

Abstract
Myeloid-derived suppressor cells (MDSCs) are a heterogenous population of immature myeloid cells whose numbers dramatically increase in chronic and acute inflammatory diseases, including cancer, autoimmune disease, trauma, burns and sepsis. Studied originally in cancer, these cells are potently immunosuppressive, particularly in their ability to suppress antigen-specific CD8+ and CD4+ T-cell activation through multiple mechanisms, including depletion of extracellular arginine, nitrosylation of regulatory proteins, and secretion of interleukin 10, prostaglandins and other immunosuppressive mediators. However, additional properties of these cells, including increased reactive oxygen species and inflammatory cytokine production, as well as their universal expansion in nearly all inflammatory conditions, suggest that MDSCs may be more of a normal component of the inflammatory response (“emergency myelopoiesis”) than simply a pathological response to a growing tumor. Recent evocative data even suggest that the expansion of MDSCs in acute inflammatory processes, such as burns and sepsis, plays a beneficial role in the host by increasing immune surveillance and innate immune responses. Although clinical efforts are currently underway to suppress MDSC numbers and function in cancer to improve antineoplastic responses, such approaches may not be desirable or beneficial in other clinical conditions in which immune surveillance and antimicrobial activities are required.
INTRODUCTION
Over the last decade, a novel heterogeneous population of immature myeloid cells with immunosuppressive properties has been described, and these cells have recently been coined myeloid-derived suppressor cells (MDSCs) (1–3). Much of the early work on the origins and functions of these cells has been in experimental and human cancer, in which these populations are known to be immunosuppressive and to result in both reduced immune surveillance and antitumor cytotoxicity (2). However, more recent findings suggest that expansion of these immature myeloid cell populations may not be limited to cancer, and that they are linked to most if not all chronic and acute inflammatory processes (3). Therefore, should MDSCs be viewed solely from the context of an anomalous and pathologic response to cancer or could the expansion of these cell populations be considered an integral component of the host response to any inflammatory stimuli? Rather than an adverse immunosuppressive response, the expansion of this cell population(s) more than likely represents a complex balance between increased immune surveillance and dampened adaptive immune responses common to many inflammatory responses.
In this review we explore the origins of these cell populations during inflammation, focusing on their role in acute inflammatory processes such those that occur during trauma and sepsis. We propose that the overall role of MDSCs involves much more than simply being an immunosuppressive population unique to some cancers. Rather, MDSC expansion is a common response to all inflammatory processes, and the functions of MDSCs are highly dependent on the circumstances in which their expansion occurs. Like much of the host response to inflammation, the expansion of the MDSC population poses both beneficial opportunities as well as potential damaging costs to the host. MDSCs have potent innate immune effector cell function, and during periods of systemic insult (that is, cancer growth, sepsis) may actually serve to protect the host from opportunistic infectious insults. Manipulation of MDSC expansion and function offers unique opportunities, but also poses risks and uncertainties.
All supplementary materials are available online at www.molmed.org.
MDSCs: HETEROGENOUS AND POORLY DESCRIBED
MDSCs have been known for several decades under a number of different monikers, ranging from “natural suppressor cells” to “immature myeloid cells” to “suppressor macrophages” (4,5). These cells have been defined predominantly by their functional properties, and little is known about the specific identity of these cell populations. In mice, MDSCs have been characterized as an inducible cell population that expresses cell-surface CD11b and GR-1 antigens, does not or only weakly expresses other markers of mature myeloid cells (such as CD14 and MHC class II antigens), has increased expression of arginase (ARG) and inducible nitric oxide synthetase (iNOS), and produces large quantities of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (6). These cells have the capacity to suppress predominantly antigen-specific CD8+ and CD4+ T-cell responses. Although these criteria are well accepted in the cancer literature, they are by no means highly specific or inclusive, and this ambiguity has often led to conflicting descriptions of their population and the argument that MDSCs originating in cancer may be different from those expanding during other acute and chronic inflammatory diseases, such as in trauma, burns, sepsis and autoimmune diseases (7).
Other markers and phenotypes have been used to further classify these cell populations, as well as to discriminate them from other myeloid cells with suppressor cell function, such as tumor-associated macrophages (8). As shown in Table 1, many investigators have attempted to further classify MDSCs on the basis of several strategies: their relative expressions of CD11b+ and the Ly6 superfamily (Ly6G and Ly6C), which is recognized by the GR-1 antibody (RB6-8C5); their overall immaturity; and their suppressive activity (6,9). In terms of their immaturity, most investigators use expression of the adhesion molecule PECAM-1 (CD31), because this marker is present on progenitor and blast myeloid cells, in addition to poor expression of MHC II and costimulatory molecules CD80/86 (10,11). For example, we have reported that after sepsis, approximately 30% of the CD11b+GR-1+ splenocyte population expresses CD31, and less than 3% of the population expresses MHC II (12). The suppressor activity within MDSC populations has also been associated with multiple markers including macrophage colony-stimulating factor (M-CSF) receptor (CD115) and interleukin 4 (IL-4) receptor-α (CD124) (9). To add to this heterogeneity, MDSC populations from a variety of inflammatory states also have varying numbers of mature myeloid cells, such as CD11c+ and F4/80+ populations, depending on the experimental model.
Table 1.
Cell surface phenotyping of murine MDSC populations.a
| Study | Disease | MDSC classification | Additional markers used |
|---|---|---|---|
| Gabrilovich et al., J. Immunol. 2001 (57) | Cancer | Gr-1+, CD11b+ | MHCII |
| Sinha et al., J. Immunol. 2005 (58) | Cancer | Gr-1+, CD11b+ | MHC II, CD80/86, F4/80, CD11c, |
| Makarenkova et al., J. Immunol. 2006 (48) | Trauma | Gr-1+, CD11b+ | MHC II, CD80/86, F4/80, CD11c, CD31 |
| Delano et al., J. Exp. Med. 2007 (12) | Sepsis | Gr-1+, CD11b+ | MHCII, CD31 |
| Zhu et al., J. Immunol. 2007 (56) | Experimental autoimmune encephalomyelitis | Ly6C+, Ly6G+, Gr-1+, CD11b+ | MHCII, CD80/86, F4/80, CD11c, CD31 |
| Noel et al., Shock 2005 (31) | Burn | Gr-1+, CD11b+ | F4/80, CD115, CD117 |
| Youn et al., J. Immunol. 2008 (14) | Cancer | Ly6C+, Ly6G+, Gr-1+, CD11b+ | CD80, CD115, CD124, PD-L1 |
| Dolcetti et al., Eur. J. Immunol. 2010 (59) | Cancer | Ly6C+, Ly6G+, Gr-1+, CD11b+ | F4/80, CD11c, CD31, CD124 |
All investigators use both CD11b and GR-1 or Ly6C/Ly6G staining to characterize their MDSC population. However, it is evident that the populations may differ significantly in their expression of other cell surface markers.
Within the CD11b+GR-1+ MDSC population, there have been several efforts by investigators to identify more highly enriched MDSC subpopulations that possess the immunosuppressive phenotype. Although the results have often been conflicting and remain controversial, several investigators have subdivided murine MDSCs into two subpopulations, termed polymorphonuclear (PMN)-MDSCs and mononuclear (MO)-MDSCs, on the basis of their relative expression of CD11b, Ly6G and Ly6C (13). MO-MDSCs are generally classified as CD11b+Gr-1intLy6G−Ly6Chigh cells, and they frequently express higher levels of F4/80, CD115 and CCR2 compared with PMN-MDSCs (13,14). These MDSCs are potently immunosuppressive, blocking antigen-specific CD8+ T-lymphocyte proliferation through an iNOS–mediated mechanism. In contrast, the PMN-MDSCs, classified as being CD11b+GR-1highLy6ClowLy6G+, are also immunosuppressive, but their mechanism(s) of action are thought to be more dependent on arginase and interferon-γ (6). Along the same lines, other investigators have subdivided MDSCs predominantly on the basis of the intensity of their GR-1 staining. For example, CD11b+GR-1high/bright subpopulations of MDSCs are primarily composed of Ly6G+Ly6C− populations and are the most mildly immunosuppressive, producing low levels of both iNOS and arginase (13,14). Conversely, the CD11b+GR-1intermed/dim population is comprised of mostly the MO-MDSCs with Ly6CbrightLy6G− expression and potent immunosuppressive phenotypes.
A picture is often worth a thousand words, and the heterogeneity of the murine MDSC population is best revealed in cytospin preparations from GR-1+-enriched splenocytes from healthy, septic, traumatized, tumor-bearing and other inflamed mice (Figure 1). Enriched for GR-1+ cells, these splenic MDSCs reflect the true heterogeneity of the population, ranging from what appear to be nearly mature PMNs to the classic ringed MDSCs (thought to be CD11b+GR-1+Ly6G+Ly6C−), to the more monocyte-ringed (doughnut), to the more immature-appearing monocyte blast-like cell population. What is noticeable, however, is the vastly different appearance of similar GR-1+–enriched MDSC populations obtained from tissues affected with different inflammatory conditions. Although all are GR-1+ splenocytes, the MDSC populations from tissues affected by trauma, sepsis and advanced tumor growth are very different in their physical appearance, with varying numbers of the classic ringed MDSC and the more immature-appearing blast cell population. Even among the same inflammatory processes, phenotypic differences in the MDSC population appear over time. We have observed in both septic mice and in tumor-bearing animals that as the inflammatory process progresses, the numbers of more mature PMN-like and ringed MDSCs appear to decline, and are replaced by more immature cell populations (unpublished observations). This observation is confirmed by increased expression of CD31+ and decreased MHC class II expression in GR-1+ splenocyte populations associated with prolonged sepsis or tumor growth. The implications are considerable, suggesting that the term MDSC may not reflect the same cell populations with the same functionality in different clinical conditions.
Figure 1.
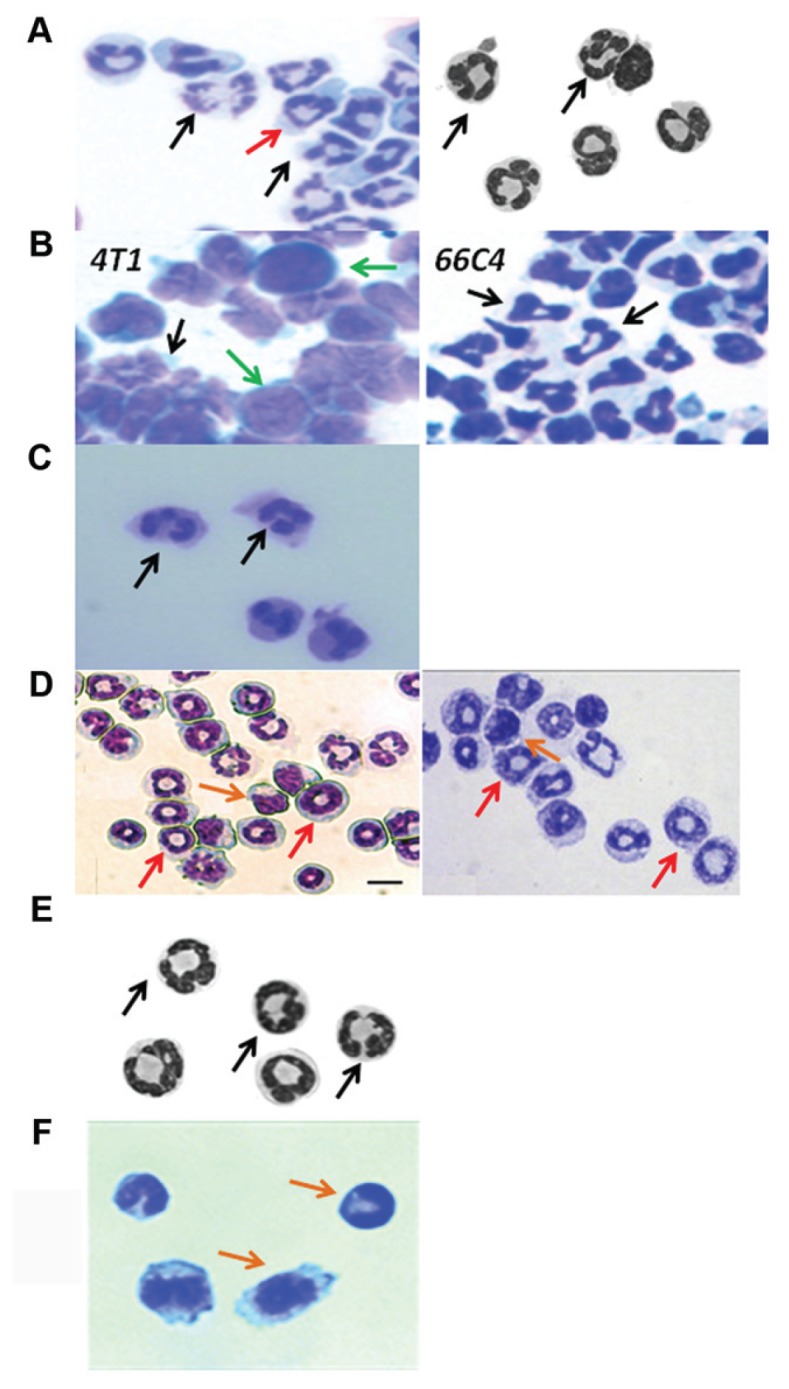
Appearance of MDSCs obtained by cytospin from different clinical conditions. (A) Murine control; (B) murine cancer; (C) human cancer; (D) murine sepsis; (E) murine trauma; (F) murine autoimmunity. GR-1+ splenocytes were obtained from a variety of experimental conditions, including healthy controls (Left, unpublished data; Right [48]), animals with transplantable tumors (Left and Right, unpublished data), human cancer (18), after trauma (48), sepsis (Left [12] and Right [28]) and experimental autoimmune disease (56). Although the photomicrographs were obtained under different experimental conditions, the populations of GR-1+ cells differ dramatically not only within the same condition, but also between conditions. MDSCs phenotypically vary from more mature-appearing neutrophils, to ringed cells, to more immature blast-like–appearing cells. Arrows indicate different subsets of MDSCs: granulocytic (black), ringlike (red), monocytic (orange) and blast-like (green).
In humans, the problem of identifying MDSCs is exponentially greater. The absence of GR-1 in humans has made the identification of human MDSCs much more challenging. In addition, reduced access to secondary lymphoid organs and reticuloendothelial tissues has forced greater reliance on identifying these homologous populations in human blood, not the ideal location. Investigators have tried to recapitulate the PMN-MDSC and MO-MDSC phenotypes in humans by identifying CD11b+CD14−CD15+ cells with a PMN granulocyte morphology as the former (15). MO-MDSCs have been identified from a peripheral blood mononuclear cell fraction as being CD14+CD11b+HLA-DRlow/neg and secreting transforming growth factor-β (16). Other investigators have identified human MDSCs as CD11b+CD14−CD15+CD33+ or as Lineagelow/−HLA-DR−CD33+CD11b+ with suppressor phenotypes (2,17,18). Yet, as is obvious, these human criteria are even less specific than those used in mice, and the search for the elusive human MDSC continues.
MDSCs ARE MORE THAN ADAPTIVE IMMUNE SUPPRESSOR CELLS
Both human and murine tumor studies have demonstrated that MDSCs produce increased levels of iNOS, arginase (ARG), and ROS, all of which have been associated with antigen- specific T-cell suppression (19–21) (Table 2). The first two enzymes are linked via l-arginine, which is not only the substrate for both enzymes, but is also a conditionally essential amino acid that must be exogenously provided to an organism during times of stress for proper T-cell function (22) (that is, during advanced cancer, trauma or sepsis). Multiple in vitro and in vivo experiments have shown that MDSCs consume l-arginine from the microenvironment, more than likely as a result of increased ARG and iNOS expression (8,23,24). l-arginine depletion has been shown to induce dysregulation of T-cell receptor (TCR)-mediated signaling as well as cell-cycle arrest (24). This mechanism has been described in both human and murine studies (17,18,21,24). In addition, Nagaraj and colleagues have demonstrated that exaggerated production of peroxynitrates, but not necessarily NO or ROS, leads to direct nitrosylation of MHC molecules, thereby preventing the interaction with CD8 TCRs and subsequent T-cell anergy (25). The same group has also demonstrated that MDSC contact with T cells causes dissociation of the TCR-signaling complex, specifically CD3ζand the TCR, further impairing T-cell responses (25).
Table 2.
Functional properties of MDSCs.a
| Pathologic process | Mechanism/function | Reference(s) | |
|---|---|---|---|
| Upregulated MDSC function | |||
| Arginase (ARG) | Cancer | l-arginine depletion and NO scavenging | (2,24) |
| Inducible nitric oxide synthetase | Cancer | l-arginine depletion and bactericidal | (2,24) |
| NADPH oxidase | Cancer | ROS/RNS production, nitrosylation of TCR, and bactericidal | (20,25) |
| M-CSF R (CD115) | Cancer | Associated with suppressive activity | (23,60) |
| IL-4rα (CD124) | Cancer | Th2 skewing and associated with suppressive activity | (9,14,23) |
| Proinflammatory cytokines | |||
| IL-1β | Cancer | Inflammation | (61) |
| TNF-α | Cancer, Sepsis | Inflammation | (12,27,61) |
| LTA4H | Cancer | Inflammation | (27) |
| MIP-1β | Sepsis | Inflammation; chemokine | (12) |
| CXCL10 | Cancer | Inflammation; chemokine | (61) |
| RANTES | Sepsis | Inflammation; chemokine | (12) |
| T-helper cell 2 skewing/antiinflammatory cytokines | |||
| IL-10 | Sepsis | Antiinflammatory; inhibits T-cell responses; causes TRC expansion | (12) |
| IL-13 | Cancer | Th2-polarizing cytokine | (9) |
| Immunosuppression | |||
| Direct antigen-specific CD8+ T-cell anergy | Cancer | MHC restricted dissociation of CD3ζ/TCR signaling | (25) |
| Indirect antigen-specific CD8+ T-cell anergy | Cancer | ARG, ROS, iNOS | (2,14,26) |
| Indirect antigen-specific CD4+ T-cell anergy | Cancer | ARG | (2,26) |
| TRC expansion | Cancer | CD40, IL-10 | (62) |
| Suppression of graft-versus-host disease | Cancer | IL-10 and TCR activation | (63) |
| Protection against infection | |||
| Protection against primary infection | Sepsis | Regulation of innate inflammation | (28) |
| Protection against secondary infection | Burn | Increases PMNs and inflammatory monocytes | (30) |
MDSCs are known to be pluripotent, affecting both innate and adaptive immunity through direct contact and the secretion of various soluble factors (including iNOS, ROS and cytokines). These properties are responsible for both the immunosuppressive function of MDSCs as well as their protective role against primary and secondary infection.
Although the expansion of MDSCs in tumor-bearing hosts ultimately leads to T-cell inhibition, MDSCs possess machinery that would enhance their innate immune effector function. Corzo et al. have demonstrated that MDSCs from tumor-bearing mice have upregulated p47phox and p91phox compared with their tumor-free counterparts, thereby increasing their ROS production versus non–tumor-bearing or naïve animals (20). In addition, there is evidence that increasing the inflammatory milieu in tumor-bearing mice further augments ROS production from MDSCs (26). Pande and colleagues have also shown that CD11b+GR-1+ cells isolated from tumor-bearing hosts produce increased myeloperoxidase and eosinophil peroxidase (27). These observations, coupled with the fact that both iNOS and ROS are vital components of innate immune effector-cell function and the massive expansion of these cells during tumor growth, sepsis and burns (12,28–32), indicate that MDSCs are poised to be excellent sentinels against infection in the face of systemic stress.
MDSCs obtained from tumor-bearing animals and patients produce increased amounts of the antiinflammatory and immunosuppressive cytokine IL-10 (33,34). This finding has led to the speculation that MDSCs may potentiate IL-10–dependent immune suppression and polarization of the T-helper 2 (Th2) adaptive immune response, as well as stimulate the development of regulatory T cells. However, we noted that MDSCs obtained from septic mice not only produced increased amounts of IL-10, but also produced increased amounts of the proinflammatory cytokine tumor necrosis factor α (TNFα), as well as the chemokines RANTES (regulated on activation normal T cell expressed and secreted) and MIP1α (macrophage inflammatory protein 1α) (12). Noel and Ogle also demonstrated that MDSCs isolated from the spleens of burned mice exhibited elevated production of TNFα, monocyte chemotactic protein-1 and stromal cell-derived factor 1 (SDF-1) (32). Thus, these MDSCs are eminently capable of producing a wide variety of inflammatory mediators, as well as the antiinflammatory IL-10.
These observations highlight several properties of MDSCs that have not received as much attention as their immunosuppressive properties on CD8+ and CD4+ T cells, which are of primary interest in the cancer literature. The biological role of MDSCs has only recently been explicitly associated with pathogen-surveillance or septic shock, although this tumor-induced myelopoietic expansion is in part regulated by inflammation and by the release of proinflammatory proteins such as S100A8/9 proteins and IL-1β (26,35). This hematopoietic link (described below) is critical to the host stress response because inflammation serves as a beacon for the primary innate immune response. Therefore, in settings of an uncontrolled systemic insult, such as metastatic tumor growth and sepsis, MDSCs and their unique properties may ultimately serve a role in protecting the host from infection through the production of increased antimicrobial products and by reducing the magnitude of the septic response (28).
ORIGINS OF MDSCs ARE IN “EMERGENCY MYELOPOIESIS”
Our evolving understanding of the role that MDSCs play in chronic and acute inflammatory processes suggests that MDSC expansion is not simply a pathologic response to a growing tumor, but rather is a programmed response to inflammation, regardless of its source. Mature myeloid cells are a relatively diverse population with half-lives ranging from a few hours for blood neutrophils to months and years for terminally differentiated macrophages and dendritic cells. However, during infection and inflammation, the requirements for and the consumption of these cells increases dramatically as the host responds to both exogenous microbial products, the presence of non-self, and endogenous danger signals, alarmins, and DAMPs (damage-associated molecular patterns) with a reorganization of myelopoiesis (36,37). Myeloid cells are vital to this process as both direct effectors of innate immunity, involved in the phagocytosis and/or killing of microbial products or transformed cells, and through the release of inflammatory mediators and communication with adaptive immunity (38). Normally, physiologic numbers of mature neutrophils and monocytes are maintained by a steady-state myelopoietic pathway, whereas either acute infection or an endogenous inflammatory process triggers the mobilization of mature neutrophils and more immature populations from the bone marrow and blood to inflammatory sites (36,37). The result of this mobilization is an early depletion of bone marrow reserves, creation of niche space, and the release of local mediators, which will drive accelerated or emergency myelopoiesis in the bone marrow (Figure 2).
Figure 2.

Emergency myelopoiesis and the expansion of the MDSC population in sepsis. The figure demonstrates the changes in the early populations of HSCs and multipotent progenitors, which are the progenitors for MDSCs. Although expansion of the MDSC population can be explained in part through increased myelopoiesis, defects in the maturation and differentiation of these populations also contributes to their expansion and their immature phenotype.
MDSCs, like all other leukocytes, originate from self-renewing, long-term hematopoietic stem cells (HSCs). We previously found that polymicrobial sepsis causes myeloid cell expansion in the bone marrow, spleen, and lymph nodes (39). Because HSCs are the precursors for MDSCs, it was not surprising that we observed a tripling in the percentage and a doubling in the absolute number of the total HSC population within 36 hours of sepsis, and both the percentage and absolute number of HSCs remained elevated for at least 7 days. We also observed an increase in both the long-term and short-term HSC subpopulations by 36 hours post sepsis.
This expansion of HSCs in sepsis did not appear to be driven by traditional innate immune-signaling pathways. Mice deficient in toll-like receptor 4 (TLR4), type I interferon signaling, MyD88 and TRIF (TIR-domain-containing adaptor protein producing IFN [interferon] type I) signaling all had a perfectly normal expansion of their HSC populations in response to sepsis (39). Rather, the signals for HSC expansion appeared to originate at least partly in the bone marrow itself. Associated with the creation of niche space, there was a marked increased expression of c-kit, and passive immunization of mice with a blocking antibody to the c-kit receptor, prevented this expansion of HSCs and ultimately, MDSCs (manuscript in review, Scumpia and Kelly-Scumpia).
By asymmetrical division, long-term HSC give rise to short-term HSC that possess a limited capacity for self-renewal and finally develop into multi-potent progenitors and then further into common myeloid progenitors that differentiate into megakaryocyte/erythroid progenitors or granulocyte/macrophage progenitors. We have also shown that during sepsis, there is a marked expansion (4- to 10-fold) in the numbers of these multipotent progenitor populations (manuscript in review).
Ostrand-Rosenberg and colleagues have recently argued that the inflammatory mediators IL-1, IL-6, prostaglandins and proinflammatory S100 proteins all drive this accumulation of MDSCs (3). Sander and Trautwein have demonstrated that MDSC expansion was dependent on IL-1R, IL-6 and gp130 (IL-6 superfamily signaling unit) signaling in sepsis (28). We also showed dependence on nuclear kB (factor NF-kB) signaling for a complete MDSC expansion (12). Such complexity and redundancy are not surprising given that the increased expression of these pathways is common to most if not all inflammatory processes.
Thus, it is not surprising that these MDSCs, which arise from intermediates between myeloid progenitors and mature myeloid cell populations, would increase in chronic inflammatory conditions. It is an oversimplification to suggest, however, that MDSCs are simply immature myeloid cells whose numbers expand during increased myelopoiesis. The dramatic increases in MDSC numbers cannot be simply explained by increased emergency myelopoiesis. Furthermore, it is not clear whether MDSCs in the spleen, liver and peripheral lymph nodes originate strictly from the bone marrow, but may also expand directly in organs owing to extra-medullary hematopoiesis. Extramedullary hematopoiesis is frequently seen in both chronic inflammatory diseases and in cancer, and has also been reported in experimental sepsis (12). Thus, the origins of these MDSCs may not necessarily be bone marrow per se, and this has significant importance when it comes to therapeutic approaches intended to block the expansion of these populations in tumors or secondary lymphoid organs.
The other unresolved question is not what drives the myelopoiesis that provides the MDSC precursors during sepsis, cancer and chronic inflammatory events, but rather, what prevents the further differentiation and terminal disposition of these immature myeloid populations, and their resulting dramatic accumulation. Why are MDSCs accumulating in the spleen while the numbers of terminally differentiated macrophages, and dendritic cells either constant or declining? Furthermore, these cells possess an immunosuppressive phenotype not seen by similar GR-1+CD11b+ cells obtained from healthy animals (12,28). We have shown that if GR-1+ cells are removed from the spleens of septic mice and cultured in vitro with growth factors present in sepsis, they will readily differentiate into dendritic cells and macrophages (12). More importantly, Kusmartsev et al. harvested GR-1+ MDSCs from tumor-bearing animals and injected them into healthy hosts and observed that the injected cells rapidly lost their MDSC phenotype and could not be distinguished from endogenous, terminally differentiated macrophages and dendritic cells (40). Such data suggest that MDSCs retain the potential to further differentiate and mature, but in the host during chronic inflammation, associated with either an actively growing tumor or sepsis, the environmental milieu has ‘trapped’ these cells in an MDSC phenotype. Such cells are remarkably resistant to maturation signals. Although Gabrilovich and Kusmartsev et al. have shown that they can drive maturation of MDSCs from tumor bearing mice with all trans-retinoic acid (40,41), we were unable to do so in the presence of sepsis (unpublished observations). There have been a number of proposed mechanisms that can explain the failure of these cell populations to further mature and terminally differentiate. Lutz and colleagues have argued that the same factors involved in promoting the increased production of MDSC precursors are also responsible for their inability to differentiate into dendritic cells (42). One suggestion has been that elevated levels of vascular endothelial growth factor (VEGF) can prevent MDSCs from differentiating further into dendritic cells (43). It has been shown that administration of recombinant VEGF to healthy animals resulted in suppressed dendritic cell development associated with accumulation of GR-1+ immature MDSCs (44). Along the same lines, treatment of tumor-bearing animals and cancer patients with sunitinib, a tyrosine kinase inhibitor that targets the VEGF receptor among others, significantly reduces the numbers of MDSCs, although it does not appear to affect their maturation in vitro (45,46). Interestingly, sunitinib treatment also appears to reduce the immunosuppressive phenotype of MDSCs, by reducing the expression of the negative costimulatory molecule PDL-1 on MDSC (46). Whether this phenotypic change is associated with merely a greater differentiation of the cell population or a specific target of the tyrosine kinase inhibitor is still unknown.
Another circulating mediator, heat shock protein (HSP) 27, is associated with tumor progression and increased post-injury infection. Laudanski et al. have reported that elevated levels of HSP27 blocked the differentiation of myeloid precursors to dendritic cells and macrophages through a p38 MAP kinase–dependent pathway (47). These findings suggest that there are a number of inflammatory mediators that regulate the maturation of MDSCs, suggesting that therapeutic interventions may be possible but challenging.
MDSCs IN TRAUMA AND SEPSIS: PATHOPHYSIOLOGIC OR BENEFICIAL?
Not surprisingly, MDSCs have been reported in the trauma and sepsis literature for several years. In 2004, Sherwood and colleagues reported on the presence of a “suppressor macrophage” population in the spleens of mice after burn injury (4). But the real breakthrough in linking MDSCs to trauma and sepsis came from the laboratory of Juan Ochoa and his colleagues at the University of Pittsburgh and Louisiana State University (48). These investigators observed a marked increase in a CD11b+GR-1+ population in the spleens of mice 24 hours after a traumatic injury. The investigators classified these cells as being MDSCs on the basis of their ability to suppress CD4+ T-cell proliferation, TCRζchain and IL-2 expression, as well as their ability to produce large quantities of arginase. However, there were a number of physical characteristics of these cells that suggested that they differed at least qualitatively from MDSCs found in cancer and sepsis. These MDSCs had markedly higher MHC class II expression and lower class I expression, and very few cells expressed the immaturity marker, CD31. As shown in Figure 1, cytospins of this population revealed a predominantly “ringed-cell” phenotype consistent with a more mature PMN-MDSC phenotype.
We stumbled onto MDSCs in murine sepsis models fortuitously. Most sepsis models based on the cecal ligation and puncture method are designed to produce early, high mortality, and to use pharmacologic attempts to mitigate outcomes (49,50). We were interested not so much in the initial host response to sepsis, but in whether the septic animal had altered innate and adaptive immune processes that could explain their increased susceptibility to secondary infectious challenges: the theoretical basis behind the “second-hit phenomenon.” Investigation of this required that we develop a sepsis model that more accurately recapitulated outcomes to human sepsis that are on the order of 20–30% mortality and develop over several days. Developing a less lethal cecal ligation and puncture model, we observed, in animals surviving more than 3 days, a massive expansion of the CD11b+GR-1+ population not only in the spleen, but also in the peripheral lymph nodes and in the bone marrow (12). By 7 to 10 days after sepsis, almost 40% of the spleen was CD11b+GR-1+; in the bone marrow, the percentages increased to close to 90%, and in peripheral lymph nodes, the percentage increased to 3–5%. More dramatically, as the sepsis continued, splenomegaly developed, and the total numbers of these cells in the spleen increased more than 50-fold from healthy control animals.
Clearly, these cells were heterogenous (Figure 1), and included both immature and mature myeloid populations, as well as the putative MO-MDSC and PMN-MDSC subpopulations. The cells were clearly immunosuppressive and could both block a CD8+ T-cell proliferation as well as promote a Th1/Th2 shift in CD4+ T-cell cytokine production in vivo (12). Although less than 3% of the cells expressed MHC class II, almost 40% expressed CD31. More importantly, when GR-1+ splenocytes were harvested from septic mice and cultured ex vivo with growth factors (granulocyte-macrophage–colony stimulating factor and IL-4), they rapidly expanded into CD11c+ (dendritic) and F4/80+ (macrophage) populations. Surprisingly, when similar GR-1+ splenocytes were harvested from healthy control mice, these populations could not be encouraged to proliferate or differentiate into terminal cell populations.
What surprised us most were the relative kinetics of MDSC appearance after sepsis. Results of the studies of Makarenkova and Ochoa in trauma suggested that within 24 hours of trauma, there should be an expansion of the MDSC population (48). In sepsis, we saw a very different response (Figure 3). Twenty-four hours after sepsis, there were no changes in either splenocyte or peripheral lymph node CD11b+GR-1+ numbers, and there was a significant decrease in the bone marrow CD11b+ GR-1+ populations. It is generally assumed that the loss of these cells early after sepsis represents the mobilization of predominantly mature and immature neutrophils from the bone marrow in response to the microbial challenge. Surprisingly, it took 3 to 5 days for the numbers of CD11b+GR-1+ cells to expand in the spleen and peripheral lymph nodes, and concentrations did not start to plateau for 10–14 days. Expansion of these cells was not dependent on either TLR4 or IFN-α/ IFN-β signaling, but was modestly dependent on MyD88 (12). We followed septic animals out for periods exceeding 12 weeks and saw no real diminution in the numbers and proportions of MDSCs. There was a trend towards increased immaturity of these cell populations as time progressed. In a very recently reported study, Sander and Trautwein confirmed these findings and demonstrated the requirement for gp130-dependent JAK-STAT signaling for their expansion (28).
Figure 3.

Kinetics of MDSC appearance in the spleen relative to the changes in regulatory T cells and myeloid dendritic cells during sepsis. During sepsis, there are dramatic dynamic changes in the numbers of different regulatory cell populations. In the spleen of mice that survive a polymicrobial sepsis (cecal ligation and puncture), there is a rapid and transient increase in both the relative and absolute numbers of regulatory T cells (CD4+CD25+FoxP3+), with a simultaneous loss of myeloid dendritic cells (CD11b+CD11c+). The response is transient, unlike the increases in both relative and absolute numbers of MDSCs (CD11b+GR-1+) which occur later and are much more sustained.
Expansion of this cell population is not limited strictly to trauma and sepsis, but is also seen in other acute inflammatory conditions. We showed that a very modest endotoxin administration produced a significant but transient increase in MDSC numbers in the spleen and lymph nodes (12). Endotoxin and IFN-γ administration produce even greater expansions (42). Noel and Ogle have reported that after a burn injury, MDSC numbers expand dramatically, and these splenic CD11b+GR-1+ cells are metabolically active, secreting large quantities of proinflammatory cytokines when stimulated ex vivo (29,31,32). We have looked at splenic and lymph node expansion of the MDSC populations after a 10–20% body surface area scald burn, Pseudomonas infection, or a Pseudomonas infection superimposed on a scald burn. The latter tends to be lethal when the surface area of the burn is 40%. As shown in Figure 4, both a burn and Pseudomonas infection were associated with an increase in the MDSC populations in both the spleens and lymph nodes at 7 days. Not surprisingly, combining the Pseudomonas infection with the burn injury resulted in marked increased expansion of this population.
Figure 4.

Expansion of Gr-1+CD11b+ cells in spleen and lymph nodes of burned and infected mice. (A), Flow cytometric scattergram showing populations of GR-1+CD11b+ cells in a spleen from a control mouse. Details concerning the experimental methods are contained in the Supplementary Methods. (B), Flow cytometric scattergram showing expansion of GR-1+CD11b+ cells in a spleen from a mouse that underwent a 10% burn and Pseudomonas infection harvested on d 7. (C), Summary data of the effect of the burn and Pseudomonas infection on splenic GR-1+CD11b+ cells. (D), Summary data of the effect of the burn and Pseudomonas infection on lymph node GR-1+CD11b+ cells. In both lymphoid organs, there was a significant increase in the expansion of GR-1+CD11b+ cells from the Pseudomonas infection alone experimental group as well as the 10% burn + infection group as compared with controls. Values represent the median and estimated quartiles of five animals per group, using a box plot representation, since the values failed tests of normality and equal variance. * indicates that the control group differed only from the groups of mice receiving the Pseudomonas infection, either alone or superimposed on the burn; P < 0.05.
We have asked whether the increased expansion of MDSCs is beneficial or adverse to the septic host. When we originally observed that blocking the expansion of MDSCs with a GR-1+ antibody prevented the Th2 polarization of the immune response and reversed the suppression of CD4+ and CD8+ T cells, we assumed that those components of the MDSC response were generally detrimental to the septic host (12). But it has been difficult to translate those immunological changes into differences in outcome to severe sepsis. As shown in Figure 5, we have attempted several approaches to prevent the expansion of MDSCs in sepsis, using gemcitabine and GR-1+ antibodies, as well as blocking SDF-1 (unpublished observations). In every case, we could prevent or attenuate the expansion of MDSCs, but none of the approaches improved outcome as would be expected if these cells were net immunosuppressive. Rather, in most cases, failure to expand the MDSC population was generally associated with dramatically worsened outcomes. Many of the adverse effects could be explained by the lack of specificity of the approaches: gemcitabine kills all actively replicating cell populations, and in addition to preventing MDSC expansion, kills rapidly—dividing epithelial cells and bone marrow progenitors. GR-1+ antibodies are effective at depleting MDSCs, but also deplete mature neutrophils, which are known to be required for a successful sepsis response (51). The studies with gemcitabine have been repeated by Ogle and colleagues in burned mice, and although it is extremely effective at preventing the expansion of MDSCs in burns, the animals are more susceptible to a secondary Pseudomonas pneumonia (30).
Figure 5.

Effect of different approaches to blocking MDSC expansion on survival to sepsis. Mice were pretreated with either gemcitabine or anti–GR-1 monoclonal antibodies, or antibodies to SDF-1 and subjected to an LD30 cecal ligation and puncture, as described in detail in the Supplemental Methods. Survival to CLP was dramatically reduced in mice treated with either gemcitabine (A, n = 20 in each group) or anti–GR-1 antibodies (C, n = 20 in each group). In surviving animals, there was evidence that the expansion of the MDSC population at 7 d was markedly suppressed (*P < 0.01).
Sander and Trautwein have convincingly argued that in septic conditions in which an exaggerated inflammatory response is lethal, blocking the MDSC expansion may also worsen outcome by promoting the inflammatory component. They showed that mice lacking gp130 and unable to signal through IL-6 failed to expand their MDSC population and had markedly higher mortalities to sepsis associated with increased inflammatory cytokine production (28). Furthermore, they showed that they could improve survival to sepsis when they reconstituted mice with CD11b+GR-1+ cells in their deficient animals. Thus, it is by no means clear whether the expansion in MDSCs contributes to sepsis immune suppression or prevents it. The answer is probably both, depending on the conditions.
The subsequent question is whether human trauma, burns and sepsis are also associated with expansion of homologous human MDSC populations. This is not known, for several reasons. First, there is little agreement about what constitutes an MDSC population in humans, in which the primary criterion of MDSC is a myeloid cell with T-cell suppressor function. Humans do not express the GR-1 antigen, and ring-shaped cells are generally not observed outside of leukemic conditions. Second, the blood compartment is not the most sensitive compartment to assess MDSC expansion, because blood appears to be a transient intermediate from either bone marrow expansion to secondary lymphoid and reticuloendothelial organs where they accumulate, or to where they are generated locally from extramedullary hematopoiesis. The processes of MDSC populations in organs and tissues of trauma and septic patients are technically challenging.
However, we have obtained formalin-fixed, paraffin embedded spleen samples from patients who have experienced either traumatic injury or have died from severe sepsis. These samples were originally analyzed for CD4+ and dendritic cell apoptosis (52). The samples were then restained with a novel antibody that is expressed on activated myeloid cells: myeloid DAP12-associating lectin-1 (MDL-1), a cell-surface receptor which has been shown to regulate myeloid cell–associated inflammatory responses (53,54). Although this antibody is not specific for MDSCs, Phillips and colleagues did find that human and mouse bone marrow cells express the highest levels of Mdl1 under normal physiological conditions, and it is highly upregulated on the cell surface of myeloid cells during chronic inflammation (J. Philips, personal communication). Correspondingly, MDL-1 protein is expressed on murine CD11b+Ly6G+ and CD11b+Ly6G−from bone marrow and the peripheral blood (54). As shown in Figure 6, the pattern of MDL1 expression on human spleens increased dramatically with trauma and sepsis. In particular, patients who died from sepsis had marked expansion of MDL1+ populations that replaced the dissolution of the lymphoid follicles, which is characteristic of severe sepsis. Although trauma itself was associated with increased numbers of MDL1+ cells, the distribution was more normal, with cells residing primarily in the mantel surrounding the follicles. Although the data represent a limited number of patients and the staining suffers from a general lack of specificity, the data are consistent with human sepsis being associated with a marked expansion of myeloid cells in the spleen.
Figure 6.

MDL1 expression in spleens of patients after trauma or who have died from sepsis. Paraffin-embedded sections from spleens of patients who had either died from sepsis (C and D), or had suffered blunt trauma and undergone splenectomy (B), or had undergone splenectomy for nontrauma or sepsis indications (A) were stained with antibodies to MDL-1. Details about the patient population can be found in (52). Sepsis was associated with the dissolution of lymphoid follicles and the dramatic increase in MDL-1+ cells. Trauma was also associated with more modest increases, although the pattern of MDL-1+ cell expression was markedly different. Staining protocol is provided in the Supplemental Methods. Magnification 100×.
CAN MDSC EXPANSION AND FUNCTION BE MANIPULATED, AND SHOULD IT BE?
The mechanisms that control the expansion and activity of MDSCs are influenced by multiple factors including cytokine/chemokine production from tumor, tumor stroma, and infiltrating T cells. NF-κB and JAK/STAT activation, in particular STAT3, are associated with both proliferation and survival of MDSCs, as well as the production of the S100 calcium-binding proteins, S100A8/9. These proteins subsequently bind and potentiate the NADPH complex and have also been shown to bind RAGE receptors (receptor for advanced glycosylation end-production) and TLR4. The net effect of this signaling cascade leads to the increased production of ROS and proinflammatory cytokines.
Because of the negative impact these increases in proinflammatory and innate immune responses have on T-cell immunity and tumor immunotherapy, strategies to modulate or inhibit the expansion of these cells have been actively pursued (2). One strategy that has been tested in the clinical setting is approached through maturation of MDSCs using the vitamin A metabolite, all-trans retinoic acid (41). Other potential strategies involve inhibiting the expansion of these cells through targeting of the vascular tumor stroma (anti-VEGF or sunitinib) (43) or hematopoiesis through targeting stem cell factor (55). However, the question remains: Is this expansion of MDSCs deleterious?
Certainly, in our hands and in the hands of others, these cells obtained from septic hosts have immunosuppressive activities on adaptive immunity, but if the expansion of MDSCs are the host’s attempt to correct and control the systemic insult, will we sacrifice enhanced innate immune responses at the expense of improved tumor immunity? Perhaps. In the setting of tumor growth, the net benefit of controlling the expansion of these cells will probably improve some aspects of clinical outcome, as dysregulated activity and expansion of these cells contribute to negative effects on the host. However, such actions are not without some theoretical risk of reducing immune surveillance. It is important to fully understand their role in host immunity, because attempts to control and prevent MDSC expansion in burn and sepsis preclinical models may actually increase susceptibility to both primary and secondary infection (Figure 4). As described above, Ogle’s group has demonstrated that after burn injury, mice are protected against secondary Pseudomonas aeruginosa infection, and depletion of MDSCs with gemcitabine blocks this effect (30). In addition, immunoneutralization of GR-1+ cells worsens survival to experimental sepsis (Figure 4) (28). Regardless, the goal of these therapies should be to strike a balance between improving adaptive immune responses and preserving innate immune function.
FUTURE DIRECTION AND GOALS
Though it is well accepted that a myelopoietic response to inflammation, infection and sepsis occurs, this response may be misunderstood. As described above, evidence from our laboratory and others suggest that this expansion of MDSCs may actually serve to protect the host through increased innate immunity and secondarily through suppression of cytokine/inflammatory responses (28,30). However, despite the demonstration of adaptive immune suppression in some of these studies, there is still significant controversy as to whether or not the MDSCs that arise in sepsis are truly MDSCs or a myelopoietic variant with similar characteristics (12). The greatest barrier to fully describing these cells despite differences between disease models and further, between species, lies in the lack of appropriate mechanistic studies, phenotypic markers and/or genomic signatures that can sufficiently unify scientific consensus. How similar are the MDSCs described in cancer to the MDSCs found to expand in autoimmunity or infection? Do the neutrophils described in clinical studies as CD11b+CD15+CD33+ have similar genomic/proteomic signatures as murine MDSCs? Or more importantly, does the expansion of MDSCs represent a conserved hematopoietic/ myelopoietic response to systemic stress that is shaped by the disease process, but is driven to create a similar, albeit heterogenous, cell population? Mechanistic studies designed to address these concerns are critically important to our understanding not only of the biology of these cells but also the myelopoietic response to systemic insult.
CONCLUSIONS AND CLINICAL RELEVANCE
Though largely dismissed before this decade, the MDSC has garnished tremendous scrutiny, despite the inability of investigators to identify a specific cell population. This examination has forced a reevaluation of the role that MDSCs play in the primary immune response and how they shape adaptive immunity. If the expansion of MDSCs were observed only as a solitary phenomenon in tumor studies, it would be clear how the interplay of the tumor and the normal inflammatory response would support the dramatic observations that have been described over the last decade. However, given the increasing knowledge regarding the myelopoioetic response to inflammation and the numerous pathological conditions in which very similar populations are expanding, the purpose of these cells cannot be defined so narrowly. Although T cells play undeniable roles in tumor and pathogen immuno-surveillance, we cannot discount the importance of the innate immune response and the possible role that T cells play in this process. Indeed, we and others have shown that as the host is compromised owing to some systemic pathologic insult, be it tumor growth, lupus or sepsis—the emergency hematopoietic response shifts toward myeloid lineages and expansion of myeloid cell precursors. Moreover, there are likely important differences in the immunosuppressive, proinflammatory mediators expressed and stored within various MDSCs, depending on the nature of the chronic inflammation. Some of these mediators may even have the potential to induce acute changes in hemodynamic responses through modulation of endothelial cells as shown by Pande et al. (27). The expansion of MDSCs is a critical component of this response that can have obvious harmful effects. Therefore, there remains a compelling need to further understand the role MDSCs play before we consider manipulating them in the diverse clinical settings in which they arise.
Supplemental Data
ACKNOWLEDGEMENTS
Supported in part by grants R01 GM-40586-22 and R01 GM-81923-03 from the National Institute of General Medical Sciences. AG Cuenca, MJ Delano, C Moreno and KM Kelly-Scumpia were supported by a T32 training grant in burns and trauma (T32 GM-08721-11). AG Cuenca was also supported by an individual National Research Service Award (F32 GM-093665-01) from the National Institute of General Medical Sciences.
The authors acknowledge the contributions of R Hotchkiss, Washington University in St. Louis, for providing the deidentified and anonymous tissue blocks. In addition, we thank J Phillips, P Hey-worth and D LaFace from DNAX/Merck Biopharma for the MDL-1 staining, as well as the fruitful discussions.
The authors have no financial conflicts with the submission.
Footnotes
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Online address: http://www.molmed.org
REFERENCES
- 1.Bronte V. Myeloid-derived suppressor cells in inflammation: uncovering cell subsets with enhanced immunosuppressive functions. Eur J Immunol. 2009;39:2670–2. doi: 10.1002/eji.200939892. [DOI] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphey ED, et al. Diminished bacterial clearance is associated with decreased IL-12 and interferon-gamma production but a sustained proinflammatory response in a murine model of postseptic immunosuppression. Shock. 2004;21:415–25. doi: 10.1097/00024382-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Young MR, Newby M, Wepsic HT. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res. 1987;47:100–5. [PubMed] [Google Scholar]
- 6.Peranzoni E, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–44. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 7.Nagaraj S, et al. Regulatory myeloid suppressor cells in health and disease. Cancer Res. 2009;69:7503–6. doi: 10.1158/0008-5472.CAN-09-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–91. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 9.Gallina G, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–90. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angulo I, et al. Nitric oxide-producing CD11b(+)Ly-6G(Gr-1)(+)CD31(ER-MP12)(+) cells in the spleen of cyclophosphamide-treated mice: implications for T-cell responses in immunosuppressed mice. Blood. 2000;95:212–20. [PubMed] [Google Scholar]
- 11.Rossner S, et al. Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol. 2005;35:3533–44. doi: 10.1002/eji.200526172. [DOI] [PubMed] [Google Scholar]
- 12.Delano MJ, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–74. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Movahedi K, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 14.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zea AH, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 16.Filipazzi P, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–53. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 17.Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–6s. doi: 10.1158/1078-0432.CCR-06-2197. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez PC, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–60. doi: 10.1158/0008-5472.CAN-08-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol. 2007;601:213–23. doi: 10.1007/978-0-387-72005-0_22. [DOI] [PubMed] [Google Scholar]
- 20.Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009;182(9):5693–701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–99. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 22.Popovic PJ, Zeh HJ, 3rd, Ochoa JB. Arginine and immunity. J Nutr. 2007;137:1681S–6S. doi: 10.1093/jn/137.6.1681S. [DOI] [PubMed] [Google Scholar]
- 23.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 24.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 25.Nagaraj S, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007;13(7):828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006;176(1):284–90. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 27.Pande K, et al. Cancer-induced expansion and activation of CD11b+ Gr-1+ cells predispose mice to adenoviral-triggered anaphylactoid-type reactions. Mol Ther. 2009;17:508–15. doi: 10.1038/mt.2008.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sander LE, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med. 2010;207:1453–64. doi: 10.1084/jem.20091474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noel G, et al. Postburn monocytes are the major producers of TNF-alpha in the heterogeneous splenic macrophage population. Shock. 2007;27:312–9. doi: 10.1097/01.shk.0000239753.75088.5e. [DOI] [PubMed] [Google Scholar]
- 30.Noel G, et al. A ribonucleotide reductase inhibitor reverses burn induced inflammatory defects. Shock. 2010;34:535–44. doi: 10.1097/SHK.0b013e3181e14f78. [DOI] [PubMed] [Google Scholar]
- 31.Noel JG, et al. Effect of thermal injury on splenic myelopoiesis. Shock. 2005;23:115–22. doi: 10.1097/01.shk.0000154239.00887.18. [DOI] [PubMed] [Google Scholar]
- 32.Noel JG, et al. Thermal injury elevates the inflammatory monocyte subpopulation in multiple compartments. Shock. 2007;28:684–93. doi: 10.1097/shk.0b013e31805362ed. [DOI] [PubMed] [Google Scholar]
- 33.Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal advance: tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE2 catabolism in myeloid cells. J Leukoc Biol. 2010;88:839–48. doi: 10.1189/jlb.1209821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vuk-Pavlovic S, et al. Immunosuppressive CD14+HLA-DRlow/-monocytes in prostate cancer. Prostate. 2010;70:443–55. doi: 10.1002/pros.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng P, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–49. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelsoe G. IL-1R type I-dependent hemopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J Immunol. 2009;182:6477–84. doi: 10.4049/jimmunol.0803961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med. 2005;201(11):1771–80. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–3. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 39.Scumpia PO, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. 2010;184:2247–51. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kusmartsev S, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–9. [PubMed] [Google Scholar]
- 41.Kusmartsev S, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008;14:8270–8. doi: 10.1158/1078-0432.CCR-08-0165. [DOI] [PubMed] [Google Scholar]
- 42.Greifenberg V, Ribechini E, Rossner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. 2009;39:2865–76. doi: 10.1002/eji.200939486. [DOI] [PubMed] [Google Scholar]
- 43.Fricke I, et al. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007;13:4840–8. doi: 10.1158/1078-0432.CCR-07-0409. [DOI] [PubMed] [Google Scholar]
- 44.Gabrilovich D, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66. [PubMed] [Google Scholar]
- 45.Khan SY, et al. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood. 2006;108:2455–62. doi: 10.1182/blood-2006-04-017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozao-Choy J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laudanski K, De A, Miller-Graziano C. Exogenous heat shock protein 27 uniquely blocks differentiation of monocytes to dendritic cells. Eur J Immunol. 2007;37:2812–24. doi: 10.1002/eji.200636993. [DOI] [PubMed] [Google Scholar]
- 48.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176:2085–94. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 49.Scumpia PO, et al. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood. 2007;110:3673–81. doi: 10.1182/blood-2007-04-087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scumpia PO, et al. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol. 2005;175:3282–6. doi: 10.4049/jimmunol.175.5.3282. [DOI] [PubMed] [Google Scholar]
- 51.Opal SM, et al. Additive effects of human recombinant interleukin-11 and granulocyte colony-stimulating factor in experimental gram-negative sepsis. Blood. 1999;93:3467–72. [PubMed] [Google Scholar]
- 52.Hotchkiss RS, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Bakker AB, Baker E, Sutherland GR, Phillips JH, Lanier LL. Myeloid DAP12-associating lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. Proc Nat Acad Sci U S A. 1999;96:9792–6. doi: 10.1073/pnas.96.17.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joyce-Shaikh B, et al. Myeloid DAP12-associating lectin (MDL)-1 regulates synovial inflammation and bone erosion associated with autoimmune arthritis. J Exp Med. 2010;207:579–89. doi: 10.1084/jem.20090516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan PY, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111:219–28. doi: 10.1182/blood-2007-04-086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu B, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5228–37. doi: 10.4049/jimmunol.179.8.5228. [DOI] [PubMed] [Google Scholar]
- 57.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 58.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–45. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 59.Dolcetti L, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40:22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 60.Huang B, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 61.Umemura N, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1-and M2-type characteristics. Journal of leukocyte biology. 2008;83:1136–44. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- 62.Pan PY, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou Z, et al. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells. 2010;28:620–32. doi: 10.1002/stem.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.