

Abstract
Accumulating evidence indicates that immune impairment in persistent viral infections could lead to T-cell exhaustion. To evaluate the potential contribution of induction of negative costimulatory molecules to impaired T-cell responses, we primed naïve T cells with mature monocyte-derived dendritic cells (MDDCs) pulsed with HIV-1 in vitro. We used quantitative real-time polymerase chain reaction and flow cytometry, respectively, to compare the gene and surface-protein expression profiles of naïve T cells primed with HIV-pulsed or mock-pulsed DCs. We detected elevated expressions of negative costimulatory molecules, including lymphocyte activation gene-3 (LAG-3), CD160, cytolytic T-lymphocyte antigen-4 (CTLA-4), T-cell immunoglobulin mucin-containing domain-3 (TIM-3), programmed death-1 (PD-1) and TRAIL (tumor necrosis-factor–related apoptosis-inducing ligand) in T cells primed by HIV-pulsed DCs. The PD-1+ T-cell population also coexpressed TIM-3, LAG-3, and CTLA-4. Interestingly, we also found an increase in gene expression of the transcriptional repressors Blimp-1 (B-lymphocyte–induced maturation protein-1) and Foxp3 (forkhead transcription factor) in T-cells primed by HIV-pulsed DCs; Blimp-1 expression was directly proportional to the expression of the negative costimulatory molecules. Furthermore, levels of the effector cytokines interleukin-2, tumor necrosis factor-α and interferon-γ, and perforin and granzyme B were decreased in T-cell populations primed by HIV-pulsed DCs. In conclusion, in vitro priming of naïve T-cells with HIV-pulsed DC leads to expansion of T cells with coexpression of a broad array of negative costimulatory molecules and Blimp-1, with potential deleterious consequences for T-cell responses.
INTRODUCTION
Persistent viral infections (PVIs) are characterized by impaired T-cell responses and futile viral control attributes (1,2). Functional impairment of T cells is the key feature of HIV-1 and certain other viral infections (3–5). A myriad of genes have been found to be upregulated or downregulated in exhausted CD8+ T cells in PVIs, suggesting a role for negative costimulatory molecules (6). It is now clear that impaired immune effector and proliferative functions seen in T cells due to HIV infection are multifactorial (7) and that upregulation of negative costimulatory molecules on HIV-specific T cells can contribute to rapid disease progression and systemic immune dysfunction (8). It has been shown that HIV results in suppressor T-cell expansion in vivo (9). We recently showed that HIV impairs the priming of naïve T cells in vitro and gives rise to contact-dependent suppressor T cells (10).
Dendritic cells (DCs), the professional antigen-presenting cells (APCs), are required for the priming of antigen-specific naïve T cells. Immature DCs (IDCs) sense pathogens by means of pattern-recognition receptors (PRRs). IDCs also express enhanced maturation markers, for example CD83, major histocompatibility complex (MHC) class I and II and costimulatory (CD40, CD80 and CD86) molecules, and migrate to peripheral lymph nodes to present pathogen-derived peptides to T cells (11). The nature of the immune response depends on competitive bidirectional binding of ligands/receptors to molecules expressed on DCs and specific T cells. The immune outcome is determined by the binding amplitude of the T-cell receptor (TCR) to MHC-peptide complexes formed from a given episode of antigen presentation, and subsequent binding of positive (CD28) or negative costimulatory molecules to their corresponding receptors/ligands (12). As well as providing critical positive signals, the costimulatory pathways could also generate key negative signals that downregulate the ensuing T-cell responses. The CD80/CD86–CD28/cytotoxic T-lymphocyte–associated antigen–4 (CTLA-4; CD152) represents a dual pathway, specific for both receptors expressed on T cells. Intriguingly, CTLA-4 binds B7 lig-ands with higher affinity than CD28 and hence, minimal CTLA-4 binding is adequate to generate efficient negative responses (13). Moreover, activation of naïve T cells requires greater CD28 signaling than is required for memory T cells (14).
A plethora of other stimulatory molecules have also been described: lymphocyte activation gene-3 (LAG-3; CD223), an MHC II ligand belonging to the immunoglobulin super-family; T-cell immunoglobulin mucin-containing domain-3 (TIM-3) with natural ligands galectin-9 and phosphatidylserine (15); tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL); programmed death-1 (PD-1; CD279), which interacts with PD-1L (CD274) and PD-2L (CD273); CD160 (BY55) (16,17); and more recently, B- and T-lymphocyte attenuator (BTLA; CD272), which binds to the herpes virus entry mediator (HVEM) on APCs and regulatory T cells (Tregs) (16,18). Functionally, CTLA-4 (13), PD-1 (19) and LAG-3 (20), along with BTLA (21) and CD160, negatively regulate the cell cycle. Furthermore, impaired CD28 expression (22) and upregulation of expression of negative costimulatory molecules (23) directly correlated with rapid HIV disease progression. Recent microar-ray experiments conducted on day-2 co-cultivated naïve T cells and DCs in vitro showed that HIV increased the coexpression of TIM-3, TRAIL, galectin-9, and LAG-3 (M Larsson, 2010, unpublished data) in T cells. Likewise, other investigators showed that HIV-specific T cells display surface inhibitory molecules, for example, PD-1 and CTLA-4 (4), TIM-3 (24) and LAG-3 (25). Whereas a few of the mechanisms regulating effector T-cell activation are understood, the molecular factors underlying the fate of naïve T cells when primed with HIV-pulsed DCs remain an area of intense interest. Herein, we report results that show that naïve T cells primed with monocyte-derived DCs (MDDCs) pulsed with HIV in vitro had relatively higher expression of certain negative costimulatory molecules compared with naïve T cells primed with mock DCs, possibly leading to decreased immune activation.
MATERIALS AND METHODS
Culture Medium, Cytokines and Reagents
RPMI1640 was supplemented with 10 mmol/L HEPES, 20 μg/mL gentamicin (Fisher Scientific, Leicestershire, UK), 2 mmol/L L-glutamine (Sigma-Aldrich, St. Louis, MO, USA), and 1% plasma or 5% heat-inactivated pooled human serum (5% PHS). Recombinant human granulocyte-macrophage–colony-stimulating factor (rhGM-CSF) (100 IU/mL) (Immunex, Seattle, WA, USA) and recombinant human interleukin-4 (rhIL-4) (300 U/mL) (R & D Systems, Abingdon, UK) were used for the in vitro differentiation of MDDCs, as described previously (10,26).
Generation of MDDCs
Buffy coats were obtained from healthy individuals (Transfusion Laboratory, Huddinge Hospital, Karolinska Institutet, Stockholm, Sweden) and peripheral blood mononuclear cells (PBMCs) were prepared from buffy coats by density-gradient centrifugation over Ficoll-Paque™ (Amersham Pharmacia, Piscataway, NJ, USA). CD14+ cells were selected by plastic adherence by incubation of PBMCs in tissue culture dishes (BD Falcon, Franklin Lakes, NJ, USA) for 1 h at 37°C in a 5% CO2 incubator. The plates were washed thrice with RPMI1640 to remove nonadherent T cells. The adhering cells were cultured in 1% plasma and supplemented with rhGM-CSF and rhIL-4, and incubated in a 5% CO2 environment at 37°C. Cytokines (indicated above) were replenished every second day to facilitate differentiation of progenitor cells into DCs, and IDCs were harvested on day 6.
DC Maturation and Immunophenotyping
The purity and readiness of IDCs was assessed by flow cytometry (FACSCalibur, BD Immunocytometry Systems, San Jose, CA, USA). The DCs were immunophenotyped by using phycoerythrin (PE)-conjugated monoclonal antibodies (mAbs) against CD83, CD86, CD80, CD14 and HLA-DR and against the isotype controls IgG1 and IgG2a (BD Pharmingen, Franklin Lakes, NJ, USA). Data were analyzed by using FlowJo software (TreeStar, Ashland, OR, USA). The differentiated IDCs were transferred to new plates, and maturation was induced by adding 30 ng/mL polyinosinic acid:polycytidylic acid (Poly I:C) (Sigma-Aldrich) and incubated at 37°C in a 5% CO2 incubator for 24 h.
Virus Preparation
HIV-1 BaL/SUPT1-CCR5 CL.30 (lot P4143) was produced by using chronically infected cultures of the ACVP/BCP cell line (No. 204), originally derived by infecting SUPT1-CCR5 CL.30 cells (graciously provided by J Hoxie, the University of Pennsylvania, Philadelphia, PA, USA) with an infectious stock of HIV-1 BaL (NIH AIDS Research and Reference Reagent Program, Cat. No. 416, Lot 59155). The virus was purified by continuous flow centrifugation using a Beckman CF32Ti rotor at ~90,000g at a flow rate of 6 L/h, followed by banding for 30 min after sample loading. Sucrose density-gradient fractions were collected, virus-containing fractions were pooled and diluted to less than 20% sucrose, and the virus was pelleted at ~100,000g for 1 h. The virus pellet was resuspended to a concentration 1000-fold higher relative to initial cell culture filtrate.
HIV Pulsing of DCs
After incubation, HIV-1 BaL (lot number P4143) 175–750 ng/mL p24 equivalents/mL, corresponding to ~0.5–2 multiplicities of infection, doses which reportedly exist in vivo (27,28), was added to 1 × 105 DCs in a flat-bottomed 24-well plate (BD Falcon) and incubated for 24 h at 37°C in a 5% CO2 incubator. Before the DCs were used in the assays the unbound viruses were washed off twice with RPMI1640. HIV-unexposed mock MDDCs served as controls, and DC viability was examined by use of Annexin V and 0.4% trypan blue exclusion methods.
Allogeneic T-Cell Activation by DCs (Mixed Leukocyte Reaction)
Proliferation assays were performed in 96-well flat-bottom cell-culture plates in 5% pooled human serum (PHS). Naïve T cells were isolated from nonadherent T cells by a negative selection process using magnetic beads. The cells were coupled with anti-CD56, anti-CD19, anti-CD45RO and anti-CD14 magnetic tagged antibodies (Miltenyi Biotec, Auburn, CA, USA) to deplete natural killer (NK) cells, B cells, memory T cells and monocytes, respectively. A part of the naïve T-cell preparation was labeled with carboxyfluorescein succinimidyl ester (CFSE) (10) to measure proliferation by flow cytometry, and cocultured with mock or HIV-pulsed DCs at a 1:10 ratio and incubated at 37°C in a 5% CO2 incubator. A portion of the DCs was stored for restimulating the priming culture on day 7 with the addition of respective HIV-pulsed DCs, and incubated for 24 h at 37°C in a 5% CO2 incubator. On day 8, the cocultures were harvested and the T-cell proliferation and immunophenotype were measured by flow cytometry or used for quantitative reverse- transcription–polymerase chain reaction (qRT-PCR).
Flow Cytometry
On day 8, the primed T cells were collected and investigated for surface expression of negative costimulatory receptors. Direct conjugated mAbs directed against CD3–fluorescein isothiocyanate (FITC), LAG-3–phycoerythrin (PE), TIM-3-APC, CD160-APC, BTLA-PE, HVEM-PE (R & D Systems), PD-1-FITC, TRAIL-PE and CTLA-4-PE (BD Pharmingen) were used for immuno phenotyping T cells after the mixed lymphocyte reaction (MLR) experiments. Perforin and granzyme B levels were measured by in-tracellular staining. Briefly, after staining with antihuman CD3, the lymphocytes were fixed with 4% paraformaldehyde (4%), then washed and permeabilized with 0.2% saponin. The cells were subsequently stained using FITC-labeled anti-human granzyme B (BD Pharmingen), and PE-labeled anti–human perforin (BD Pharmingen) Abs and analyzed by flow cytometry. Foxp3 was measured using anti–Foxp3-FITC (eBioscience, San Diego, CA, USA) by intracellular staining. T cells were double stained with PD-1 FITC, and TIM-3-APC, LAG-3-PE, CTLA-4-APC or CD160-APC mAbs. Data were acquired on a 4-color FACSCalibur (BD Immunocytometry Systems, San Jose, CA, USA) using CellQuest software and analyzed using FlowJo software (TreeStar).
Effector Cytokine Analysis
Supernatants collected from day 8 of the priming assays were analyzed for effector cytokines, interleukin 2 (IL-2), tumor necrosis factor-α (TNF-α) and interferon γ (IFN-γ) using a commercial Bio-Plex™ Cytokine Luminex assay (Bio-Rad, Hercules, CA, USA).
mRNA Isolation, cDNA Synthesis and qRT-PCR
Cryopreserved cell lysates prepared from day 8 T cells were used for RNA extraction, cDNA synthesis and qRT-PCR analysis. Briefly, mRNA was isolated using a commercial Spin technology (Qiagen AB, Solna, Sweden) according to the manufacturer’s protocol. First-strand cDNA was made from 1 μg of mRNA in a 20-μL reaction volume. We used 20 μL of reaction mix containing 1 μL of oligo dT (50 μmol/L), deoxynucleoside-5′-triphosphate (10 mmol/L), SuperScript™ III (200 U/mL), dithiothreitol (0.1 mol/L), RNaseOUT™ (40 U/mL), 4 μL of 5× First-Strand buffer and 1 μg of mRNA in 11 μL of nuclease-free water (all reagents obtained from Invitrogen, Carlsbad, CA, USA). The primers were designed using the NCBI Primer-BLAST (Basic Local Alignment Search Tool) online software at http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome. The list of primers tested against the corresponding genes is given in Table 1. cDNA was quantified for the negative costimulatory molecules and β-actin endogenous control by qRT-PCRs using Prism 7900HT (Applied Biosystems, Foster City, CA, USA) in 96-well MicroAmp® optical fast-plates (Applied Biosystems). Each sample was run in triplicate in a 10-μL reaction mixture that contained 10 nmol/L (1 μL each) of forward and reverse primers (Cybergene AB, Stockholm, Sweden), 5 μL Fast SYBR Green® Master Mix (Applied Biosystems), 4 nmol/L (2 μL) template DNA and 1 μL of water. Results were expressed as ΔΔCt and presented as normalized values of ΔΔCt.
Table 1.
The list of primers (forward and reverse) tested against the corresponding genes for expression in T cells.
| Primer | ||
|---|---|---|
| Blimp-1 | Forward | CAGCTCGCCCACCTGCAGAA |
| Reverse | GCCGCAGCGCAGTTCCCTTT | |
| BTLA | Forward | TGCCTGGTTTGTTTTCTTCCAGGC |
| Reverse | TGGGTCATACCGCTGTTCTGCAA | |
| CD160 | Forward | GCCTTGTGGCCCTTCAAGCTTTGT |
| Reverse | TCCCCTGTGCCCTGTTGCAT | |
| CTLA-4 | Forward | GGGCATAGGCAACGGAACCCA |
| Reverse | GGGGGCATTTTCACATAGACCCCTG | |
| Foxp3 | Forward | CAGCACATTCCCAGAGTTCCTC |
| Reverse | GCGTGTGAACCAGTGGTAGATC | |
| LAG-3 | Forward | CTAGCCCAGGTGCCCAACGC |
| Reverse | GCCTGCGGAGGGTGAATCCC | |
| PD-1 | Forward | CTCAGGGTGACAGAGAGAAG |
| Reverse | GACACCAACCACCAGGGTTT | |
| TIM-3 | Forward | AGGGGACATGGCCCAGCAGA |
| Reverse | GCCAGCCCAGCACAGATCCC | |
| TRAIL | Forward | CTTTACCAACGAGCTG |
| Reverse | GTTATGTGAGCTGCTAC | |
| β-Actin | Forward | AGAGGGAAATCGTGCGTGAC |
| Reverse | CAATAGTGATGACCTGGCCGT | |
| HVEM | Forward | CCGACGTCTTGAGGCTGGTGC |
| Reverse | TGCCTGGAGGGCAGGGTTCA |
Statistical Analysis
Statistical significance was analyzed using MS Excel and GraphPad Prism 4.0 software (GraphPad, La Jolla, CA, USA) using a paired t test for comparison between the two groups. Because of the considerable degree of variability within the experiments, we obtained statistical values by normalizing/transforming the data whereby each data set was divided by the sum total of all values in the data set. Each value within a group is presented as a percentage. Differences were considered significant with P < 0.05, and the measure of significance was represented by *P < 0.05, **P < 0.005 and ***P < 0.001.
RESULTS
HIV-Pulsed Dendritic Cells Impair T-Cell Proliferation
Using an in vitro allogeneic primary cell culture system (10), we investigated whether high doses (175–750 ng p24 equivalents) of infectious HIV (HIV BaL) impaired the ability of DCs to prime naïve T cells. Briefly, mature DCs were cocultured with negatively selected CD3+CD45RA+CD62L+ bulk naïve T cells and assessed for T-cell proliferation by CFSE dilution assay. The presence of HIV impaired the T-cell proliferation by 20–60% depending on the donor (Figure 1A, B; P < 0.0001). Furthermore, HIV pulsing did not affect DC viability or maturation status, nor the viability of the T cells, as has been shown previously (10,29).
Figure 1.

HIV-pulsed DCs impair T-cell proliferation. Mature DCs were pulsed with HIV (750 ng/mL) for 24 h, washed and cocultured with CFSE-labeled naïve bulk T cells at a ratio of 1:10 (DC:T cells). Assays were restimulated after 7 d and T-cell proliferation was measured after 24 h (d 8) by flow cytometry. (A) Mock- and HIV-pulsed DCs cocultured with naïve T cells (a representative of one experiment). (B) Ten combined experiments normalized together (P < 0.001).
Upregulation of CTLA-4, PD-1 and TRAIL in T Cells Primed by HIV-Pulsed DCs
Our earlier work demonstrated induction of PD-1, CTLA-4, and TRAIL on T cells primed with HIV-pulsed DCs and showed that these molecules contributed to T-cell suppression (10). To evaluate transcriptional expression of these molecules, we used qRT-PCR to measure the profiles of CTLA-4, PD-1, and TRAIL on day-8 proliferated T cells. Others have shown that CTLA-4 and PD-1 were up-regulated on HIV-specific T cells (4,13,19) and that HIV exposure could sensitize activated T cells toward TRAIL-mediated apoptosis (30). We found that HIV-pulsed DCs, compared with mock-pulsed DCs, induced significantly higher CTLA-4 expression in T cells (P < 0.05) (Figure 2A). Likewise, the expression of messages for PD-1 and TRAIL were significantly increased in T cells primed by HIV-pulsed DCs compared with those primed by mock-pulsed DCs (P < 0.05 and P < 0.0001 respectively) (Figure 2A).
Figure 2.

HIV-pulsed DC-primed T cells with increased CTLA-4, PD-1 and TRAIL gene and protein expression. Day-8 cocultures were harvested and mRNA extracted for qPCR or immunophenotyped for CTLA-4, PD-1 and TRAIL. (A) Gene expression values normalized for CTLA-4 (P < 0.05), PD-1 (P < 0.05) and TRAIL (P < 0.001) from seven separate experiments. (B) CTLA-4, PD-1 and TRAIL surface protein expression from one representative experiment. (C) Protein expression values normalized for CTLA-4 (P < 0.001), PD-1 (P < 0.001) and TRAIL (P < 0.001).
We next investigated if changes in gene expression of CTLA-4, PD-1 and TRAIL were reflected in surface protein expression levels on the expanded T cells. The primed T cells were immunophenotyped, and mean fluorescence intensities for CTLA-4, PD-1 and TRAIL were measured 1 d after restimulation (Figure 2B, C). The elevated mRNA expression levels correlated with surface expression attributes (CTLA-4, P < 0.05; PD-1, P < 0.05; and TRAIL, P < 0.001) (Figure 2A–C).
Elevated LAG-3, TIM-3 and CD160 and Decreased BTLA mRNA Expression in T Cells Primed by HIV-Pulsed DCs
LAG-3, TIM-3 and CD160 have been implicated as negative costimulatory molecules (16,17,24,31–33). Accumulating evidence suggests that the TIM-3 facilitates T-cell tolerance (34) and T-cell dysfunction in persistent hepatitis C virus infection (35), and can be associated with rapidly progressive HIV disease (24). To delineate the relationship between HIV exposure and TIM-3 gene expression, we quantified TIM-3 in HIV-pulsed and mock-pulsed DC-primed T cells by qRT-PCR. We found that the TIM-3 message (P < 0.001) was expressed at higher levels in T cells primed by HIV-pulsed DCs compared with mock-pulsed DCs (Figure 3A) and cell-surface TIM-3 protein expression was also increased (P < 0.005) (Figure 3B). Likewise, LAG-3 was also increased both at the gene (P < 0.005) and protein levels (P < 0.05) in T cells primed with HIV-pulsed DCs compared with the levels for those primed with mock-pulsed DCs. (Figure 3A, B). Evidence suggests that CD160 and BTLA upregulation inhibits CD4+ T-cell activation (36), and CD160 upregulation correlated directly with raised Blimp-1 in T cells (37). Our studies showed a tendency towards increased CD160 gene expression in T cells primed with HIV-pulsed DCs compared with mock DCs (Figure 3A). The trend for increased gene expression (P = 0.07) was paralleled by significantly increased cell-surface CD160 expression (P < 0.05) on T cells primed by HIV-pulsed DCs (Figure 3B, C). Interestingly, we found no increased expression of BTLA in T cells primed with HIV-pulsed DCs (P = 0.6), which correlated well with the lack of increased BTLA expression on the cell surface (Figures 3A–C). In addition, the expression of HVEM was not affected or was slightly decreased in some of the experiments, both at the gene (P = 0.9) and protein levels (Figure 3A–C) in T cells primed with HIV-pulsed DCs. mRNA expression correlated with cell surface expression levels of the corresponding proteins although variations in the mean fluorescence intensities of cells expressing BTLA and HVEM were noticed.
Figure 3.

HIV-pulsed DC prime T cells with increased gene and protein expression of LAG-3, TIM-3, BTLA, CD160 and HVEM. Day-8 cocul-tures were harvested and mRNA was extracted for qPCR or immunophenotyped for surface protein expression. (A) Gene expression values normalized for LAG-3 (P < 0.001), TIM-3 (P < 0.001), BTLA (P = 0.6), CD160 and HVEM from seven separate experiments. (B) LAG-3, TIM-3, BTLA, CD160 and HVEM surface protein expression from one representative experiment. (C) Protein expression values normalized for LAG-3 (P < 0.05), TIM-3 (P < 0.005), BTLA (P = 0.6), CD160 (P < 0.05), and HVEM.
Expression of Negative Costimulatory Molecules Correlated with Blimp-1 Expression in T Cells Primed by HIV-Pulsed DCs
Blimp-1 acts as a transcriptional repressor of T-cell proliferation, and the elevated expression of Blimp-1 directly correlated with the upregulation of a myriad of cell surface inhibitory molecules in a PVI model (38). Moreover, co-expression of Foxp3 and Blimp-1 could be vital, because Foxp3 reportedly leads to activation of Blimp-1 in antigen-exposed T cells (39). Hence, we set out to compare the quantitative expression levels of Blimp-1 and Foxp3 mRNA between T cells primed by HIV-pulsed DCs and mock-pulsed DCs. Blimp-1 (P < 0.001) and Foxp3 (P < 0.005) gene expressions were significantly upregulated in T cells primed by HIV-pulsed DCs compared with T cells primed by mock-pulsed DCs (Figure 4A, B). We also found a corresponding increase in the expression of Foxp3 protein (P < 0.005) (Figure 4C, D). Intriguingly, an increase of Blimp-1 mRNA expression in T cells primed with HIV-pulsed DCs correlated with increased gene and protein expression for negative stimulatory molecules (LAG-3, TIM-3 CD160, CTLA-4, PD-1 and TRAIL). However, we found that the increase of Blimp-1 expression was not associated with an increase in the expression of BTLA.
Figure 4.

HIV-pulsed DC prime T cells with increased Blimp-1 and Foxp3 gene expression. Day 8 cocultures were harvested and mRNA was extracted for qPCR or immunophenotyped for protein expression. (A) Blimp-1 normalized gene expression (n = 7, P < 0.001); (B) normalized Foxp3 gene expression (n = 6, P < 0.001); (C) a representative figure for Foxp3 protein expression; (D) normalized Foxp3 protein expression values (n = 7, P < 0.005).
In Vitro Priming by DCs Pulsed with HIV Leads to Expression of a Broad Array of Inhibitory Molecules
Modulation of gene transcript expression is indicative of ongoing changes at the molecular level that could result in alterations in receptor protein expression, leading to altered downstream signaling events. The overall summary demonstrates (Figure 5A, B) that the expression of negative costimulatory molecules in T cells primed by DCs pulsed with HIV was significantly increased compared with those primed by mock-pulsed DCs (P = 0.002). Next, we set out to study the level of effector attributes of the primed T cells, and the levels of IL-2, TNF-α and IFN-γ in the supernatants of MLRs were assessed by a Bio-Plex™ Cy-tokine Luminex assay. We observed that IL-2 (P = 0.002), TNF-α (P = 0.005) and IFN-γ (P = 0.003) levels were lowered in assays containing T cells primed by HIV-pulsed DCs (Figure 6A, B). Furthermore, we also examined the expression of perforin and granzyme B and observed that the cytolysin levels were relatively lesser in T cells primed by HIV-pulsed DCs than those primed by mock DCs (Figure 7). This finding could be attributed to lesser T-cell activation and, thus, warrants elaborate investigation.
Figure 5.

HIV-pulsed DC-primed naïve T cells give rise to a broad spectrum of inhibitory molecules. (A) Representative gene expression values from all 10 negative stimulatory molecules studied represented in a single graph, (B) Normalized gene expression values from all 10 negative stimulatory values (n = 10, P < 0.001).
Figure 6.

HIV-pulsed DC-primed T cells with decreased TNF-α, IFN-γ and IL-2 secretions. Day-8 supernatants were harvested and analyzed by IL-2, TNF-α and IFN-γ using a commercial Bio-Plex™ Cytokine Luminex assay, and the results were normalized. (A) A representative figure for TNF-α, IFN-γ and IL-2 secretion in MLR supernatants. (B) Normalized TNF-α (n = 7, P < 0.005), IFN-γ (n = 6, P < 0.003) and IL-2 expression values (n = 6, P < 0.05). P < 0.005 and P < 0.001, paired t test.
Figure 7.
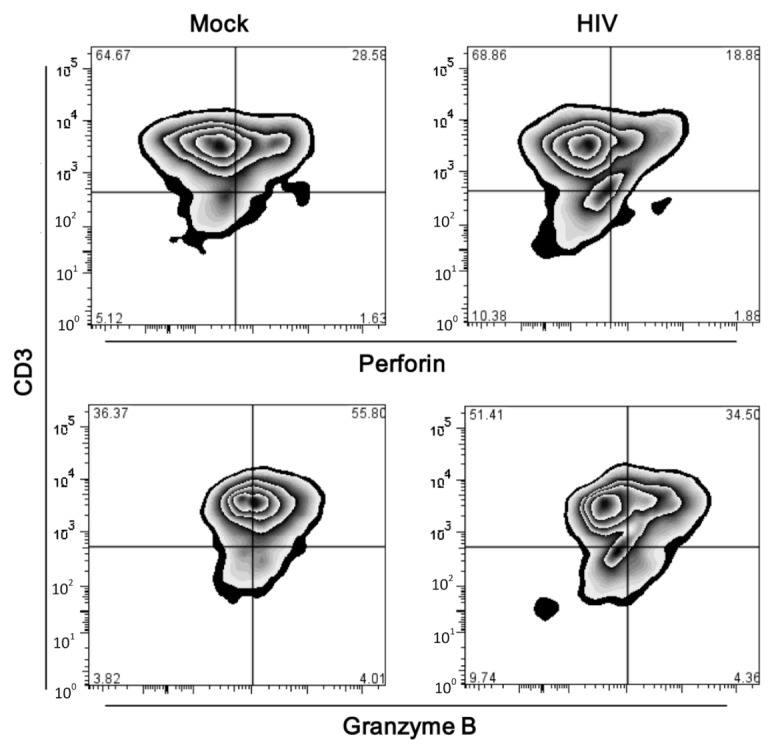
HIV-pulsed DC-primed T cells with decreased protein expression of perforin and granzyme B. Day-8 primed T cells were harvested and stained for intracellular expression of perforin and granzyme B.
PD-1+ T-Cell Priming by HIV-Pulsed DCs Coexpressed TIM-3, LAG-3 and CTLA-4 Inhibitory Molecules
PD-1 is the most widely studied inhibitory molecule in HIV-1, simian immunodeficiency virus (SIV) and certain other persistent viral infections (40–42). Hence, we investigated the coexpression of PD-1 with a few of the negative costimulatory molecules described herein by flow cytometry. PD-1 and coexpression of TIM-3, LAG-3 and CTLA-4 were detected, with much higher levels of double positive in the HIV primed assays; CD160 expression did not show any difference between the mock and HIV-primed conditions (Figure 8).
Figure 8.
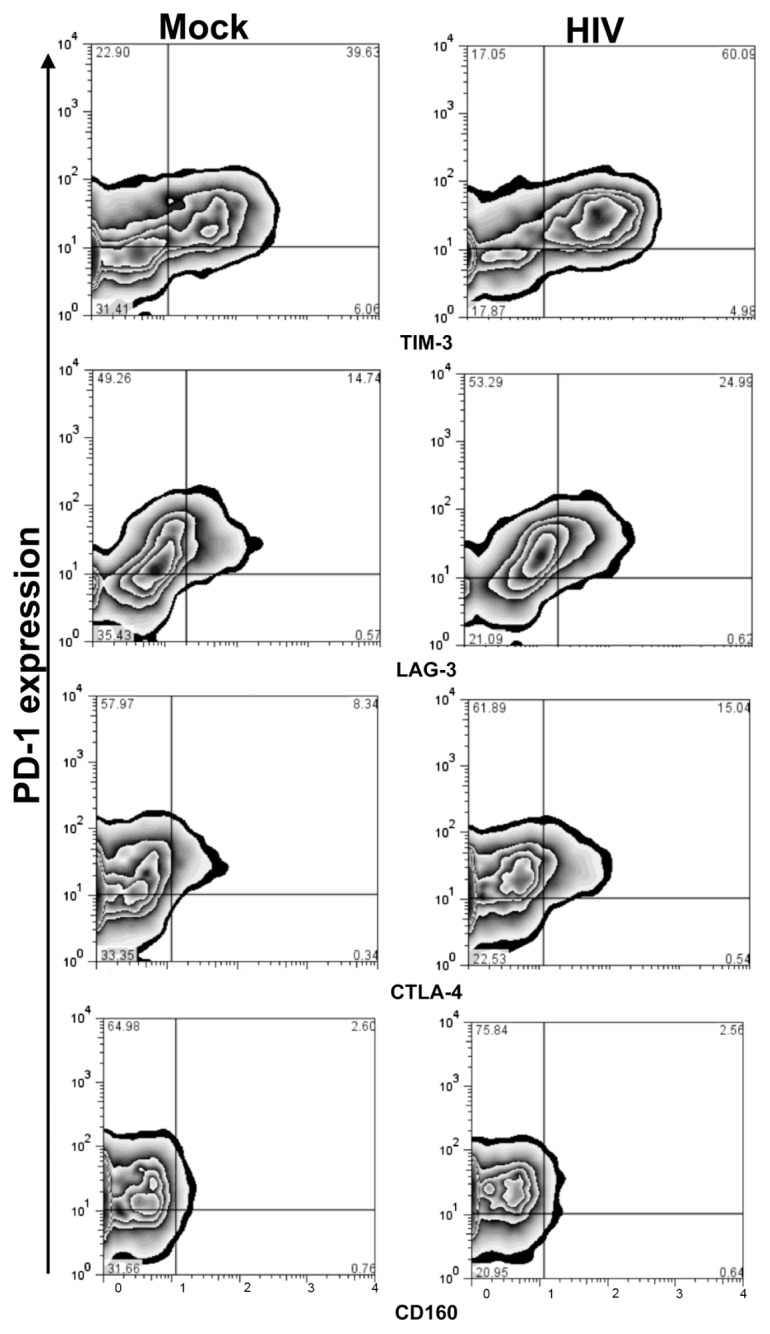
PD-1+ T cells coexpressing TIM-3-APC, LAG-3-PE, CTLA-4-APC and CD160-APC protein. Day-8 primed T cells positive for PD-1 were costained for surface protein of TIM-3, LAG-3, CTLA-4 and CD160.
DISCUSSION
The expression of negative costimulatory molecules on T cells has been proposed as a contributing factor for suboptimal T-cell responses in HIV infection (8). Hence, identification of factors controlling the expression of these molecules on T cells is of great interest to enhance our understanding of the potential mechanisms of T-cell suppression in HIV pathogenesis. We found that the priming of T cells by DCs pulsed with HIV caused an upregulation of an array of negative costimulatory molecules on T cells. Numerous studies have linked the functional role of inhibitory molecules on T cells with the onset of immune dysfunction (13,16,17,19,31,32,43–45). Tregs express in general CTLA-4 (46), CD25 (IL-2Rα), Foxp3, the GITR (glucocorticosteroid-induced TNF family- related receptor) and PD-1. Expansion of Foxp3+ T cells has been linked to ongoing HIV/SIV replication, potentially contributing to immunosuppression (47–49). Priming of T cells with HIV-pulsed DCs was associated with increased expression of CTLA-4 and Foxp3 compared with priming with mock-pulsed DCs (50). Our recent in vitro findings suggested that priming with HIV-pulsed DCs could lead to expansion of suppressor T cells characterized by upregulated expression of PD-1, CTLA-4, TRAIL and Foxp3 (10). This study is in-line with recent studies in T cells of chronic SIV-infected rhesus macaques that showed CTLA-4 and Foxp3 coexpression (48,51).
Recent reports showed that HIV-enhanced LAG-3 expression in T cells (25,52). LAG-3 reportedly is capable of maximal T-cell suppression by itself (31,32,53,54) via its cross-linking of MHC II with high affinity, which inhibits TCR-induced Ca2+ fluxes leading to T-cell inhibition (55). In addition, LAG-3 expression has been linked to functional exhaustion of CD8+ T cells in PVI models (37). Other investigators showed that LAG-3 was expressed on cells that also expressed PD-1 and TIM-3 (56). Recently it was shown that LAG-3 levels were elevated in subjects with HIV infection (52) and unrestrained viral replication (25). Our current results are consistent with such a notion of virus exposure–linked increased expression of LAG-3 playing a negative role in HIV infection (25,52).
Our finding of induction of elevated PD-1 gene and protein expression in T cells primed with HIV-pulsed DCs is particularly encouraging. PD-1 expression on HIV-specific CD8+ T cells (23) has been linked to increased viral load and reduced CD4+ T-cell levels (23,57). Results of recent studies suggested a role for the PD-1 pathway in specific T-cell exhaustion in HIV-infected individuals (5,6), as PD-1 blockade restored T-cell functions (10,23,58). The elevated TRAIL levels in T cells pulsed with HIV-pulsed DCs is intriguing because TRAIL can negatively regulate proliferation via mechanisms distinct from apoptosis (59). TRAIL can interact with DR4 and DR5 receptors, which are capable of inducing apoptosis (60), and 3 other receptors that facilitate suppression without initiating apoptosis (61). We recently showed increased TRAIL expression on T cells primed by HIV-pulsed DCs and its involvement in the suppression of T-cell proliferation (10). In addition, clinical studies also showed that TRAIL was elevated in HIV-infected compared with un-infected subjects, and that antiretroviral therapy that lowered viral load dramatically decreased TRAIL expression (30). Hence, TRAIL could be another potential negative factor contributing to T-cell suppression in HIV infection.
BTLA (CD272) inhibits TCR-mediated signaling via its immunoreceptor tyrosine-based inhibitory and switch motifs (18,62). Likewise, CD160 mediates negative signaling and is induced in a similar manner as CTLA-4 in T cells (16). CD160 is expressed on NK, NKT and in-traepithelial T cells, and in a low frequency of peripheral CD4 and CD8+ T cells. Engaging CD160 or BTLA on T cells with their ligand HVEM on Tregs inhibited effector CD4+ T-cell responses (63,64). CD160 has been shown to be increased in acute and chronic HIV infections on both total and HIV-specific CD8+ T cells (65,66), which is in-line with our in vitro observations, hence linking the role of HIV with the upregu-lation of CD160 in T cells. We herein hypothesize its role in contributing to T-cell suppression in vitro.
Our findings of elevated expression of transcription factors Foxp3 and Blimp-1 in T cells primed with HIV-pulsed DCs have opened up newer avenues in HIV infection. Recent findings associated increased Foxp3 expression with the onset of T-cell dysfunction in HIV/AIDS (67). The elevated expression of both Foxp3 and Blimp-1 in T cells primed with HIV-pulsed DCs suggests a potential direct role of Foxp3 in controlling Blimp-1 expression in antigen-exposed T cells, consistent with prior observations from a genome-wide investigation, which showed that Blimp-1 is directly activated by Foxp3, adding key dimension to the notion that Blimp-1 is necessary for accurate functioning of suppressor T cells (39). Blimp-1 is an evolutionarily conserved transcriptional repressor (44) that attenuates T-cell proliferation (68), homeostasis and CD4+ Treg functions (69) and is reportedly enhanced in antigen-experienced T cells (70). Blimp-1 was upregulated in herpes simplex virus–specific CD8+ T cells (69) and Blimp-1-deficient CD4+ T cells were hyperproliferative (71). Intriguingly, high Blimp-1 expression correlated with increased PD-1, CTLA-4 and CD160 expression in chronic HIV infection (38), similar to our findings in vitro. Furthermore, numerous Foxp3-linked genes were either up- or downregulated in T cells, indicating that Foxp3 may act both as a transcriptional activator and a repressor (39,45). Although Foxp3 is associated with suppressor T-cell development, the physiological inducers of Foxp3 have remained elusive. Foxp3 regulates CD4+ T-cell activation, and T cells upregulate Foxp3 to acquire suppressive attributes upon stimulation (72). Also, HIV/SIV (48) can induce Foxp3 expression (49,73) in T cells, and viral attachment enhances the suppressive potentials of CD4+ CD25+ Tregs (73) in HIV (9,74,75) and SIV infections (48). Interestingly, high CTLA-4 expression on Tregs also depends on Foxp3 along with the nuclear factor of activated T cells (76,77). Our findings of elevated Blimp-1 and Foxp3 coexpression in HIV-experienced T cells is in-line with the notion that Blimp-1 may be central for T-cell functions, a topic for further investigation. In addition, the likely synergistic role of Blimp-1, Foxp3 and other negative molecules at both the gene and protein levels requires a closer examination, especially in regard to Blimp-1 signaling.
Certain inhibitory molecules are linked to T-cell exhaustion, and many of these molecules coexist on T-cells in HIV infection (43). Certain immunoglobulin superfamily members and the TNF receptor (78) regulate clonal expansion, deletion and/or anergy induction. Earlier studies showed that TIM-3 expression is indicative of T-cell tolerance (24,33). Results of more recent studies of lymphocytic choriomeningitis virus infection showed the coexpression of TIM-3, LAG-3 and PD-1 (56). TIM-3 was elevated on T cells in acute and progressive chronic HIV infection (24). Our experiments showed increased TIM-3 expression when HIV was present, which could be an indication of T-cell dysfunction. The intrinsic expression of negative costimulatory molecules regulates T-cell functions and may limit antiviral capabilities (2). We have shown the induction of an array of these molecules (CD160, PD-1, LAG-3, CTLA-4, TRAIL, TIM-3, Foxp3 and Blimp-1) on T cells primed by HIV-pulsed DCs both at the molecular and protein levels. These immunoregulatory factors have been shown to suppress effective antiviral T-cell responses in the microenvironment. Furthermore, it is likely that these negative costimulatory molecules are coexpressed; for example, the PD-1+ population also expressed TIM-3, LAG-3, and CTLA-4. Indoleamine 2,3-dioxygenase (IDO) is another factor commonly linked with immune suppression (2), because we have also found increased IDO expression alongside these suppressor molecules (M Larsson, 2010, unpublished data). Therefore, the T-cell types that are described herein could broadly be considered suppressor T cells, as shown in our earlier findings (10). Thus, priming of T cells in the setting of abundant HIV could lead to increased expression of negative costimulatory molecules on T cells. It is likely not coincidental that HIV preferentially upregulated expression of inhibitory molecules likely to enable its persistence in an infected host. Ligation of CTLA-4 with B7 leads to IDO upregulation in DCs (M Larsson, 2010, unpublished data) that results in kynureine accumulation curtailing T-cell proliferation (71). HIV-pulsed DC priming of T cells leads to upregulation of Foxp3 that activates Blimp-1 expression in T cells (70), as with our in vitro experiments. Others have shown that enhanced Blimp-1 correlated directly with CD160, PD-1, CTLA-4 and other receptors (37), similar to our findings in MLRs, which supports the likely role of Blimp-1 in controlling the expression of the above negative molecules. The above cascade of events in vivo would facilitate immune hyperactivation, eventually resulting in rapid disease progression (48). Moreover, levels of perforin and granzyme B were lowered in the HIV primed T-cell populations, which could be the side effect of lower T-cell activation in these assays, and requires further investigation.
To conclude, we have shown that the negative costimulatory molecular expression possibly could account for the subsequent onset of immune impairments. This high expression, especially of the transcriptional repressor Blimp-1 in T cells and its potential association with T-cell suppression and exhaustion requires further investigation. Hence, we postulated that after priming by HIV-pulsed DCs in vitro, naïve T-cells could lead to expansion of suppressor T cells as described earlier (10) and could thus participate in the immune deficiency associated with HIV infection. Furthermore, although it is unclear as to which specific antigenic component of HIV is responsible for the generation of suppressor T-cell phenotypes, it is now clear that HIV infection in vitro could modulate DCs to generate suppressor T cells, armed by numerous inhibitory molecules with immunoimpairing abilities.
ACKNOWLEDGMENTS
We thank the Biological Products Core of the AIDS and Cancer Virus Program, SAIC Frederick, National Cancer Institute, Frederick, MD, USA, for generously providing HIV. This work has been supported by grants through: M Larsson: AI52731, the Swedish Research Council, the Swedish, Physicians against AIDS Research Foundation, the Swedish International Development Cooperation Agency; SIDA SARC, VINNMER for Vinnova, Linköping University Hospital Research Fund, CALF, and the Swedish Society of Medicine. JD Lifson was supported in part with federal funds from the National Cancer Institute, National Institutes of Health, under contracts NO1-CO-124000 and HHSN266200400088C.
Footnotes
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Online address: http://www.molmed.org
REFERENCES
- 1.Shin H, Wherry EJ. CD8 T-cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19:408–15. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 3.Boni C, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–25. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufmann DE, et al. Upregulation of CTLA-4 by HIV- specific CD4+ T-cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8:1246–54. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 5.Urbani S, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Migueles SA, et al. Defective human immunodeficiency virus-specific CD8+ T-cell poly-functionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J Virol. 2009;83:11876–89. doi: 10.1128/JVI.01153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol. 2009;182:5891–7. doi: 10.4049/jimmunol.0803771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss L, et al. Human immunodeficiency virus-driven expansion of CD4+CD25+ Tregs, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood. 2004;104:3249–56. doi: 10.1182/blood-2004-01-0365. [DOI] [PubMed] [Google Scholar]
- 10.Che KF, et al. HIV-1 impairs in vitro priming of naive T cells and gives rise to contact-dependent suppressor T cells. Eur J Immunol. 40:2248–58. doi: 10.1002/eji.201040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–78. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 12.Coyle AJ, Gutierrez-Ramos JC. The expanding B7 superfamily: increasing complexity in costimulatory signals regulating T cell function. Nat Immunol. 2001;2:203–9. doi: 10.1038/85251. [DOI] [PubMed] [Google Scholar]
- 13.Brunner MC, et al. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813–20. [PubMed] [Google Scholar]
- 14.Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J Immunol. 1998;160:1532–8. [PubMed] [Google Scholar]
- 15.Zhu C, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 16.Cai G, et al. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9:176–85. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 17.Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev. 2009;229:244–58. doi: 10.1111/j.1600-065X.2009.00783.x. [DOI] [PubMed] [Google Scholar]
- 18.Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Bio-phys Res Comm. 2003;312:1236–43. doi: 10.1016/j.bbrc.2003.11.070. [DOI] [PubMed] [Google Scholar]
- 19.Carter L, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–43. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Workman CJ, et al. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 2004;172:5450–5. doi: 10.4049/jimmunol.172.9.5450. [DOI] [PubMed] [Google Scholar]
- 21.Sedy JR, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–8. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 22.Gamberg J, Pardoe I, Bowmer MI, Howley C, Grant M. Lack of CD28 expression on HIV-specific cytotoxic T lymphocytes is associated with disease progression. Immunol Cell Biol. 2004;82:38–46. doi: 10.1111/j.1440-1711.2004.01204.x. [DOI] [PubMed] [Google Scholar]
- 23.Day CL, et al. PD-1 expression on HIV- specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 24.Jones RB, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–79. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price P, et al. CXCR4 or CCR5 tropism of human immunodeficiency virus type 1 isolates does not determine the immunological milieu in patients responding to antiretroviral therapy. Viral Immunol. 2006;19:734–40. doi: 10.1089/vim.2006.19.734. [DOI] [PubMed] [Google Scholar]
- 26.Rossio JL, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. 1998;72:7992–8001. doi: 10.1128/jvi.72.10.7992-8001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen HY, Di Mascio M, Perelson AS, Ho DD, Zhang L. Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc Nat Acad Sci U S A. 2007;104:19079–84. doi: 10.1073/pnas.0707449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimitrov DS, et al. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol. 1993;67:2182–90. doi: 10.1128/jvi.67.4.2182-2190.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubong Sabado R, et al. In vitro priming recapitulates in vivo HIV-1 specific T cell responses, revealing rapid loss of virus reactive CD4 T cells in acute HIV-1 infection. 2009;4:e4256. doi: 10.1371/journal.pone.0004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herbeuval JP, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106:3524–31. doi: 10.1182/blood-2005-03-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Workman CJ, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. 2009;182:1885–91. doi: 10.4049/jimmunol.0800185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang CT, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–13. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Sanchez-Fueyo A, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 34.Sabatos CA, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–10. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 35.Golden-Mason L, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83:9122–30. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leuk Biol. 87:223–35. doi: 10.1189/jlb.0809590. [DOI] [PubMed] [Google Scholar]
- 37.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin H, et al. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–20. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng Y, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–40. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 40.Titanji K, et al. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Invest. 2010;120:3878–90. doi: 10.1172/JCI43271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Velu V, et al. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol. 2007;81:5819–28. doi: 10.1128/JVI.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velu V, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–10. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hafler DA, Kuchroo V. TIMs: central regulators of immune responses. J Exp Med. 2008;205:2699–701. doi: 10.1084/jem.20082429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.John SA, Garrett-Sinha LA. Blimp1: a conserved transcriptional repressor critical for differentiation of many tissues. Exp Cell Res. 2009;315:1077–84. doi: 10.1016/j.yexcr.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 45.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–5. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi T, et al. Immunologic self- tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Estes JD, et al. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J Infect Dis. 2006;193:703–12. doi: 10.1086/500368. [DOI] [PubMed] [Google Scholar]
- 48.Nigam P, et al. Expansion of FOXP3+ CD8 T cells with suppressive potential in colorectal mucosa following a pathogenic simian immunodeficiency virus infection correlates with diminished antiviral T cell response and viral control. J Immunol. 2010;184:1690–701. doi: 10.4049/jimmunol.0902955. [DOI] [PubMed] [Google Scholar]
- 49.Nilsson J, et al. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood. 2006;108:3808–17. doi: 10.1182/blood-2006-05-021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saito T, Yamasaki S. Negative feedback of T cell activation through inhibitory adapters and costimulatory receptors. Immunol Rev. 2003;192:143–60. doi: 10.1034/j.1600-065x.2003.00022.x. [DOI] [PubMed] [Google Scholar]
- 51.Boasso A, et al. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood. 2007;109:3351–9. doi: 10.1182/blood-2006-07-034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li N, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007;26:494–504. doi: 10.1038/sj.emboj.7601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005;115:170–8. doi: 10.1111/j.1365-2567.2005.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. 2003;33:970–9. doi: 10.1002/eji.200323382. [DOI] [PubMed] [Google Scholar]
- 55.Hannier S, Tournier M, Bismuth G, Triebel F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J Immunol. 1998;161:4058–65. [PubMed] [Google Scholar]
- 56.Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. Int Immunol. 22:13–23. doi: 10.1093/intimm/dxp107. [DOI] [PubMed] [Google Scholar]
- 57.Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 58.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 59.Lunemann JD, et al. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J Immunol. 2002;168:4881–8. doi: 10.4049/jimmunol.168.10.4881. [DOI] [PubMed] [Google Scholar]
- 60.Chaudhary PM, et al. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–30. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- 61.Pan G, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–3. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 62.Vendel AC, et al. B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol. 2009;182:1509–17. doi: 10.4049/jimmunol.182.3.1509. [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez LC, et al. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc Nat Acad Sci U S A. 2005;102:1116–21. doi: 10.1073/pnas.0409071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watanabe N, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–9. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 65.Li Q, et al. Microarray analysis of lymphatic tissue reveals stage-specific, gene expression signatures in HIV-1 infection. J Immunol. 2009;183:1975–82. doi: 10.4049/jimmunol.0803222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peretz Y, et al. Elite controllers are enriched with HIV-specific cells expressing CD160 but lacking PD-1. Proceedings of the 16th Conference on Retroviruses and Opportunistic Infections; 2010 Feb 16–19; San Francisco, CA. Abstract nr 356. [Google Scholar]
- 67.Loxton A, Roberts T, Black G, Walzl G. Regulatory T-cells and high levels of FOXP3 mRNA leads to decreased immune responses in HIV-TB co-infection. Retrovirology. 2009;6(Suppl 3):230. [Google Scholar]
- 68.Nutt SL, Fairfax KA, Kallies A. BLIMP1 guides the fate of effector B and T-cells. Nat Rev Immunol. 2007;7:923–7. doi: 10.1038/nri2204. [DOI] [PubMed] [Google Scholar]
- 69.Kallies A, et al. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. 2006;7:466–74. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- 70.Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Ann Rev Immunol. 2008;26:133–69. doi: 10.1146/annurev.immunol.26.021607.090241. [DOI] [PubMed] [Google Scholar]
- 71.Martins GA, et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. 2006;7:457–65. doi: 10.1038/ni1320. [DOI] [PubMed] [Google Scholar]
- 72.Pillai V, Ortega SB, Wang CK, Karandikar NJ. Transient regulatory T-cells: a state attained by all activated human T-cells. Clin Immunol. 2007;123:18–29. doi: 10.1016/j.clim.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andersson J, et al. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J Immunol. 2005;174:3143–7. doi: 10.4049/jimmunol.174.6.3143. [DOI] [PubMed] [Google Scholar]
- 74.Aandahl EM, Michaelsson J, Moretto WJ, Hecht FM, Nixon DF. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J Virol. 2004;78:2454–9. doi: 10.1128/JVI.78.5.2454-2459.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kinter AL, et al. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med. 2004;200:331–43. doi: 10.1084/jem.20032069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Birebent B, et al. Suppressive properties of human CD4+CD25+ regulatory T cells are dependent on CTLA-4 expression. Eur J Immunol. 2004;34:3485–96. doi: 10.1002/eji.200324632. [DOI] [PubMed] [Google Scholar]
- 77.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 78.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]