, et al., 2007).
Collectively, these studies indicate that the amygdala is structurally and functionally abnormal in cases of pediatric anxiety, and possibly in children who have not developed anxiety but do have a behaviorally inhibited temperament. Interestingly, the alteration of amygdala activity are similar to what has been seen in children with autism. This has prompted us to suggest that the amygdala pathology in autism may be more associated with the abnormal fear and anxiety that is a common co-morbid symptom of autism (Schumann & Amaral, 2009). In addition to serving as a possible biomarker for pediatric anxiety, heightened amygdala activity may also provide predictive information regarding treatment response (Maslowsky, et al., 2010; McClure, Adler, et al., 2007). However, the relationship between amygdala activation and pediatric anxiety is likely to be complicated by the presence or absence of a number of other risk factors, including genetic influences such as the serotonin transporter (Lau, et al., 2009).
Childhood bipolar
The hallmark of bipolar disorder is mood dysregulation, which typically includes manic episodes, marked by high energy and anxiety, and depressive episodes, marked by feelings of hopelessness and isolation, that severely interfere with daily function (APA, 2000). Depression and mania can co-occur, or rapidly alternate, often resulting in paranoia; some severe manic episodes can lead to schizophrenia-like psychosis. The diagnosis of bipolar disorder in children is controversial. In fact, the APA’s DSM-IV current criterion for diagnosis is limited to adult-onset (APA, 2000). Nonetheless, population studies report that 1% of children may be considered bipolar (Hudziak, Althoff, Derks, Faraone, & Boomsma, 2005; Moreno, et al., 2007), although the etiology and neurobiology of childhood versus adult onset may differ (Garrett & Chang, 2008; Kato, 2007).
Children given a diagnosis of bipolar disorder demonstrate impaired recognition of emotions in facial expressions and social function, which are often linked to amygdala dysfunction (Jacobsen, et al., 1998; McClure, Pope, Hoberman, Pine, & Leibenluft, 2003; McClure, et al., 2005). MRI studies in bipolar children consistently show a 10–16% decrease in amygdala volume compared to typically developing children (Blumberg, et al., 2003; Chang, et al., 2005; Chen, et al., 2004; DelBello, Zimmerman, Mills, Getz, & Strakowski, 2004; Dickstein, et al., 2005; Frazier, et al., 2005; Garrett & Chang, 2008). There is some evidence that amygdala volume decreases further with age (Doty, et al., 2008). One small longitudinal study found 10 adolescents with bipolar disorder had a decrease in amygdala volume over a 2-year period (Blumberg, Fredericks, et al., 2005) that did not occur in typically developing adolescents. However, it is difficult to determine whether the decrease in amygdala volume is a primary feature of bipolar disorder or is a consequence of the high level of stress associated with the disorder, which may have a neurotoxic effect (Post & Leverich, 2006). Postmortem studies of children with bipolar disorder are scarce. However there is some evidence that the smaller amygdala volume may be due to a reduction in glial cell density (Bowley, Drevets, Ongur, & Price, 2002).
The consistent finding of reduced amygdala volumes in children is in stark contrast to studies of adult-onset bipolar disorder, which typically report that the amygdala is enlarged (Arnone, et al., 2009). Collectively, these studies suggest that child- and adult-onset bipolar disorders may have different etiologies. It is also difficult to interpret findings because the psychotropic medications that patients often take for bipolar disorder such as lithium may actually have neuroprotective effects that could alter amygdala size (Moore, et al., 2000). Adults, following their first-episode of mania and no psychotropic exposure, had decreased amygdala volume compared to typical controls (Rosso, et al., 2007). However, another study of bipolar adults with psychotropic exposure reported larger amygdala volumes relative to typical adults (Foland, et al., 2008). Chang et al. (Chang, et al., 2005) found that children with bipolar disorder and no psychotropic drug exposure had decreased amygdala volumes compared to typically-developing controls; however those with psychotropic drug exposure had amygdala volumes that did not differ from control. All of these findings are consistent with the notion that psychotropic drug administration over time leads to an increased size of the amygdala.
Among the few functional imaging studies that have been carried out in children with bipolar disorder, the amygdala seems to be hyperactive when the subject is asked to rate faces depicting fear and anger (Chang, Wagner, Garrett, Howe, & Reiss, 2008; Pavuluri, O’Connor, Harral, & Sweeney, 2007; Rich, et al., 2006). Chang and colleagues (2008) reported that adolescents with bipolar disorder display increased amygdala activation while viewing negatively-valenced pictures. Amygdala hyperactivation is also commonly seen in adults while performing a variety of emotional and face processing tasks (Altshuler, et al., 2005; Blumberg, Donegan, et al., 2005; Pavuluri, et al., 2007; Yurgelun-Todd, et al., 2000), although mood stabilizing medications may suppress hyperactivity (Blumberg, Donegan, et al., 2005). Increased amygdala activity appears to be a consistent finding in patients with bipolar disorder regardless of age, duration of illness, or amygdala size (A. Garrett & Chang, 2008).
Collectively, structural and functional imaging studies in children with bipolar disorder suggest that the amygdala is pathological, but differs from the structural pathology found in adults with the disorder. It remains unclear whether the child- and adult-onset forms are in fact similar or different disorders. Future studies of children with bipolar disorder would benefit from a longitudinal design to determine if there is a relationship between amygdala size and either the age of onset or duration of psychotropic drug exposure.
Schizophrenia
Schizophrenia is characterized by disorganized thought and an abnormal perception of reality which results in severe social and occupational dysfunction (APA, 2000). The diagnosis is based on the presence of “positive” symptoms including hallucinations and delusions and “negative” symptoms including flattened affect and disorganized speech. Patients also exhibit impairments in emotion perception and social cognition (Hooker & Park, 2002). There are behaviorally-defined subtypes of schizophrenia in which some patients exhibit marked paranoia, catatonia, disorganized behavior, or some combination thereof (APA, 2000).
Although typically not diagnosed until late adolescence or early adulthood, schizophrenia is increasingly considered a psychiatric disorder with neurodevelopmental origins (Lewis & Levitt, 2002; Murray, et al., 2004; Weinberger, 1987). In addition to genetic risk (Walsh et al., 2008; The International Schizophrenia Consortium, 2008), obstetric events (Clarke, Harley, & Cannon, 2006) such as maternal malnourishment (Susser, et al., 1996), stress (Huttunen & Niskanen, 1978), or infection (Davies, Welham, Chant, Torrey, & McGrath, 2003) in utero, may be contributory factors to developing the disease. Prospective studies have documented developmental delays and diagnoses of pervasive developmental disorder (PDD) in children prior to the onset of psychosis (Done, Crow, Johnstone, & Sacker, 1994; Isohanni, et al., 2000; P. Jones, Rodgers, Murray, & Marmot, 1994) and a specific pattern of impairments in neuromotor, receptive language, and cognitive development are seen in children who later develop schizophrenia (Cannon, et al., 2002).
To date, the extreme heterogeneity within the disorder and the often late-onset of behavioral symptoms has hampered the establishment of a consistent etiology or pathology for schizophrenia. There is good reason to suspect, from the behavioral perspective, that the amygdala may be pathological in schizophrenia given that patients are impaired in their ability to appropriately appraise and monitor emotional stimuli, including threatening or aversive stimuli, and anxiety is a common feature of the disorder. Patients show aberrant scan paths while viewing faces depicting strong emotion (Loughland, Williams, & Gordon, 2002) and tend to mislabel neutral or ambiguous stimuli as threatening (Holt & Phillips, 2009), suggesting that they have an increased sensitivity to threat (Green, Williams, & Davidson, 2003) which may be related to psychotic symptoms (Phillips et al., 2000; Taylor et al., 2002).
Recent evidence from longitudinal studies at the National Institute of Mental Health (NIMH) indicates that the etiology of childhood-onset schizophrenia may differ from the adolescent or adult-onset form (Addington & Rapoport, 2009). Children with schizophrenia who are diagnosed prior to 13 years of age show a progressive loss of cortical gray matter through the adolescent years that exceeds what is seen in typically developing adolescents (Arango & Kahn, 2008; Gogtay, et al., 2004; Thompson, et al., 2004). Reports of abnormalities in amygdala volume and function have been much less consistent but, similar to cortical abnormalities, seem to depend on time of onset of the disorder (see table 1). As with autism, the amygdala in schizophrenic patients may undergo an abnormal growth trajectory rather than remain consistently larger or smaller than controls. There is a trend for patients at high-risk for childhood or early-onset schizophrenia to have an enlarged amygdala during adolescence (Levitt, et al., 2001; Welch, et al., 2009). However, adults with chronic schizophrenia tend to show a 5–10% reduction in amygdala volume relative to controls (Ellison-Wright, Glahn, Laird, Thelen, & Bullmore, 2008; Exner, Boucsein, Degner, Irle, & Weniger, 2004; Honea, Crow, Passingham, & Mackay, 2005; Joyal, et al., 2003; Lawrie & Abukmeil, 1998; Nelson, Saykin, Flashman, & Riordan, 1998; Witthaus, et al., 2009; Wright, et al., 2000). Though, it is important to point out that almost half of the structural MRI studies carried out to date have not found a difference in amygdala volume at any age or stage of the disorder (Frazier, et al., 2008; Gibbs, et al., 2008; Giedd, et al., 1999; Jacobsen, et al., 1998; Tanskanen, et al., 2005).
Velakoulis and colleagues (Velakoulis, et al., 2006) reported amygdala enlargement in patients who recently experienced their first episode of psychosis, but not in those with chronic schizophrenia; the latter had been taking neuroleptic medications for several years. Two other studies reported a reduction in amygdala volume following an initial episode of psychosis (Joyal, et al., 2003; Witthaus, et al., 2009). These studies raise the possibility that the variability in findings may be due to when in development the onset of behavioral symptoms occur. In the study by Velakoulis (Velakoulis, et al., 2006), patients with schizophrenia experienced their first episode of psychosis in late adolescence, whereas in the study by Joyal (Joyal, et al., 2003) and by Witthaus (Witthaus, et al., 2009), the first episode of psychosis occurred when subjects were, on average, in their late 20’s. Consistent evidence of amygdala pathology has not been found in childhood schizophrenia with onset prior to 13 years of age, (Giedd, et al., 1999; Jacobsen, et al., 1998).
Postmortem studies on the amygdala in schizophrenia are scarce (Bogerts, 1984; Chance, Esiri, & Crow, 2002; Heckers, Heinsen, Heinsen, & Beckmann, 1990; Kreczmanski, et al., 2007) and generally involve a wide age range of cases with mostly adult-onset. In early postmortem studies, reductions in GABAergic levels and increases in dopamine in the amygdala were linked to psychosis (see Benes, 2010 for review). Kreczmanski, Schmitz and colleagues (Kreczmanski, et al., 2007) carried out the only postmortem study of the amygdala employing modern stereological methods to measure the volume, neuron density and total neuron number in the lateral nucleus. In their study of 13 male patients with schizophrenia ranging in age from 22 to 64 years at death and 13 age-matched controls, they found a reduced mean volume and total neuron number of the lateral nucleus of the amygdala in cases of schizophrenia. One caveat to these findings is that most patients with schizophrenia also had seizure disorder and were subjected to long-term treatment with neuroleptics, which may or may not affect amygdala pathology. It is interesting that the reduction in neuron number in the lateral nucleus of the amygdala in schizophrenia is similar to the reduced neuron numbers in the lateral nucleus reported in cases of autism (Schumann & Amaral, 2006).
Functional imaging studies to date suggest that there is no consistent pattern of amygdala dysfunction in patients with schizophrenia. Some studies have found reduced activation (Addington & Rapoport, 2009; Gur, et al., 2007; Gur, et al., 2002; Habel, et al., 2004; Hempel, Hempel, Schonknecht, Stippich, & Schroder, 2003;P. J. Johnston, Stojanov, Devir, & Schall, 2005; Paradiso, et al., 2003; Phillips, et al., 1999; Schneider, et al., 1998; Takahashi, et al., 2004; Taylor, Liberzon, Decker, & Koeppe, 2002; Williams, et al., 2004; Williams, et al., 2007) and others increased activation (Gur, et al., 2007; Hall, et al., 2008; D. J. Holt, et al., 2006; Kosaka, et al., 2002) depending on the task, the stage of the disorder studied, and whether the patient population preferentially exhibited more positive or negative symptoms. Collectively, Holt and Phillips (Holt & Phillips, 2009) suggest that the pattern of amygdala dysfunction in schizophrenia is complex and results in an impairment in appraising the level of threat in the stimulus. They note that patients tend to show increased activation to neutral or ambiguous stimuli and judge them as more threatening than do controls. Interestingly, schizophrenic patients with prominent paranoia show a tendency to mislabel neutral or ambiguous stimuli as more threatening than controls and also exhibit increased amygdala activity during this task (Phillips, Drevets, Rauch, & Lane, 2003). However, the same patient population exhibits diminished amygdala response to faces clearly depicting fear (Phillips, et al., 1999; Williams, et al., 2004; Williams, et al., 2007). Therefore it may be the uncertainty about the stimulus that leads to the elevated amygdala activation in patients with schizophrenia.
Another explanation for conflicting functional imaging findings is that, similar to autism and fragile X, people with schizophrenia avoid looking at the eye region of the face (Loughland, et al., 2002) and therefore the reduced amygdala activation noted in several studies may be because they are not looking at the same features of the face that controls are. The eye region of the face is particularly important for recognizing fear; without information from the eyes, fear can be difficult to identify (Adolphs, 2008). Patients with schizophrenia are impaired in identifying fear (Schneider, et al., 2006) and, in the majority of studies, exhibit decreased amygdala activation (Holt & Phillips, 2009). This paradox is also similar to studies of autism, in which increased amygdala activation compared to typical controls was found only when subjects were actually looking at the eye region of the face (Dalton, et al., 2005). Morris and colleagues (Morris, Weickert, & Loughland, 2009) suggest that patients with schizophrenia avoid eye contact because they find all faces more threatening.
Collectively, structural and functional imaging, as well as postmortem studies, indicate that the amygdala is pathological is schizophrenia. However, the pattern of pathology is complicated by several factors including the patient’s primary symptoms, degrees of illness severity, age of onset, illness duration, subtypes, co-morbid diagnoses, and antipsychotic medications. As mentioned above, childhood schizophrenia may have an entirely different etiology than adolescent-onset schizophrenia (Arango & Kahn, 2008) and thus potentially very different effects on brain morphology. Clearly more studies are needed of this population before firm conclusions can be drawn.
Williams Syndrome
Williams syndrome (WS) is a rare neurodevelpmental disorder resulting from a hemizygous deletion of approximately 25 genes in chromosome band 7q11.23 (Ewart, et al., 1993; Korenberg, et al., 2000). Individuals with WS have a specific craniofacial dysmorphology, as well as a variety of connective or soft tissue disorders, including cardiac abnormalities (Tassabehji, et al., 1999). In addition to these physical features, individuals with WS demonstrate a distinctive profile of cognitive, social and emotional abilities and impairments. While most individuals with WS have mild to moderate intellectual impairment (Bellugi, Lichtenberger, Jones, Lai, & St George, 2000), they demonstrate relative strengths in certain aspects of language and memory despite pronounced deficits in visuospatial skills (Mervis & John, 2010; Mervis & Klein-Tasman, 2000). Another defining feature of WS is hypersociability, characterized by uninhibited social interests and overly emotional and empathetic reasoning (W. Jones, et al., 2000). In spite of this strong social drive to interact with others (Doyle, Bellugi, Korenberg, & Graham, 2004), individuals with WS often have difficulty maintaining peer relationships (Laws & Bishop, 2004). Interestingly, the majority of WS individuals present with at least one anxiety disorder (Dykens, 2003; Leyfer, Woodruff-Borden, & Mervis, 2009; Woodruff-Borden, Kistler, Henderson, Crawford, & Mervis, 2010), though anxious behaviors are typically manifested in non-social situations (Einfeld, Tonge, & Florio, 1997).
Williams Syndrome provides a unique opportunity to study gene-brain-behavior relationships in a human condition with a known genetic basis paired with a well characterized and distinctive behavioral and cognitive profile (Meyer-Lindenberg, Mervis, & Berman, 2006). Neuroanatomical studies are a logical place to begin this endeavor. Unfortunately, there are very few comprehensive assessments of postmortem brain tissue available from individuals with WS and the cases that have been examined focus primarily on cortical neuropathology (Galaburda, Holinger, Bellugi, & Sherman, 2002; Galaburda, Wang, Bellugi, & Rossen, 1994). A single postmortem case study indicated that the overall volume of the amygdala was decreased in a WS patient compared to controls, with the most pronounced differences localized to the dorsal portion of the lateral nucleus (Galaburda & Bellugi, 2000). Additional postmortem analyses of the WS amygdala are clearly needed to interpret these findings.
Structural MRI studies of the WS brain have consistently reported an overall decrease in absolute brain volume of WS participants compared to controls, with the most pronounced reductions in occipital and parietal regions (reviewed by (Eckert, et al., 2006; Jarvinen-Pasley, et al., 2008). However, in spite of this global decrease in brain volume, specific brain regions appear to show a relative sparing or even a disproportional increase relative to the overall brain volume. For example, using both volumetric analyses and voxel-based morphometry, Reiss et al. (Reiss, et al., 2004) reported that amygdala volume was disproportionately increased in WS participants compared to age matched controls. This study was based on a relatively large sample of 43 WS participants (mean age 28.9 years; range 12–50 years) and 40 age matched controls. Manual tracing of the amygdala by Martens et al., (Martens, Wilson, Dudgeon, & Reutens, 2009) revealed a similar disproportionate enlargement of the amygdala in a younger population of WS participants (mean age 16.9; range 8–41) compared to 22 age matched controls. However, not all volumetric studies have replicated these findings of disproportionately large amygdala volumes in individuals with WS. For example, Meyer-Lindenberg used voxel-based morphometry to examine amygdala volumes in individuals with WS who have normal IQ and found no differences in grey matter density within the WS amygdala compared to controls (Meyer-Lindenberg, et al., 2004). Chiang et al. used tensor-based morphometry to automatically detect and quantify patterns of brain volume differences in adults with WS relative to control subjects (Chiang, et al., 2007). They found a consistent 10–15% overall reduction in brain volume in individuals with WS compared to controls; however, the trend of larger right amygdala volume in WS compared to control participants was not significant. These inconsistencies are likely due to methodological and sample selection differences.
Given the paucity of studies involving children with WS, we know even less about the developmental trajectory of amygdala growth at earlier time points. While some structural abnormalities found in adults with WS have also been found in children with WS (i.e., changes in parieto-occipital grey matter) (Boddaert, et al., 2006), disproportional increase in amygdala volumes have yet to be reported in younger WS samples. For example, manual tracing measures of amygdala volumes failed to reach statistical significance between WS children (mean age 13 years; range 8–16) and age and gender matched controls (Campbell, et al., 2009).
In spite of the inconsistencies from structural imaging studies, associations have been made between amygdala volume and behaviors characteristic of WS. For example, Martens et al. reported that individuals with WS, but not controls, demonstrated a positive correlation between right amygdala volume and approachability ratings for pictures of negative faces (Martens, et al., 2009). Interestingly, this study also reported that the WS individuals used a different strategy for evaluating approachability in faces. Unlike controls, the WS participants relied on features other than the eyes and mouth to determine approachability, a finding reminiscent of the unique face processing deficits observed in individuals with bilateral amygdala damage (reviewed in Adolphs, 2010).
Functional imaging studies have also provided evidence of amygdala dysfunction in WS. Meyer-Lindenberg and colleagues evaluated amygdala response to social and nonsocial threatening stimuli (i.e., faces or scenes) in adult individuals with WS (Meyer-Lindenberg, et al., 2005). Compared to controls, the WS participants showed reduced amygdala activation to faces, but increased amygdala activity to threatening scenes. A subsequent study from this same sample of high functioning WS participants replicated the finding of amygdala hyperactivation in response to non-social fearful stimuli (Munoz, et al., 2010). The findings of hypoactivation to fearful social stimuli have also been replicated. Hass et al. reported that adult WS participants demonstrate absent or attenuated amygdala reactivity to fearful faces, compared with control participants (Haas, et al., 2009). When a subset of these individuals with WS participated in a subsequent study, they found that a greater tendency to approach strangers was associated with less left (but not right) amygdala activation in response to viewing fearful faces (Haas, et al., 2010).
Collectively, the functional imaging studies suggest that individuals with WS show increased amygdala response to threatening non-social stimuli and decreased amygdala response to threatening social stimuli. These findings are opposite the pattern of amygdala activation observed in healthy controls, but are consistent with the behavioral profile of individuals with WS. It is not, however, known when changes in amygdala structure and function emerge and what consequence these changes have on other aspects of neural development. The structural and functional changes in the WS amygdala likely alter other aspects of brain circuitry, as evidenced by a reduction in functional connectivity of face processing centers of the brain (i.e., the fusiform face area) with the amygdala and prefrontal cortex in adults with WS (Sarpal, et al., 2008). Additional studies of pediatric WS populations will play a critical role in determining the role of the amygdala in the unique behavioral profile of individuals with WS.
Fragile X
Fragile X (FXS) is the most commonly inherited form of intellectual disability, affecting approximately 1 in 4,000 males and 1 in 8,000 females (Crawford, Acuna, & Sherman, 2001). This disorder is caused by the disrupted expression of the fragile X mental retardation (FMR1) gene located on the long arm of the X chromosome (Verkerk, et al., 1991). This region includes a trinucleotide CGG repeat expansion that results in ~30 copies of the trinucleotide repeat in typically developing individuals, 50–200 repeats in pre-mutation carriers, and over 200 repeats in individuals with the full mutation. The full mutation result in hypermethylation of the FMR1 gene promoter and the subsequent reduction of FMRP protein, which in turn has a profound impact on brain development. Individuals with FXS generally have intellectual disability, with other behavioral features including gaze aversion, social difficulties, impulsivity, anxiety and hyper arousal (Reiss & Freund, 1992).
Consistent changes in the brains of individuals with the full mutation include enlargement of the caudate nucleus, a decrease of the cerebellar vermis and superior temporal gyrus volume, with less consistent changes reported in amygdala volumes (reviewed in (Hessl, Rivera, & Reiss, 2004; Lightbody & Reiss, 2009; Reiss & Dant, 2003). Using manual tracing protocols Kates et al. first reported a 10% reduction in amygdala volume in children with FXS (boys mean age 6.3 years; girls mean age 9 years); however, these volumes were not significantly different from matched controls (Kates, Abrams, Kaufmann, Breiter, & Reiss, 1997). Statistically significant decreases in amygdala volumes were, however, reported in a volumetric analysis of younger children with fragile X compared to matched controls (mean age 11.7; range 1.1–22.7) (Gothelf, et al., 2008). Brain regions that best distinguished subjects with FXS from typically developing controls included an enlarged caudate, followed by decreased posterior vermis and decreased amygdala. Of the subjects that had all three of these characteristics, 91.5% were in the FXS group, indicating that the combination of these three neuroanatomical abnormities is highly characteristic of children with FXS. Decreased amygdala volumes were also reported in a recent study comparing very young boys (18–42 months) with FXS (with and without autism) to boys with idiopathic autism (without FXS) and controls (Hazlett, et al., 2009). Manual tracing of the amygdala revealed that children with FXS had a smaller amygdala volume regardless of whether the child also met criteria for autistic disorder. However, not all studies in young children with FXS have found significant differences in amygdala volume. A recent study of 51 young boys with FXS (average age 35 months) failed to find significant differences in amygdala volumes with voxel-based or volumetric analyses (Hoeft, et al., 2008). A pattern classification analysis of these data revealed that individuals with FXS demonstrate an unusual pattern of increased grey matter volumes in the medial portion of the amygdala paired with decreased grey matter volume in the lateral amygdala. These findings indicate that structural changes in the amygdala are a common feature of FXS.
Functional MRI has also been used to examine the neural basis of behaviors associated with FXS, focusing on the pronounced gaze aversion characteristic of subjects with this disorder. Studies in adults with FXS have linked gaze aversion with abnormal activity in face processing regions of the brain (Dalton, Holsen, Abbeduto, & Davidson, 2008;A. S. Garrett, Menon, MacKenzie, & Reiss, 2004; Holsen, Dalton, Johnstone, & Davidson, 2008), though specific differences in amygdala activation were not observed in these paradigms. Functional imaging studies in younger children with FXS have, however, found differences in amygdala function. When viewing stimuli of faces with direct eye gaze, young boys with FXS show greater amygdala activation than do typically developing controls (Watson, Hoeft, Garrett, Hall, & Reiss, 2008). When shown successive images of direct gaze, the boys with FXS responded with sustained left amygdala activation, indicating that unlike controls, the FXS participants were responding with increased rather than habituated amygdala activity. Moreover, a significant negative correlation between left amygdala sensitization and task accuracy suggests that repeated exposure to direct gaze stimuli may have increased anxiety and had a negative impact on test performance in children with FXS compared to controls. The authors suggest that this pattern of greater, sustained amygdala activation in response to direct gaze may underlie the striking gaze aversion observed in FXS populations.
Although many questions remain regarding the role of amygdala function in FXS, there is compelling evidence for a reduction in amygdala volume combined with reports of amygdala dysfunction in FXS. Interestingly, there is preliminary evidence suggesting that alterations in amygdala structure and function may even be present in individuals with the permutation (Hessl, et al., 2007; Moore, et al., 2004). Thus, future studies exploring the relationship between FMRP levels and specific profiles of amygdala dysfunction may provide insight into the mechanisms of the disorder.
Conclusions
The amygdaloid complex is a brain region that emerged early in phylogeny for the main purpose of detecting dangers in the environment. Through its myriad connections with other brain regions, it is capable, once having detected a potential danger, of coordinating an appropriate response leading to escape. The response is complex and includes influencing cortical sensory processing to focus attention on the environmental threat to mobilizing bodily resources to mount an effective escape. One end result of this process is the conscious appreciation of fear upon facing an imminent threat or of anxiety in contemplating a future, potential threat. This system, which is designed to detect dangers and modulate all subsequent behavior, can have a profound influence on human activities. If the threshold is to low, normally benign aspects of the environment are perceived as dangers and interactions are limited. If the set point for danger detection is too high, risk taking is increased and inappropriate sociality may occur. Given that many of the neurodevelopmental disorders that we have reviewed involve too little or too much fear or anxiety or too little of too much social interaction, it is not surprising that the amygdala has been implicated in the pathophysiology of many of them.
Much of the evidence for pathology of the amygdala in neurodevelopmental disorders comes from rather coarse levels of analysis. To find that the amygdala is smaller or larger in individuals with a neurodevelopmental disorder does not provide much insight into the underlying mechanism for potential dysfunction. As we have reviewed above, both enlarged and decreased size of the amygdala can be associated with increased anxiety! We have also learned from studies of autism, that when during development the measurements are made is critically important. There is now substantial evidence that, during early postnatal development, the amygdala grows larger faster in a sizable group of children with autism. But, the ultimate size of the amygdala is the same as typically developing children. Thus, if one studies a group of adolescents, the size of the amygdala in individuals with autism and age-matched controls is about the same and there is no indication of the abnormal trajectory of amygdala development in the autistic individuals. Given the paucity of postmortem data that are available from young individuals with autism, there is currently no information on the cellular basis for the precocious growth of the amygdala. On the one hand, normal progressive processes, such as dendritic growth and synaptogenesis, could simply be happening at a more rapid pace. On the other hand, an abnormal process such as glial proliferation, perhaps as part of an inflammatory process, could underlie the precocious growth. Our inability to support or discount these possibilities belies the need for substantially greater effort at examining brains from individuals with autism and for developing high-resolution, noninvasive techniques for evaluating cellular and metabolic processes in the human brain.
In adults with autism, there are some data indicating that the amygdala is smaller. One can only speculate currently whether this is part of the evolving disease process or is a repercussion of earlier stages of abnormal development. If, for example, the early hypertrophy of the amygdala in autism leads to abnormal anxiety with concomitant activation of stress mechanisms, decreasing size of the amygdala could be a result of allostatic load and feedback degenerative processes as seen in long term depression (Frodl, et al., 2008; McEwen, 2005).
Functional magnetic resonance imaging studies are an important validation of amygdala pathology in neurodevelopmenal disorders. Generally speaking, hyperactivity of the amygdala is associated with increased anxiety. But, the correlation between the size of the amygdala and increased activation is not particularly clear. Moreover, given that the amygdaloid complex comprises 13 different nuclei and cortical regions with a variety of different neuronal cell types, what if any implication does an increased bold signal have for understanding the neural systems of cellular dysregulation that underlies the behavioral alterations associated with neurodevelopmental disorders? Clearly, animal models will need to be developed to provide insight into potential targets of therapeutic interactions.
Finally, it has been known for many years that the amygdaloid complex is highly epileptogenic (Gloor, 1990; Goddard, McIntyre, & Leech, 1969). It is equally clear that the amygdala undergoes various pathological changes in individuals with epilepsy (Pitkanen, et al., 1998). Co-morbid seizure disorders are common in autism and other neurodevelopmental disorders. At least some of the variability in reported findings that we reviewed above may be due to the inconsistent inclusion of subjects with or without seizure disorders in both structural and functional MRI studies and in postmortem analyses.
The amygdala is a common site for neuropathology in neurodevelopmental disorders and is therefore a potential target for the alleviation of core or co-morbid symptoms. The speed with which therapeutic interventions will be developed and applied may depend, in part, on the availability of much more intensive information on the normal development of the amygdala both in humans and in animal models. Knowledge of genes that are preferentially expressed in the amygdala (Zirlinger & Anderson, 2003; Zirlinger, Kreiman, & Anderson, 2001) and of physiological mechanisms underlying evaluation of environmental threat, will undoubtedly suggest targets for the alleviation of abnormal fears and anxiety. While treatment of these symptoms may not deal directly with the core features of neurodevelopmental disorders, it will nonetheless improve the quality of life of affected individuals and facilitate other therapies that are more targeted at the core symptoms.
Acknowledgments
Original research reviewed here was supported by NIMH Grant MH 041479 and NINDS grant NS 16980.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addington AM, Rapoport JL. The genetics of childhood-onset schizophrenia: when madness strikes the prepubescent. Curr Psychiatry Rep. 2009;11(2):156–161. doi: 10.1007/s11920-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolphs R. Fear, faces, and the human amygdala. Curr Opin Neurobiol. 2008;18(2):166–172. doi: 10.1016/j.conb.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolphs R. The social brain: neural basis of social knowledge. Annu Rev Psychol. 2009;60:693–716. doi: 10.1146/annurev.psych.60.110707.163514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolphs R. What does the amygdala contribute to social cognition? Ann N Y Acad Sci. 2010;1191(1):42–61. doi: 10.1111/j.1749-6632.2010.05445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolphs R, Gosselin F, Buchanan TW, Tranel D, Schyns P, Damasio AR. A mechanism for impaired fear recognition after amygdala damage. Nature. 2005;433(7021):68–72. doi: 10.1038/nature03086. [DOI] [PubMed] [Google Scholar]
- Adolphs R, Tranel D, Damasio H, Damasio A. Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature. 1994;372(6507):669–672. doi: 10.1038/372669a0. [DOI] [PubMed] [Google Scholar]
- Altshuler L, Bookheimer S, Proenza MA, Townsend J, Sabb F, Firestine A, et al. Increased amygdala activation during mania: a functional magnetic resonance imaging study. Am J Psychiatry. 2005;162(6):1211–1213. doi: 10.1176/appi.ajp.162.6.1211. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Price JL, Pitkänen A, Carmichael T. Anatomical organization of the primate amygdaloid complex. In: Aggleton J, editor. The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. New York: Wiley–Liss; 1992. pp. 1–66. [Google Scholar]
- Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31(3):137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Arango C, Kahn R. Progressive brain changes in schizophrenia. Schizophr Bull. 2008;34(2):310–311. doi: 10.1093/schbul/sbm166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone D, Cavanagh J, Gerber D, Lawrie SM, Ebmeier KP, McIntosh AM. Magnetic resonance imaging studies in bipolar disorder and schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(3):194–201. doi: 10.1192/bjp.bp.108.059717. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Minshew NJ, Goldstein G, Honeycutt NA, Augustine AM, Yates KO, et al. MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology. 1999;53(9):2145–2150. doi: 10.1212/wnl.53.9.2145. [DOI] [PubMed] [Google Scholar]
- Bachevalier J. Medial temporal lobe structures and autism: a review of clinical and experimental findings. Neuropsychologia. 1994;32(6):627–648. doi: 10.1016/0028-3932(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Bachevalier J, Alvarado MC, Malkova L. Memory and socioemotional behavior in monkeys after hippocampal damage incurred in infancy or in adulthood. Biol Psychiatry. 1999;46(3):329–339. doi: 10.1016/s0006-3223(99)00123-7. [DOI] [PubMed] [Google Scholar]
- Bachevalier J, Ungerleider LG, O’Neill JB, Friedman DP. Regional distribution of [3H]naloxone binding in the brain of a newborn rhesus monkey. Brain Res. 1986;390(2):302–308. doi: 10.1016/s0006-8993(86)80240-2. [DOI] [PubMed] [Google Scholar]
- Baird AA, Gruber SA, Fein DA, Maas LC, Steingard RJ, Renshaw PF, et al. Functional magnetic resonance imaging of facial affect recognition in children and adolescents. J Am Acad Child Adolesc Psychiatry. 1999;38(2):195–199. doi: 10.1097/00004583-199902000-00019. [DOI] [PubMed] [Google Scholar]
- Barger N, Stefanacci L, Semendeferi K. A comparative volumetric analysis of the amygdaloid complex and basolateral division in the human and ape brain. Am J Phys Anthropol. 2007;134(3):392–403. doi: 10.1002/ajpa.20684. [DOI] [PubMed] [Google Scholar]
- Barton RA, Aggleton JP, Grenyer R. Evolutionary coherence of the mammalian amygdala. Proc Biol Sci. 2003;270(1514):539–543. doi: 10.1098/rspb.2002.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman MD, Lavenex P, Mason WA, Capitanio JP, Amaral DG. The development of mother–infant interactions after neonatal amygdala lesions in rhesus monkeys. J Neurosci. 2004a;24(3):711–721. doi: 10.1523/JNEUROSCI.3263-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman MD, Lavenex P, Mason WA, Capitanio JP, Amaral DG. The development of social behavior following neonatal amygdala lesions in rhesus monkeys. J Cogn Neurosci. 2004b;16(8):1388–1411. doi: 10.1162/0898929042304741. [DOI] [PubMed] [Google Scholar]
- Beesdo K, Lau JY, Guyer AE, McClure-Tone EB, Monk CS, Nelson EE, et al. Common and distinct amygdala-function perturbations in depressed vs anxious adolescents. Arch Gen Psychiatry. 2009;66(3):275–285. doi: 10.1001/archgenpsychiatry.2008.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellugi U, Lichtenberger L, Jones W, Lai Z, St George M. I. The neurocognitive profile of Williams Syndrome: a complex pattern of strengths and weaknesses. J Cogn Neurosci. 2000;12(Suppl 1):7–29. doi: 10.1162/089892900561959. [DOI] [PubMed] [Google Scholar]
- Bishop SJ. Neurocognitive mechanisms of anxiety: an integrative account. Trends Cogn Sci. 2007;11(7):307–316. doi: 10.1016/j.tics.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Blumberg HP, Donegan NH, Sanislow CA, Collins S, Lacadie C, Skudlarski P, et al. Preliminary evidence for medication effects on functional abnormalities in the amygdala and anterior cingulate in bipolar disorder. Psychopharmacology (Berl) 2005;183(3):308–313. doi: 10.1007/s00213-005-0156-7. [DOI] [PubMed] [Google Scholar]
- Blumberg HP, Fredericks C, Wang F, Kalmar JH, Spencer L, Papademetris X, et al. Preliminary evidence for persistent abnormalities in amygdala volumes in adolescents and young adults with bipolar disorder. Bipolar Disord. 2005;7(6):570–576. doi: 10.1111/j.1399-5618.2005.00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg HP, Kaufman J, Martin A, Whiteman R, Zhang JH, Gore JC, et al. Amygdala and hippocampal volumes in adolescents and adults with bipolar disorder. Arch Gen Psychiatry. 2003;60(12):1201–1208. doi: 10.1001/archpsyc.60.12.1201. [DOI] [PubMed] [Google Scholar]
- Boddaert N, Mochel F, Meresse I, Seidenwurm D, Cachia A, Brunelle F, et al. Parieto-occipital grey matter abnormalities in children with Williams syndrome. Neuroimage. 2006;30(3):721–725. doi: 10.1016/j.neuroimage.2005.10.051. [DOI] [PubMed] [Google Scholar]
- Bogerts B. Neuropathology of schizophrenias. Fortschr Neurol Psychiatr. 1984;52(12):428–437. doi: 10.1055/s-2007-1002212. [DOI] [PubMed] [Google Scholar]
- Bowley MP, Drevets WC, Ongur D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry. 2002;52(5):404–412. doi: 10.1016/s0006-3223(02)01404-x. [DOI] [PubMed] [Google Scholar]
- Brothers L, Ring B. Mesial temporal neurons in the macaque monkey with responses selective for aspects of social stimuli. Behavioural Brain Research. 1993;57(1):53–61. doi: 10.1016/0166-4328(93)90061-t. [DOI] [PubMed] [Google Scholar]
- Brothers L, Ring B, Kling A. Response of neurons in the macaque amygdala to complex social stimuli. Behav Brain Res. 1990;41(3):199–213. doi: 10.1016/0166-4328(90)90108-q. [DOI] [PubMed] [Google Scholar]
- Campbell LE, Daly E, Toal F, Stevens A, Azuma R, Karmiloff-Smith A, et al. Brain structural differences associated with the behavioural phenotype in children with Williams syndrome. Brain Res. 2009;1258:96–107. doi: 10.1016/j.brainres.2008.11.101. [DOI] [PubMed] [Google Scholar]
- Cannon M, Caspi A, Moffitt TE, Harrington H, Taylor A, Murray RM, et al. Evidence for early-childhood, pan-developmental impairment specific to schizophreniform disorder: results from a longitudinal birth cohort. Arch Gen Psychiatry. 2002;59(5):449–456. doi: 10.1001/archpsyc.59.5.449. [DOI] [PubMed] [Google Scholar]
- Chance SA, Esiri MM, Crow TJ. Amygdala volume in schizophrenia: postmortem study and review of magnetic resonance imaging findings. Br J Psychiatry. 2002;180:331–338. doi: 10.1192/bjp.180.4.331. [DOI] [PubMed] [Google Scholar]
- Chang K, Karchemskiy A, Barnea-Goraly N, Garrett A, Simeonova DI, Reiss A. Reduced amygdalar gray matter volume in familial pediatric bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(6):565–573. doi: 10.1097/01.chi.0000159948.75136.0d. [DOI] [PubMed] [Google Scholar]
- Chang KD, Wagner C, Garrett A, Howe M, Reiss A. A preliminary functional magnetic resonance imaging study of prefrontal-amygdalar activation changes in adolescents with bipolar depression treated with lamotrigine. Bipolar Disord. 2008;10(3):426–431. doi: 10.1111/j.1399-5618.2007.00576.x. [DOI] [PubMed] [Google Scholar]
- Charney DS, Deutch A. A functional neuroanatomy of anxiety and fear: implications for the pathophysiology and treatment of anxiety disorders. Crit Rev Neurobiol. 1996;10(3–4):419–446. doi: 10.1615/critrevneurobiol.v10.i3-4.70. [DOI] [PubMed] [Google Scholar]
- Chen BK, Sassi R, Axelson D, Hatch JP, Sanches M, Nicoletti M, et al. Cross-sectional study of abnormal amygdala development in adolescents and young adults with bipolar disorder. Biol Psychiatry. 2004;56(6):399–405. doi: 10.1016/j.biopsych.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Reiss AL, Lee AD, Bellugi U, Galaburda AM, Korenberg JR, et al. 3D pattern of brain abnormalities in Williams syndrome visualized using tensor–based morphometry. Neuroimage. 2007;36(4):1096–1109. doi: 10.1016/j.neuroimage.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choate JV, Slayden OD, Resko JA. Immunocytochemical localization of androgen receptors in brains of developing and adult male rhesus monkeys. Endocrine. 1998;8(1):51–60. doi: 10.1385/ENDO:8:1:51. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Harley M, Cannon M. The role of obstetric events in schizophrenia. Schizophr Bull. 2006;32(1):3–8. doi: 10.1093/schbul/sbj028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, et al. Mapping early brain development in autism. Neuron. 2007;56(2):399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3(5):359–371. doi: 10.1097/00125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton KM, Holsen L, Abbeduto L, Davidson RJ. Brain function and gaze fixation during facial-emotion processing in fragile X and autism. Autism Res. 2008;1(4):231–239. doi: 10.1002/aur.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton KM, Nacewicz BM, Johnstone T, Schaefer HS, Gernsbacher MA, Goldsmith HH, et al. Gaze fixation and the neural circuitry of face processing in autism. Nat Neurosci. 2005;8(4):519–526. doi: 10.1038/nn1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Welham J, Chant D, Torrey EF, McGrath J. A systematic review and meta-analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophr Bull. 2003;29(3):587–593. doi: 10.1093/oxfordjournals.schbul.a007030. [DOI] [PubMed] [Google Scholar]
- Dawson G, Meltzoff AN, Osterling J, Rinaldi J. Neuropsychological correlates of early symptoms of autism. Child Development. 1998;69(5):1276–1285. [PMC free article] [PubMed] [Google Scholar]
- De Bellis MD, Casey BJ, Dahl RE, Birmaher B, Williamson DE, Thomas KM, et al. A pilot study of amygdala volumes in pediatric generalized anxiety disorder. Biol Psychiatry. 2000;48(1):51–57. doi: 10.1016/s0006-3223(00)00835-0. [DOI] [PubMed] [Google Scholar]
- DelBello MP, Zimmerman ME, Mills NP, Getz GE, Strakowski SM. Magnetic resonance imaging analysis of amygdala and other subcortical brain regions in adolescents with bipolar disorder. Bipolar Disord. 2004;6(1):43–52. doi: 10.1046/j.1399-5618.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- Dickstein DP, Milham MP, Nugent AC, Drevets WC, Charney DS, Pine DS, et al. Frontotemporal alterations in pediatric bipolar disorder: results of a voxel-based morphometry study. Arch Gen Psychiatry. 2005;62(7):734–741. doi: 10.1001/archpsyc.62.7.734. [DOI] [PubMed] [Google Scholar]
- Done DJ, Crow TJ, Johnstone EC, Sacker A. Childhood antecedents of schizophrenia and affective illness: social adjustment at ages 7 and 11. BMJ. 1994;309(6956):699–703. doi: 10.1136/bmj.309.6956.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doty TJ, Payne ME, Steffens DC, Beyer JL, Krishnan KR, LaBar KS. Age-dependent reduction of amygdala volume in bipolar disorder. Psychiatry Res. 2008;163(1):84–94. doi: 10.1016/j.pscychresns.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TF, Bellugi U, Korenberg JR, Graham J. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am J Med Genet A. 2004;124(3):263–273. doi: 10.1002/ajmg.a.20416. [DOI] [PubMed] [Google Scholar]
- Dykens EM. Anxiety, fears, and phobias in persons with Williams syndrome. Dev Neuropsychol. 2003;23(1–2):291–316. doi: 10.1080/87565641.2003.9651896. [DOI] [PubMed] [Google Scholar]
- Eckert MA, Galaburda AM, Mills DL, Bellugi U, Korenberg JR, Reiss AL. The neurobiology of Williams syndrome: cascading influences of visual system impairment? Cell Mol Life Sci. 2006;63(16):1867–1875. doi: 10.1007/s00018-005-5553-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einfeld SL, Tonge BJ, Florio T. Behavioral and emotional disturbance in individuals with Williams syndrome. Am J Ment Retard. 1997;102(1):45–53. doi: 10.1352/0895-8017(1997)102<0045:BAEDII>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Ellison-Wright I, Glahn DC, Laird AR, Thelen SM, Bullmore E. The anatomy of first-episode and chronic schizophrenia: an anatomical likelihood estimation meta-analysis. Am J Psychiatry. 2008;165(8):1015–1023. doi: 10.1176/appi.ajp.2008.07101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5(1):11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- Exner C, Boucsein K, Degner D, Irle E, Weniger G. Impaired emotional learning and reduced amygdala size in schizophrenia: a 3-month follow-up. Schizophr Res. 2004;71(2–3):493–503. doi: 10.1016/j.schres.2004.02.023. [DOI] [PubMed] [Google Scholar]
- Foland LC, Altshuler LL, Sugar CA, Lee AD, Leow AD, Townsend J, et al. Increased volume of the amygdala and hippocampus in bipolar patients treated with lithium. Neuroreport. 2008;19(2):221–224. doi: 10.1097/WNR.0b013e3282f48108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier JA, Ahn MS, DeJong S, Bent EK, Breeze JL, Giuliano AJ. Magnetic resonance imaging studies in early-onset bipolar disorder: a critical review. Harv Rev Psychiatry. 2005;13(3):125–140. doi: 10.1080/10673220591003597. [DOI] [PubMed] [Google Scholar]
- Frazier JA, Hodge SM, Breeze JL, Giuliano AJ, Terry JE, Moore CM, et al. Diagnostic and sex effects on limbic volumes in early-onset bipolar disorder and schizophrenia. Schizophr Bull. 2008;34(1):37–46. doi: 10.1093/schbul/sbm120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodl TS, Koutsouleris N, Bottlender R, Born C, Jager M, Scupin I, et al. Depression-related variation in brain morphology over 3 years: effects of stress? Arch Gen Psychiatry. 2008;65(10):1156–1165. doi: 10.1001/archpsyc.65.10.1156. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Bellugi U. V. Multi-level analysis of cortical neuroanatomy in Williams syndrome. J Cogn Neurosci. 2000;12(Suppl 1):74–88. doi: 10.1162/089892900561995. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Holinger DP, Bellugi U, Sherman GF. Williams syndrome: neuronal size and neuronal-packing density in primary visual cortex. Arch Neurol. 2002;59(9):1461–1467. doi: 10.1001/archneur.59.9.1461. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Wang PP, Bellugi U, Rossen M. Cytoarchitectonic anomalies in a genetically based disorder: Williams syndrome. Neuroreport. 1994;5(7):753–757. doi: 10.1097/00001756-199403000-00004. [DOI] [PubMed] [Google Scholar]
- Garrett A, Chang K. The role of the amygdala in bipolar disorder development. Dev Psychopathol. 2008;20(4):1285–1296. doi: 10.1017/S0954579408000618. [DOI] [PubMed] [Google Scholar]
- Garrett AS, Menon V, MacKenzie K, Reiss AL. Here’s looking at you, kid: neural systems underlying face and gaze processing in fragile X syndrome. Arch Gen Psychiatry. 2004;61(3):281–288. doi: 10.1001/archpsyc.61.3.281. [DOI] [PubMed] [Google Scholar]
- Gibbs AA, Dazzan P, Morgan KD, Naudts KH, Morgan C, Hutchinson G, et al. Sexually dimorphic changes in the amygdala in relation to delusional beliefs in first episode psychosis. J Psychiatr Res. 2008;42(11):913–919. doi: 10.1016/j.jpsychires.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Giedd JN. Normal development. Child and Adolescent Psychiatric Clinics of North America. 1997;6(2):265–282. [Google Scholar]
- Giedd JN, Jeffries NO, Blumenthal J, Castellanos FX, Vaituzis AC, Fernandez T, et al. Childhood-onset schizophrenia: progressive brain changes during adolescence. Biol Psychiatry. 1999;46(7):892–898. doi: 10.1016/s0006-3223(99)00072-4. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Snell JW, Lange N. Quantitative magnetic resonance imaging of human brain development: Ages 4–18. Cerebral Cortex. 1996;6:551–560. doi: 10.1093/cercor/6.4.551. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Snell JW, Lange N, et al. Quantitative magnetic resonance imaging of human brain development: Ages 4–18. Cerebral Cortex. 1996;6:551–560. doi: 10.1093/cercor/6.4.551. [DOI] [PubMed] [Google Scholar]
- Gloor P. Experiential phenomena of temporal lobe epilepsy. Facts and hypotheses. Brain. 1990;113(Pt 6):1673–1694. doi: 10.1093/brain/113.6.1673. [DOI] [PubMed] [Google Scholar]
- Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25(3):295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- Gogtay N, Sporn A, Clasen LS, Nugent TF, 3rd, Greenstein D, Nicolson R, et al. Comparison of progressive cortical gray matter loss in childhood-onset schizophrenia with that in childhood-onset atypical psychoses. Arch Gen Psychiatry. 2004;61(1):17–22. doi: 10.1001/archpsyc.61.1.17. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O’Hara R, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Ann Neurol. 2008;63(1):40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MJ, Williams LM, Davidson D. Visual scanpaths to threat-related faces in deluded schizophrenia. Psychiatry Res. 2003;119(3):271–285. doi: 10.1016/s0165-1781(03)00129-x. [DOI] [PubMed] [Google Scholar]
- Grelotti DJ, Gauthier I, Schultz RT. Social interest and the development of cortical face specialization: what autism teaches us about face processing. Dev Psychobiol. 2002;40(3):213–225. doi: 10.1002/dev.10028. [DOI] [PubMed] [Google Scholar]
- Gur RE, Loughead J, Kohler CG, Elliott MA, Lesko K, Ruparel K, et al. Limbic activation associated with misidentification of fearful faces and flat affect in schizophrenia. Arch Gen Psychiatry. 2007;64(12):1356–1366. doi: 10.1001/archpsyc.64.12.1356. [DOI] [PubMed] [Google Scholar]
- Gur RE, McGrath C, Chan RM, Schroeder L, Turner T, Turetsky BI, et al. An fMRI study of facial emotion processing in patients with schizophrenia. Am J Psychiatry. 2002;159(12):1992–1999. doi: 10.1176/appi.ajp.159.12.1992. [DOI] [PubMed] [Google Scholar]
- Guyer AE, Lau JY, McClure-Tone EB, Parrish J, Shiffrin ND, Reynolds RC, et al. Amygdala and ventrolateral prefrontal cortex function during anticipated peer evaluation in pediatric social anxiety. Arch Gen Psychiatry. 2008;65(11):1303–1312. doi: 10.1001/archpsyc.65.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BW, Hoeft F, Searcy YM, Mills D, Bellugi U, Reiss A. Individual differences in social behavior predict amygdala response to fearful facial expressions in Williams syndrome. Neuropsychologia. 2010;48(5):1283–1288. doi: 10.1016/j.neuropsychologia.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BW, Mills D, Yam A, Hoeft F, Bellugi U, Reiss A. Genetic influences on sociability: heightened amygdala reactivity and event-related responses to positive social stimuli in Williams syndrome. J Neurosci. 2009;29(4):1132–1139. doi: 10.1523/JNEUROSCI.5324-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habel U, Klein M, Shah NJ, Toni I, Zilles K, Falkai P, et al. Genetic load on amygdala hypofunction during sadness in nonaffected brothers of schizophrenia patients. Am J Psychiatry. 2004;161(10):1806–1813. doi: 10.1176/ajp.161.10.1806. [DOI] [PubMed] [Google Scholar]
- Hall J, Whalley HC, McKirdy JW, Romaniuk L, McGonigle D, McIntosh AM, et al. Overactivation of fear systems to neutral faces in schizophrenia. Biol Psychiatry. 2008;64(1):70–73. doi: 10.1016/j.biopsych.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Hamann SB, Adolphs R. Normal recognition of emotional similarity between facial expressions following bilateral amygdala damage. Neuropsychologia. 1999;37(10):1135–1141. doi: 10.1016/s0028-3932(99)00027-5. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe MD, Lighbody AA, Gerig G, MacFall JR, Ross AK, et al. Teasing apart the heterogeneity of autism: Same behavior, different brains in toddlers with fragile X syndrome and autism. J Neurodev Disord. 2009;1:1. doi: 10.1007/s11689-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haznedar MM, Buchsbaum MS, Wei TC, Hof PR, Cartwright C, Bienstock CA, et al. Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am J Psychiatry. 2000;157(12):1994–2001. doi: 10.1176/appi.ajp.157.12.1994. [DOI] [PubMed] [Google Scholar]
- Heckers S, Heinsen H, Heinsen YC, Beckmann H. Limbic structures and lateral ventricle in schizophrenia. A quantitative postmortem study. Arch Gen Psychiatry. 1990;47(11):1016–1022. doi: 10.1001/archpsyc.1990.01810230032006. [DOI] [PubMed] [Google Scholar]
- Hempel A, Hempel E, Schonknecht P, Stippich C, Schroder J. Impairment in basal limbic function in schizophrenia during affect recognition. Psychiatry Res. 2003;122(2):115–124. doi: 10.1016/s0925-4927(02)00126-9. [DOI] [PubMed] [Google Scholar]
- Hessl D, Rivera S, Koldewyn K, Cordeiro L, Adams J, Tassone F, et al. Amygdala dysfunction in men with the fragile X premutation. Brain. 2007;130(Pt 2):404–416. doi: 10.1093/brain/awl338. [DOI] [PubMed] [Google Scholar]
- Hessl D, Rivera SM, Reiss AL. The neuroanatomy and neuroendocrinology of fragile X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10(1):17–24. doi: 10.1002/mrdd.20004. [DOI] [PubMed] [Google Scholar]
- Hill SY, Tessner K, Wang S, Carter H, McDermott M. Temperament at 5years of age predicts amygdala and orbitofrontal volume in the right hemisphere in adolescence. Psychiatry Res. 2010;182(1):14–21. doi: 10.1016/j.pscychresns.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeft F, Lightbody AA, Hazlett HC, Patnaik S, Piven J, Reiss AL. Morphometric spatial patterns differentiating boys with fragile X syndrome, typically developing boys, and developmentally delayed boys aged 1 to 3 years. Arch Gen Psychiatry. 2008;65(9):1087–1097. doi: 10.1001/archpsyc.65.9.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsen LM, Dalton KM, Johnstone T, Davidson RJ. Prefrontal social cognition network dysfunction underlying face encoding and social anxiety in fragile X syndrome. Neuroimage. 2008;43(3):592–604. doi: 10.1016/j.neuroimage.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt DJ, Kunkel L, Weiss AP, Goff DC, Wright CI, Shin LM, et al. Increased medial temporal lobe activation during the passive viewing of emotional and neutral facial expressions in schizophrenia. Schizophr Res. 2006;82(2–3):153–162. doi: 10.1016/j.schres.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Holt DJ, Phillips ML. The Human Amygdala in Schizophrenia. New York: Guilford Press; 2009. [Google Scholar]
- Honea R, Crow TJ, Passingham D, Mackay CE. Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am J Psychiatry. 2005;162(12):2233–2245. doi: 10.1176/appi.ajp.162.12.2233. [DOI] [PubMed] [Google Scholar]
- Hooker C, Park S. Emotion processing and its relationship to social functioning in schizophrenia patients. Psychiatry Res. 2002;112(1):41–50. doi: 10.1016/s0165-1781(02)00177-4. [DOI] [PubMed] [Google Scholar]
- Hudziak JJ, Althoff RR, Derks EM, Faraone SV, Boomsma DI. Prevalence and genetic architecture of Child Behavior Checklist-juvenile bipolar disorder. Biol Psychiatry. 2005;58(7):562–568. doi: 10.1016/j.biopsych.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Hulshoff Pol HE, Schnack HG, Mandl RC, van Haren NE, Koning H, Collins DL, et al. Focal gray matter density changes in schizophrenia. Arch Gen Psychiatry. 2001;58(12):1118–1125. doi: 10.1001/archpsyc.58.12.1118. [DOI] [PubMed] [Google Scholar]
- Humphrey T, editor. The development of the human amygdaloid complex. New York: Plenum; 1972. [Google Scholar]
- Huttunen MO, Niskanen P. Prenatal loss of father and psychiatric disorders. Arch Gen Psychiatry. 1978;35(4):429–431. doi: 10.1001/archpsyc.1978.01770280039004. [DOI] [PubMed] [Google Scholar]
- Isohanni M, Jones P, Kemppainen L, Croudace T, Isohanni I, Veijola J, et al. Childhood and adolescent predictors of schizophrenia in the Northern Finland 1966 birth cohort--a descriptive life-span model. Eur Arch Psychiatry Clin Neurosci. 2000;250(6):311–319. doi: 10.1007/s004060070006. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Giedd JN, Castellanos FX, Vaituzis AC, Hamburger SD, Kumra S, et al. Progressive reduction of temporal lobe structures in childhood-onset schizophrenia. Am J Psychiatry. 1998;155(5):678–685. doi: 10.1176/ajp.155.5.678. [DOI] [PubMed] [Google Scholar]
- Jarvinen-Pasley A, Bellugi U, Reilly J, Mills DL, Galaburda A, Reiss AL, et al. Defining the social phenotype in Williams syndrome: a model for linking gene, the brain, and behavior. Dev Psychopathol. 2008;20(1):1–35. doi: 10.1017/S0954579408000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB. Further contributions to the study of the evolution of the forebrain. J Comp Neurol. 1923;35:337–481. [Google Scholar]
- Johnston PJ, Stojanov W, Devir H, Schall U. Functional MRI of facial emotion recognition deficits in schizophrenia and their electrophysiological correlates. Eur J Neurosci. 2005;22(5):1221–1232. doi: 10.1111/j.1460-9568.2005.04294.x. [DOI] [PubMed] [Google Scholar]
- Jones P, Rodgers B, Murray R, Marmot M. Child development risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet. 1994;344(8934):1398–1402. doi: 10.1016/s0140-6736(94)90569-x. [DOI] [PubMed] [Google Scholar]
- Jones W, Bellugi U, Lai Z, Chiles M, Reilly J, Lincoln A, et al. II. Hypersociability in Williams Syndrome. J Cogn Neurosci. 2000;12(Suppl 1):30–46. doi: 10.1162/089892900561968. [DOI] [PubMed] [Google Scholar]
- Joyal CC, Laakso MP, Tiihonen J, Syvalahti E, Vilkman H, Laakso A, et al. The amygdala and schizophrenia: a volumetric magnetic resonance imaging study in first-episode, neuroleptic-naive patients. Biol Psychiatry. 2003;54(11):1302–1304. doi: 10.1016/s0006-3223(03)00597-3. [DOI] [PubMed] [Google Scholar]
- Kagan J, Reznick JS, Snidman N, Gibbons J, Johnson MO. Childhood derivatives of inhibition and lack of inhibition to the unfamiliar. Child Dev. 1988;59(6):1580–1589. doi: 10.1111/j.1467-8624.1988.tb03685.x. [DOI] [PubMed] [Google Scholar]
- Kagan J, Snidman N, McManis M, Woodward S. Temperamental contributions to the affect family of anxiety. Psychiatr Clin North Am. 2001;24(4):677–688. doi: 10.1016/s0193-953x(05)70257-4. [DOI] [PubMed] [Google Scholar]
- Kates WR, Abrams MT, Kaufmann WE, Breiter SN, Reiss AL. Reliability and validity of MRI measurement of the amygdala and hippocampus in children with fragile X syndrome. Psychiatry Res. 1997;75(1):31–48. doi: 10.1016/s0925-4927(97)00019-x. [DOI] [PubMed] [Google Scholar]
- Kato T. Molecular genetics of bipolar disorder and depression. Psychiatry Clin Neurosci. 2007;61(1):3–19. doi: 10.1111/j.1440-1819.2007.01604.x. [DOI] [PubMed] [Google Scholar]
- Kennedy DP, Glascher J, Tyszka JM, Adolphs R. Personal space regulation by the human amygdala. Nat Neurosci. 2009;12(10):1226–1227. doi: 10.1038/nn.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killgore WD, Oki M, Yurgelun-Todd DA. Sex-specific developmental changes in amygdala responses to affective faces. Neuroreport. 2001;12(2):427–433. doi: 10.1097/00001756-200102120-00047. [DOI] [PubMed] [Google Scholar]
- Kilts CD, Kelsey JE, Knight B, Ely TD, Bowman FD, Gross RE, et al. The neural correlates of social anxiety disorder and response to pharmacotherapy. Neuropsychopharmacology. 2006;31(10):2243–2253. doi: 10.1038/sj.npp.1301053. [DOI] [PubMed] [Google Scholar]
- Klein RG, Pine DS. Anxiety disorders. In: Rutter M, Taylor E, Hersov L, editors. Child and Adolescent Psychiatry. New York: Elsevier; 2002. pp. 486–509. [Google Scholar]
- Kleinhans NM, Johnson LC, Richards T, Mahurin R, Greenson J, Dawson G, et al. Reduced neural habituation in the amygdala and social impairments in autism spectrum disorders. Am J Psychiatry. 2009;166(4):467–475. doi: 10.1176/appi.ajp.2008.07101681. [DOI] [PubMed] [Google Scholar]
- Kling A, Green PC. Effects of neonatal amygdalectomy in the maternally reared and maternally deprived monkey. Nature. 1967;213(5077):742–743. doi: 10.1038/213742b0. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Piecinski P, Rakic P. Neurogenesis of the amygdaloid nuclear complex in the rhesus monkey. Brain Res Dev Brain Res. 1992;68(1):9–15. doi: 10.1016/0165-3806(92)90242-o. [DOI] [PubMed] [Google Scholar]
- Korenberg JR, Chen XN, Hirota H, Lai Z, Bellugi U, Burian D, et al. VI. Genome structure and cognitive map of Williams syndrome. J Cogn Neurosci. 2000;12(Suppl 1):89–107. doi: 10.1162/089892900562002. [DOI] [PubMed] [Google Scholar]
- Kosaka H, Omori M, Murata T, Iidaka T, Yamada H, Okada T, et al. Differential amygdala response during facial recognition in patients with schizophrenia: an fMRI study. Schizophr Res. 2002;57(1):87–95. doi: 10.1016/s0920-9964(01)00324-3. [DOI] [PubMed] [Google Scholar]
- Kreczmanski P, Heinsen H, Mantua V, Woltersdorf F, Masson T, Ulfig N, et al. Volume, neuron density and total neuron number in five subcortical regions in schizophrenia. Brain. 2007;130(Pt 3):678–692. doi: 10.1093/brain/awl386. [DOI] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20(5):937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- Laberge F, Muhlenbrock-Lenter S, Grunwald W, Roth G. Evolution of the amygdala: new insights from studies in amphibians. Brain Behav Evol. 2006;67(4):177–187. doi: 10.1159/000091119. [DOI] [PubMed] [Google Scholar]
- Lau JY, Goldman D, Buzas B, Fromm SJ, Guyer AE, Hodgkinson C, et al. Amygdala function and 5-HTT gene variants in adolescent anxiety and major depressive disorder. Biol Psychiatry. 2009;65(4):349–355. doi: 10.1016/j.biopsych.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie SM, Abukmeil SS. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br J Psychiatry. 1998;172:110–120. doi: 10.1192/bjp.172.2.110. [DOI] [PubMed] [Google Scholar]
- Laws G, Bishop D. Pragmatic language impairment and social deficits in Williams syndrome: a comparison with Down’s syndrome and specific language impairment. Int J Lang Commun Disord. 2004;39(1):45–64. doi: 10.1080/13682820310001615797. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. The amygdala. Curr Biol. 2007;17(20):R868–874. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Leonard CM, Rolls ET, Wilson FAW, Baylis GC. Neurons in the amydgala of the monkey with responses selective for faces. Behavioural Brain Research. 1985;1985:159–176. doi: 10.1016/0166-4328(85)90062-2. [DOI] [PubMed] [Google Scholar]
- Levitt JG, Blanton RE, Caplan R, Asarnow R, Guthrie D, Toga AW, et al. Medial temporal lobe in childhood-onset schizophrenia. Psychiatry Res. 2001;108(1):17–27. doi: 10.1016/s0925-4927(01)00108-1. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Leyfer O, Woodruff-Borden J, Mervis CB. Anxiety Disorders in Children with Williams Syndrome, Their Mothers, and Their Siblings: Implications for the Etiology of Anxiety Disorders. J Neurodev Disord. 2009;1(1):4–14. doi: 10.1007/s11689-009-9003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightbody AA, Reiss AL. Gene, brain, and behavior relationships in fragile X syndrome: evidence from neuroimaging studies. Dev Disabil Res Rev. 2009;15(4):343–352. doi: 10.1002/ddrr.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughland CM, Williams LM, Gordon E. Schizophrenia and affective disorder show different visual scanning behavior for faces: a trait versus state-based distinction? Biol Psychiatry. 2002;52(4):338–348. doi: 10.1016/s0006-3223(02)01356-2. [DOI] [PubMed] [Google Scholar]
- Martens MA, Wilson SJ, Dudgeon P, Reutens DC. Approachability and the amygdala: insights from Williams syndrome. Neuropsychologia. 2009;47(12):2446–2453. doi: 10.1016/j.neuropsychologia.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Maslowsky J, Mogg K, Bradley BP, McClure-Tone E, Ernst M, Pine DS, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105–111. doi: 10.1089/cap.2009.0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure EB, Adler A, Monk CS, Cameron J, Smith S, Nelson EE, et al. fMRI predictors of treatment outcome in pediatric anxiety disorders. Psychopharmacology (Berl) 2007;191(1):97–105. doi: 10.1007/s00213-006-0542-9. [DOI] [PubMed] [Google Scholar]
- McClure EB, Monk CS, Nelson EE, Parrish JM, Adler A, Blair RJ, et al. Abnormal attention modulation of fear circuit function in pediatric generalized anxiety disorder. Arch Gen Psychiatry. 2007;64(1):97–106. doi: 10.1001/archpsyc.64.1.97. [DOI] [PubMed] [Google Scholar]
- McClure EB, Pope K, Hoberman AJ, Pine DS, Leibenluft E. Facial expression recognition in adolescents with mood and anxiety disorders. Am J Psychiatry. 2003;160(6):1172–1174. doi: 10.1176/appi.ajp.160.6.1172. [DOI] [PubMed] [Google Scholar]
- McClure EB, Treland JE, Snow J, Schmajuk M, Dickstein DP, Towbin KE, et al. Deficits in social cognition and response flexibility in pediatric bipolar disorder. Am J Psychiatry. 2005;162(9):1644–1651. doi: 10.1176/appi.ajp.162.9.1644. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54(5 Suppl 1):20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Mervis CB, John AE. Cognitive and behavioral characteristics of children with Williams syndrome: implications for intervention approaches. Am J Med Genet C Semin Med Genet. 2010;154C(2):229–248. doi: 10.1002/ajmg.c.30263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mervis CB, Klein-Tasman BP. Williams syndrome: cognition, personality, and adaptive behavior. Ment Retard Dev Disabil Res Rev. 2000;6(2):148–158. doi: 10.1002/1098-2779(2000)6:2<148::AID-MRDD10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Hariri AR, Munoz KE, Mervis CB, Mattay VS, Morris CA, et al. Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci. 2005;8(8):991–993. doi: 10.1038/nn1494. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Kohn P, Mervis CB, Kippenhan JS, Olsen RK, Morris CA, et al. Neural basis of genetically determined visuospatial construction deficit in Williams syndrome. Neuron. 2004;43(5):623–631. doi: 10.1016/j.neuron.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci. 2006;7(5):380–393. doi: 10.1038/nrn1906. [DOI] [PubMed] [Google Scholar]
- Milham MP, Nugent AC, Drevets WC, Dickstein DP, Leibenluft E, Ernst M, et al. Selective reduction in amygdala volume in pediatric anxiety disorders: a voxel-based morphometry investigation. Biol Psychiatry. 2005;57(9):961–966. doi: 10.1016/j.biopsych.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Monk CS, McClure EB, Nelson EE, Zarahn E, Bilder RM, Leibenluft E, et al. Adolescent immaturity in attention-related brain engagement to emotional facial expressions. Neuroimage. 2003;20(1):420–428. doi: 10.1016/s1053-8119(03)00355-0. [DOI] [PubMed] [Google Scholar]
- Monk CS, Nelson EE, McClure EB, Mogg K, Bradley BP, Leibenluft E, et al. Ventrolateral prefrontal cortex activation and attentional bias in response to angry faces in adolescents with generalized anxiety disorder. Am J Psychiatry. 2006;163(6):1091–1097. doi: 10.1176/ajp.2006.163.6.1091. [DOI] [PubMed] [Google Scholar]
- Monk CS, Telzer EH, Mogg K, Bradley BP, Mai X, Louro HM, et al. Amygdala and ventrolateral prefrontal cortex activation to masked angry faces in children and adolescents with generalized anxiety disorder. Arch Gen Psychiatry. 2008;65(5):568–576. doi: 10.1001/archpsyc.65.5.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CJ, Daly EM, Schmitz N, Tassone F, Tysoe C, Hagerman RJ, et al. A neuropsychological investigation of male premutation carriers of fragile X syndrome. Neuropsychologia. 2004;42(14):1934–1947. doi: 10.1016/j.neuropsychologia.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Moore CM, Breeze JL, Gruber SA, Babb SM, Frederick BB, Villafuerte RA, et al. Choline, myo-inositol and mood in bipolar disorder: a proton magnetic resonance spectroscopic imaging study of the anterior cingulate cortex. Bipolar Disord. 2000;2(3 Pt 2):207–216. doi: 10.1034/j.1399-5618.2000.20302.x. [DOI] [PubMed] [Google Scholar]
- Moreno C, Laje G, Blanco C, Jiang H, Schmidt AB, Olfson M. National trends in the outpatient diagnosis and treatment of bipolar disorder in youth. Arch Gen Psychiatry. 2007;64(9):1032–1039. doi: 10.1001/archpsyc.64.9.1032. [DOI] [PubMed] [Google Scholar]
- Morris RW, Weickert CS, Loughland CM. Emotional face processing in schizophrenia. Curr Opin Psychiatry. 2009;22(2):140–146. doi: 10.1097/YCO.0b013e328324f895. [DOI] [PubMed] [Google Scholar]
- Mosconi MW, Cody-Hazlett H, Poe MD, Gerig G, Gimpel-Smith R, Piven J. Longitudinal study of amygdala volume and joint attention in 2- to 4-year-old children with autism. Arch Gen Psychiatry. 2009;66(5):509–516. doi: 10.1001/archgenpsychiatry.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, O’Rahilly R. The amygdaloid complex and the medial and lateral ventricular eminences in staged human embryos. J Anat. 2006;208(5):547–564. doi: 10.1111/j.1469-7580.2006.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz KE, Meyer-Lindenberg A, Hariri AR, Mervis CB, Mattay VS, Morris CA, et al. Abnormalities in neural processing of emotional stimuli in Williams syndrome vary according to social vs. non-social content. Neuroimage. 2010;50(1):340–346. doi: 10.1016/j.neuroimage.2009.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson J, Dawson G, Abbott R, Faja S, Webb SJ, Friedman SD, et al. Amygdalar volume and behavioral development in autism. Arch Gen Psychiatry. 2006;63(6):686–693. doi: 10.1001/archpsyc.63.6.686. [DOI] [PubMed] [Google Scholar]
- Murray RM, Sham P, Van Os J, Zanelli J, Cannon M, McDonald C. A developmental model for similarities and dissimilarities between schizophrenia and bipolar disorder. Schizophr Res. 2004;71(2–3):405–416. doi: 10.1016/j.schres.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Nacewicz BM, Dalton KM, Johnstone T, Long MT, McAuliff EM, Oakes TR, et al. Amygdala volume and nonverbal social impairment in adolescent and adult males with autism. Arch Gen Psychiatry. 2006;63(12):1417–1428. doi: 10.1001/archpsyc.63.12.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MD, Saykin AJ, Flashman LA, Riordan HJ. Hippocampal volume reduction in schizophrenia as assessed by magnetic resonance imaging: a meta-analytic study. Arch Gen Psychiatry. 1998;55(5):433–440. doi: 10.1001/archpsyc.55.5.433. [DOI] [PubMed] [Google Scholar]
- Nikolic I, Kostovic I. Development of the lateral amygdaloid nucleus in the human fetus: transient presence of discrete cytoarchitectonic units. Anat Embryol (Berl) 1986;174(3):355–360. doi: 10.1007/BF00698785. [DOI] [PubMed] [Google Scholar]
- Nordahl CW, Scholz R, Simon T, Rogers S, Amaral DG. Subgroups of abnormal growth trajectories: A longitudinal analysis of amygdala growth in young children with autism. Paper presented at the International Meeting for Autism Research; Philadelphia, PA. 2009. [Google Scholar]
- Ostby Y, Tamnes CK, Fjell AM, Westlye LT, Due-Tonnessen P, Walhovd KB. Heterogeneity in subcortical brain development: A structural magnetic resonance imaging study of brain maturation from 8 to 30 years. J Neurosci. 2009;29(38):11772–11782. doi: 10.1523/JNEUROSCI.1242-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso S, Andreasen NC, Crespo-Facorro B, O’Leary DS, Watkins GL, Boles Ponto LL, et al. Emotions in unmedicated patients with schizophrenia during evaluation with positron emission tomography. Am J Psychiatry. 2003;160(10):1775–1783. doi: 10.1176/appi.ajp.160.10.1775. [DOI] [PubMed] [Google Scholar]
- Pavuluri MN, O’Connor MM, Harral E, Sweeney JA. Affective neural circuitry during facial emotion processing in pediatric bipolar disorder. Biol Psychiatry. 2007;62(2):158–167. doi: 10.1016/j.biopsych.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Perez-Edgar K, Fox NA. Temperament and anxiety disorders. Child Adolesc Psychiatr Clin N Am. 2005;14(4):681–706. viii. doi: 10.1016/j.chc.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Perez-Edgar K, Roberson-Nay R, Hardin MG, Poeth K, Guyer AE, Nelson EE, et al. Attention alters neural responses to evocative faces in behaviorally inhibited adolescents. Neuroimage. 2007;35(4):1538–1546. doi: 10.1016/j.neuroimage.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan KL, Fitzgerald DA, Nathan PJ, Tancer ME. Association between amygdala hyperactivity to harsh faces and severity of social anxiety in generalized social phobia. Biol Psychiatry. 2006;59(5):424–429. doi: 10.1016/j.biopsych.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48(2):175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 2003;54(5):515–528. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Williams L, Senior C, Bullmore ET, Brammer MJ, Andrew C, et al. A differential neural response to threatening and non-threatening negative facial expressions in paranoid and non-paranoid schizophrenics. Psychiatry Res. 1999;92(1):11–31. doi: 10.1016/s0925-4927(99)00031-1. [DOI] [PubMed] [Google Scholar]
- Pierce K, Haist F, Sedaghat F, Courchesne E. The brain response to personally familiar faces in autism: findings of fusiform activity and beyond. Brain. 2004;127(Pt 12):2703–2716. doi: 10.1093/brain/awh289. [DOI] [PubMed] [Google Scholar]
- Pierce K, Müller RA, Ambrose J, Allen G, Courchesne E. Face processing occurs outside the fusiform ‘face area’ in autism: Evidence from fMRI. Brain. 2001;124:2059–2073. doi: 10.1093/brain/124.10.2059. [DOI] [PubMed] [Google Scholar]
- Pine DS. Research review: a neuroscience framework for pediatric anxiety disorders. J Child Psychol Psychiatry. 2007;48(7):631–648. doi: 10.1111/j.1469-7610.2007.01751.x. [DOI] [PubMed] [Google Scholar]
- Pine DS, Guyer AE, Leibenluft E. Functional magnetic resonance imaging and pediatric anxiety. J Am Acad Child Adolesc Psychiatry. 2008;47(11):1217–1221. doi: 10.1097/CHI.0b013e318185dad0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Amaral DG. Organization of the intrinsic connections of the monkey amygdaloid complex: projections originating in the lateral nucleus. J Comp Neurol. 1998;398(3):431–458. doi: 10.1002/(sici)1096-9861(19980831)398:3<431::aid-cne9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Kelly JL, Amaral DG. Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the entorhinal cortex in the macaque monkey. Hippocampus. 2002;12(2):186–205. doi: 10.1002/hipo.1099. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Tuunanen J, Kalviainen R, Partanen K, Salmenpera T. Amygdala damage in experimental and human temporal lobe epilepsy. Epilepsy Res. 1998;32(1–2):233–253. doi: 10.1016/s0920-1211(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Post RM, Leverich GS. The role of psychosocial stress in the onset and progression of bipolar disorder and its comorbidities: the need for earlier and alternative modes of therapeutic intervention. Dev Psychopathol. 2006;18(4):1181–1211. doi: 10.1017/S0954579406060573. [DOI] [PubMed] [Google Scholar]
- Prather MD, Amaral DG. Abstract Viewer and Itinerary Planner. Washington DC: Society for Neuroscience; 2000. The development and distribution of serotonergic fibers in the macaque monkey amygdala. 2003. Online. [Google Scholar]
- Prather MD, Lavenex P, Mauldin-Jourdain ML, Mason WA, Capitanio JP, Mendoza SP, et al. Increased social fear and decreased fear of objects in monkeys with neonatal amygdala lesions. Neuroscience. 2001;106(4):653–658. doi: 10.1016/s0306-4522(01)00445-6. [DOI] [PubMed] [Google Scholar]
- Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18(9):383–388. doi: 10.1016/0166-2236(95)93934-p. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB, et al. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry. 2000;47(9):769–776. doi: 10.1016/s0006-3223(00)00828-3. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Dant CC. The behavioral neurogenetics of fragile X syndrome: analyzing gene-brain-behavior relationships in child developmental psychopathologies. Dev Psychopathol. 2003;15(4):927–968. doi: 10.1017/s0954579403000464. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Eckert MA, Rose FE, Karchemskiy A, Kesler S, Chang M, et al. An experiment of nature: brain anatomy parallels cognition and behavior in Williams syndrome. J Neurosci. 2004;24(21):5009–5015. doi: 10.1523/JNEUROSCI.5272-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AL, Freund L. Behavioral phenotype of fragile X syndrome: DSM-III-R autistic behavior in male children. Am J Med Genet. 1992;43(1–2):35–46. doi: 10.1002/ajmg.1320430106. [DOI] [PubMed] [Google Scholar]
- Reynolds GP. Increased Concentrations and Lateral Asymmetry of Amygdala Dopamine in Schizophrenia. Nature. 1983;305(5934):527–529. doi: 10.1038/305527a0. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Czudek C, Andrews HB. Deficit and hemispheric asymmetry of GABA uptake sites in the hippocampus in schizophrenia. Biol Psychiatry. 1990;27(9):1038–1044. doi: 10.1016/0006-3223(90)90039-5. [DOI] [PubMed] [Google Scholar]
- Rice C. Prevalence of Autism Spectrum Disorders. National Center on Birth Defects and Developmental Disabilities, Center for Disease Control; 2006. [Google Scholar]
- Rich BA, Vinton DT, Roberson-Nay R, Hommer RE, Berghorst LH, McClure EB, et al. Limbic hyperactivation during processing of neutral facial expressions in children with bipolar disorder. Proc Natl Acad Sci U S A. 2006;103(23):8900–8905. doi: 10.1073/pnas.0603246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodman HR. Development of inferior temporal cortex in the monkey. Cereb Cortex. 1994;4(5):484–498. doi: 10.1093/cercor/4.5.484. [DOI] [PubMed] [Google Scholar]
- Rolls ET. Neurons in the cortex of the temporal lobe and in the amygdala of the monkey with responses selective for faces. Human Neurobiology. 1984;3:209–222. [PubMed] [Google Scholar]
- Rosso IM, Killgore WD, Cintron CM, Gruber SA, Tohen M, Yurgelun-Todd DA. Reduced amygdala volumes in first-episode bipolar disorder and correlation with cerebral white matter. Biol Psychiatry. 2007;61(6):743–749. doi: 10.1016/j.biopsych.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Sarpal D, Buchsbaum BR, Kohn PD, Kippenhan JS, Mervis CB, Morris CA, et al. A genetic model for understanding higher order visual processing: functional interactions of the ventral visual stream in Williams syndrome. Cereb Cortex. 2008;18(10):2402–2409. doi: 10.1093/cercor/bhn004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Gur RC, Koch K, Backes V, Amunts K, Shah NJ, et al. Impairment in the specificity of emotion processing in schizophrenia. Am J Psychiatry. 2006;163(3):442–447. doi: 10.1176/appi.ajp.163.3.442. [DOI] [PubMed] [Google Scholar]
- Schneider F, Weiss U, Kessler C, Salloum JB, Posse S, Grodd W, et al. Differential amygdala activation in schizophrenia during sadness. Schizophr Res. 1998;34(3):133–142. doi: 10.1016/s0920-9964(98)00085-1. [DOI] [PubMed] [Google Scholar]
- Schultz RT. Developmental deficits in social perception in autism: the role of the amygdala and fusiform face area. Int J Dev Neurosci. 2005;23:125–141. doi: 10.1016/j.ijdevneu.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Amaral DG. Stereological analysis of amygdala neuron number in autism. J Neurosci. 2006;26(29):7674–7679. doi: 10.1523/JNEUROSCI.1285-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Amaral DG, editors. The Human Amygdala in Autism. New York: Guilford Press; 2009. [Google Scholar]
- Schumann CM, Barnes CC, Lord C, Courchesne E. Amygdala Enlargement in Toddlers with Autism Related to Severity of Social and Communication Impairments. Biol Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Hamstra J, Goodlin-Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, et al. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. Journal of neuroscience. 2004;24:6392–6401. doi: 10.1523/JNEUROSCI.1297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz CE, Wright CI, Shin LM, Kagan J, Rauch SL. Inhibited and uninhibited infants “grown up”: adult amygdalar response to novelty. Science. 2003;300(5627):1952–1953. doi: 10.1126/science.1083703. [DOI] [PubMed] [Google Scholar]
- Setzer M, Ulfig N. Differential expression of calbindin and calretinin in the human fetal amygdala. Microsc Res Tech. 1999;46(1):1–17. doi: 10.1002/(SICI)1097-0029(19990701)46:1<1::AID-JEMT1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Shaw P, Lawrence EJ, Radbourne C, Bramham J, Polkey CE, David AS. The impact of early and late damage to the human amygdala on ‘theory of mind’ reasoning. Brain. 2004;127(Pt 7):1535–1548. doi: 10.1093/brain/awh168. [DOI] [PubMed] [Google Scholar]
- Simpson MD, Slater P, Deakin JF, Royston MC, Skan WJ. Reduced GABA uptake sites in the temporal lobe in schizophrenia. Neurosci Lett. 1989;107(1–3):211–215. doi: 10.1016/0304-3940(89)90819-7. [DOI] [PubMed] [Google Scholar]
- Sparks BF, Friedman SD, Shaw DW, Aylward E, Echelard D, Artru AA, et al. Brain sructural abnormalities in young children with autism spectrum disorder. Neurology. 2002;59(2):184–192. doi: 10.1212/wnl.59.2.184. [DOI] [PubMed] [Google Scholar]
- Spezio ML, Adolphs R, Hurley RS, Piven J. Abnormal use of facial information in high-functioning autism. J Autism Dev Disord. 2007;37(5):929–939. doi: 10.1007/s10803-006-0232-9. [DOI] [PubMed] [Google Scholar]
- Spokes EG, Garrett NJ, Rossor MN, Iversen LL. Distribution of GABA in post-mortem brain tissue from control, psychotic and Huntington’s chorea subjects. J Neurol Sci. 1980;48(3):303–313. doi: 10.1016/0022-510x(80)90103-3. [DOI] [PubMed] [Google Scholar]
- Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry. 2006;188:510–518. doi: 10.1192/bjp.188.6.510. [DOI] [PubMed] [Google Scholar]
- Stein MB, Goldin PR, Sareen J, Zorrilla LT, Brown GG. Increased amygdala activation to angry and contemptuous faces in generalized social phobia. Arch Gen Psychiatry. 2002;59(11):1027–1034. doi: 10.1001/archpsyc.59.11.1027. [DOI] [PubMed] [Google Scholar]
- Stephan H, Frahm HD, Baron G. Comparison of brain structure volumes in Insectivora and primates. VII. Amygdaloid components. J Hirnforsch. 1987;28(5):571–584. [PubMed] [Google Scholar]
- Straube T, Mentzel HJ, Miltner WH. Neural mechanisms of automatic and direct processing of phobogenic stimuli in specific phobia. Biol Psychiatry. 2006;59(2):162–170. doi: 10.1016/j.biopsych.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, et al. Schizophrenia after prenatal famine. Further evidence. Arch Gen Psychiatry. 1996;53(1):25–31. doi: 10.1001/archpsyc.1996.01830010027005. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Petrovich GD. What is the amygdala? Trends Neurosci. 1998;21(8):323–331. doi: 10.1016/s0166-2236(98)01265-x. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Koeda M, Oda K, Matsuda T, Matsushima E, Matsuura M, et al. An fMRI study of differential neural response to affective pictures in schizophrenia. Neuroimage. 2004;22(3):1247–1254. doi: 10.1016/j.neuroimage.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Tanskanen P, Veijola JM, Piippo UK, Haapea M, Miettunen JA, Pyhtinen J, et al. Hippocampus and amygdala volumes in schizophrenia and other psychoses in the Northern Finland 1966 birth cohort. Schizophr Res. 2005;75(2–3):283–294. doi: 10.1016/j.schres.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Metcalfe K, Karmiloff-Smith A, Carette MJ, Grant J, Dennis N, et al. Williams syndrome: use of chromosomal microdeletions as a tool to dissect cognitive and physical phenotypes. Am J Hum Genet. 1999;64(1):118–125. doi: 10.1086/302214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SF, Liberzon I, Decker LR, Koeppe RA. A functional anatomic study of emotion in schizophrenia. Schizophr Res. 2002;58(2–3):159–172. doi: 10.1016/s0920-9964(01)00403-0. [DOI] [PubMed] [Google Scholar]
- Thomas KM, Drevets WC, Dahl RE, Ryan ND, Birmaher B, Eccard CH, et al. Amygdala response to fearful faces in anxious and depressed children. Archives of General Psychiatry. 2001;58(11):1057–1063. doi: 10.1001/archpsyc.58.11.1057. [DOI] [PubMed] [Google Scholar]
- Thomas KM, Drevets WC, Whalen PJ, Eccard CH, Dahl RE, Ryan ND, et al. Amygdala response to facial expressions in children and adults. Biol Psychiatry. 2001;49(4):309–316. doi: 10.1016/s0006-3223(00)01066-0. [DOI] [PubMed] [Google Scholar]
- Thompson CI. Social Development of Infant Rhesus Monkeys Following Amygdaloid Lesions. U. Wisconsin; 1968. [Google Scholar]
- Thompson CI. Long-term behavioral development of rhesus monkeys after amygdalectomy in infancy. In: Ben-Ari Y, editor. The Amygdaloid Complex. Amsterdam: Elsevier; 1981. pp. 259–270. [Google Scholar]
- Thompson CI, Bergland RM, Towfighi JT. Social and nonsocial behaviors of adult rhesus monkeys after amygdalectomy in infancy or adulthood. Journal of Comparative & Physiological Psychology. 1977;91(3):533–548. doi: 10.1037/h0077352. [DOI] [PubMed] [Google Scholar]
- Thompson CI, Schwartzbaum JS, Harlow HF. Development of Social Fear After Amygdalectomy in Infant Rhesus Monkeys. Physiology & Behavior. 1969;4(2):249–254. [Google Scholar]
- Thompson PM, Hayashi KM, Sowell ER, Gogtay N, Giedd JN, Rapoport JL, et al. Mapping cortical change in Alzheimer’s disease, brain development, and schizophrenia. Neuroimage. 2004;23(Suppl 1):S2–18. doi: 10.1016/j.neuroimage.2004.07.071. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Vidal C, Giedd JN, Gochman P, Blumenthal J, Nicolson R, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci U S A. 2001;98(20):11650–11655. doi: 10.1073/pnas.201243998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfig N, Setzer M, Bohl J. Transient architectonic features in the basolateral amygdala of the human fetal brain. Acta Anat (Basel) 1998;163(2):99–112. doi: 10.1159/000046489. [DOI] [PubMed] [Google Scholar]
- Ulfig N, Setzer M, Bohl J. Distribution of GAP-43-immunoreactive structures in the human fetal amygdala. Eur J Histochem. 1999;43(1):19–28. [PubMed] [Google Scholar]
- Ulfig N, Setzer M, Bohl J. Ontogeny of the human amygdala. Ann N Y Acad Sci. 2003;985:22–33. doi: 10.1111/j.1749-6632.2003.tb07068.x. [DOI] [PubMed] [Google Scholar]
- Velakoulis D, Wood SJ, Wong MT, McGorry PD, Yung A, Phillips L, et al. Hippocampal and amygdala volumes according to psychosis stage and diagnosis: a magnetic resonance imaging study of chronic schizophrenia, first-episode psychosis, and ultra-high-risk individuals. Arch Gen Psychiatry. 2006;63(2):139–149. doi: 10.1001/archpsyc.63.2.139. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Watson C, Hoeft F, Garrett AS, Hall SS, Reiss AL. Aberrant brain activation during gaze processing in boys with fragile X syndrome. Arch Gen Psychiatry. 2008;65(11):1315–1323. doi: 10.1001/archpsyc.65.11.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MJ, Bachevalier J, Ungerleider LG. Connections of inferior temporal areas TEO and TE with parietal and frontal cortex in macaque monkeys. Cereb Cortex. 1994;4(5):470–483. doi: 10.1093/cercor/4.5.470. [DOI] [PubMed] [Google Scholar]
- Webster MJ, Ungerleider LG, Bachevalier J. Connections of inferior temporal areas TE and TEO with medial temporal-lobe structures in infant and adult monkeys. J Neurosci. 1991;11(4):1095–1116. doi: 10.1523/JNEUROSCI.11-04-01095.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44(7):660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Welch KA, Stanfield AC, Moorhead TW, Haga K, Owens DC, Lawrie SM, et al. Amygdala volume in a population with special educational needs at high risk of schizophrenia. Psychol Med. 2009:1–10. doi: 10.1017/S0033291709990870. [DOI] [PubMed] [Google Scholar]
- Werner E, Dawson G, Osterling J, Dinno N. Brief report: Recognition of autism spectrum disorder before one year of age: a retrospective study based on home videotapes. J Autism Dev Disord. 2000;30(2):157–162. doi: 10.1023/a:1005463707029. [DOI] [PubMed] [Google Scholar]
- Williams LM, Das P, Harris AW, Liddell BB, Brammer MJ, Olivieri G, et al. Dysregulation of arousal and amygdala-prefrontal systems in paranoid schizophrenia. Am J Psychiatry. 2004;161(3):480–489. doi: 10.1176/appi.ajp.161.3.480. [DOI] [PubMed] [Google Scholar]
- Williams LM, Das P, Liddell BJ, Olivieri G, Peduto AS, David AS, et al. Fronto-limbic and autonomic disjunctions to negative emotion distinguish schizophrenia subtypes. Psychiatry Res. 2007;155(1):29–44. doi: 10.1016/j.pscychresns.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Witthaus H, Kaufmann C, Bohner G, Ozgurdal S, Gudlowski Y, Gallinat J, et al. Gray matter abnormalities in subjects at ultra-high risk for schizophrenia and first-episode schizophrenic patients compared to healthy controls. Psychiatry Res. 2009;173(3):163–169. doi: 10.1016/j.pscychresns.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Woodruff-Borden J, Kistler DJ, Henderson DR, Crawford NA, Mervis CB. Longitudinal course of anxiety in children and adolescents with Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(2):277–290. doi: 10.1002/ajmg.c.30259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright IC, Rabe-Hesketh S, Woodruff PW, David AS, Murray RM, Bullmore ET. Meta-analysis of regional brain volumes in schizophrenia. Am J Psychiatry. 2000;157(1):16–25. doi: 10.1176/ajp.157.1.16. [DOI] [PubMed] [Google Scholar]
- Yoon KL, Fitzgerald DA, Angstadt M, McCarron RA, Phan KL. Amygdala reactivity to emotional faces at high and low intensity in generalized social phobia: a 4-Tesla functional MRI study. Psychiatry Res. 2007;154(1):93–98. doi: 10.1016/j.pscychresns.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Yurgelun-Todd DA, Gruber SA, Kanayama G, Killgore WD, Baird AA, Young AD. fMRI during affect discrimination in bipolar affective disorder. Bipolar Disord. 2000;2(3 Pt 2):237–248. doi: 10.1034/j.1399-5618.2000.20304.x. [DOI] [PubMed] [Google Scholar]
- Zirlinger M, Anderson D. Molecular dissection of the amygdala and its relevance to autism. Genes Brain Behav. 2003;2(5):282–294. doi: 10.1034/j.1601-183x.2003.00039.x. [DOI] [PubMed] [Google Scholar]
- Zirlinger M, Kreiman G, Anderson DJ. Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei. Proc Natl Acad Sci U S A. 2001;98(9):5270–5275. doi: 10.1073/pnas.091094698. [DOI] [PMC free article] [PubMed] [Google Scholar]