

Abstract
Defective interfering particles (DIP) of equine herpesvirus 1 (EHV-1) inhibit standard virus replication and mediate persistent infection. The DIP genome is comprised of only three genes: UL3, UL4, and a hybrid gene composed of portions of the IR4 (EICP22) and UL5 (EICP27) genes. The hybrid gene is important for DIP interference, but the function(s) of the UL3 and UL4 genes are unknown. Here, we show that UL4 is an early gene activated solely by the immediate early protein. The UL4 protein (UL4P) was detected at 4 hours post-infection, was localized throughout the nucleus and cytoplasm, and was not present in purified virions. EHV-1 lacking UL4P expression was infectious and displayed cell tropism and pathogenic properties in the mouse model similar to those of parental and revertant viruses. Reporter assays demonstrated that the UL4P has a broad inhibitory function, suggesting a potential role in establishing and/or maintaining DIP-mediated persistent infection.
INTRODUCTION
Equine herpesvirus 1 (EHV-1), a member of the Alphaherpesvirinae subfamily, is a major pathogen of equines worldwide, resulting in severe respiratory, neurological, and abortigenic disease (Allen and Bryans, 1986; Mettenleiter et al., 2008; O’Callaghan and Osterrieder, 2008). Viral replication requires the gene program to be regulated in a coordinated and temporal fashion, following a progression from immediate early (IE) to early (E) to late (L) gene expression (Caughman et al., 1985; Gray et al., 1987). EHV-1 encodes six regulatory proteins that govern the viral gene program, five being important in promoter activation and one serving as a negative regulatory protein. The sole IE protein (IEP) trans-activates early and some late EHV-1 gene promoters and is capable of trans-repressing its own promoter (Smith et al., 1992). Early proteins IR4P and UL5P function synergistically with the IEP to enhance expression of early and late promoters (Albrecht et al., 2004; Holden et al., 1995; Kim et al., 1997; Zhao et al., 1995). EICP0P is an early regulatory protein that independently serves as a potent and promiscuous trans-activator of EHV-1 genes of all temporal classes (Bowles et al., 1997). The late ETIF protein is a tegument protein responsible for activating expression of the sole IE gene (Lewis et al., 1993; Purewal et al., 1994) and is essential for secondary envelopment and virus egress (von Einem et al., 2006). The early IR2 protein is a truncated form of the IE protein (Harty and O’Callaghan, 1991) and serves to down-regulate the expression from all classes of viral promoters (Kim et al., 2006). Lastly, the EHV-1 unique IR3 gene encodes a transcript that is antisense to the IE mRNA (Holden et al., 1992), is not translated to a detectable protein product (Ahn et al., 2007), and functions to down-regulate expression of the IE gene (Ahn et al., 2010).
Like many viruses (Huang and Baltimore, 1970), EHV-1 passaged at high multiplicity will form defective interfering particles (DIP) that can mediate a state of persistent infection (Campbell et al., 1976; Dauenhauer et al., 1982; Ebner et al., 2008; Ebner and O’Callaghan, 2006). Present within the DIP genome (~7.5 kbp; Fig. 1B) are the origin of replication, cis elements for cleavage and packaging, and only three genes: UL3 and UL4 conserved perfectly from the left terminus of the standard genome (~155 kbp; Fig. 1A) and a unique hybrid gene formed by a recombination event that joins portions of the IR4 and UL5 regulatory genes (Chen et al., 1996, 1999; Ebner and O’Callaghan, 2006). The IR4/UL5 hybrid gene (Hyb) negatively affects expression of many viral genes during DIP-mediated persistent infection (Chen et al., 1999). Recent studies demonstrated that the IR4 portion of the Hyb protein is important for mediating this negative regulation as well as interfering with standard virus replication (Ebner et al., 2008). To date, no function has been reported for the UL3 or UL4 genes during persistent or lytic infection. In the present study, we identify the UL4 gene as an early gene and demonstrate that the UL4 protein (UL4P) has a nuclear and cytoplasmic localization and is not a component of purified EHV-1 virions. A possible regulatory function of the UL4P was examined through transient transfection assays which revealed that the UL4P inhibited the expression of reporter genes under the control of EHV-1 promoters of all gene classes as well as the promoters of heterologous viruses and cell genes tested to date. In addition, the absence of the UL4P resulted in an increase in the amount of viral transcripts of all gene classes during infection. Studies with a mutant EHV-1 lacking UL4 expression showed that the UL4P was not essential for viral replication in cell culture or a pathogenic phenotype in the CBA mouse model.
Fig. 1.

Genomes of the standard EHV-1 and EHV-1 defective interfering particles. (A) Organization of the EHV-1 standard (STD) genome and location/orientation of the regulatory genes and genes conserved within the DIP genome. UL unique long region; US unique short segment; IR inverted repeat segment. Numbers show size of viral proteins in amino acid residues. (B) DIP genome comprised of a 7.5 kbp repeat. CPS cleavage/packaging sequence, ORI origin of DNA replication; Hyb hybrid gene of IR4/UL5 sequences.
RESULTS
The UL4 gene belongs to the early class in the EHV-1 gene program
A previous report described the location of the 5’ and 3’ termini of the UL4 mRNA in relation to a TATA box and polyadenylation signal, respectively (Harty et al., 1993). We set out to characterize UL4 gene transcription and assign the UL4 gene to a temporal class in the EHV-1 gene program. Northern blot analysis with a nucleotide probe specific for the UL4 transcript first detected a ~0.9 kb mRNA at 2 hours post-infection (hpi) which reached maximal expression levels by 7 hpi (Fig. 2A). These data suggest that UL4 is an early gene. Additionally, metabolic inhibitor studies demonstrated that the UL4 gene is not transcribed when protein synthesis is inhibited by cycloheximide (CHX; Fig. 2B); whereas, IE mRNA, as expected, is detected in the presence and absence of CHX. Furthermore, the UL4 transcript like that of the early thymidine kinase (TK) transcript was synthesized when viral DNA replication was inhibited by phosphonoacetic acid (PAA). These data confirm that UL4 belongs to the early gene class, a finding further supported by the absence of a TAATGARAT motif within the UL4 promoter. This motif is present within the promoters of immediate early genes of other alphaherpesviruses (Lewis et al., 1997; Misra et al., 1994; Moriuchi et al., 1995), including the sole IE gene of EHV-1 (Elliott and O’Hare, 1995; Grundy et al., 1989), and is the target sequence for binding by the viral α-trans-inducing factor (Elliot and O’Hare, 1995). Furthermore, in reporter assays employing the UL4 promoter region inserted upstream of a luciferase reporter gene, the EHV-1 α-trans-inducing factor (ETIF) failed to activate the UL4 promoter, whereas the IEP strongly trans-activated the UL4 promoter (Fig. 2C), confirming that UL4 is an early gene. Additional studies concerning the activation of the UL4 promoter by combinations of plasmids that express EHV-1 regulatory proteins indicated that the IEP alone trans-activated the UL4 promoter maximally, and that no synergistic activation occurred when the IEP was co-expressed with other EHV-1 regulatory proteins (Fig. 2D).
Fig. 2.
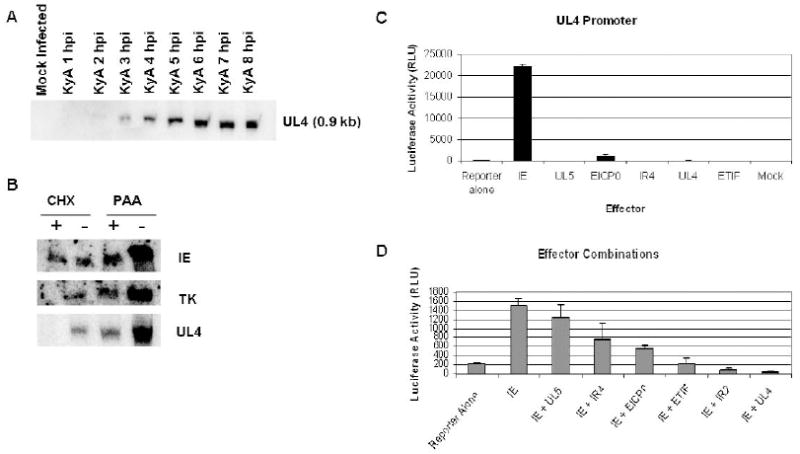
Characterizing UL4 as an early gene through metabolic inhibitor studies, northern blotting, and luciferase assays. (A) Northern blot analysis of RK13 cells infected with EHV-1 with a UL4 specific oligonucleotide indicates that UL4 is an early gene. (B) Metabolic inhibitors were used to confirm that UL4 is an early gene. Cycloheximide (CHX) and phosphonoacetic acid (PAA) were used to treat RK13 cells infected with EHV-1 to inhibit protein synthesis or viral DNA replication, respectively. RNA was isolated from treated (+) and untreated (-) cells at 4 hpi for CHX and 8 hpi for PAA, and northern blot analysis was performed using probes for the IE, early thymidine kinase (TK), and UL4 genes. (C and D) Luciferase reporter assays were performed to examine the activation of the UL4 promoter by EHV-1 regulatory proteins. RK13 cells seeded in 24-well plates were transfected with 1 pmol of the pUL4-Luc reporter plasmid and 0.5 pmol of the effector plasmids pSV-IE, pSV-UL5, pSV-EICP0, pCMV-IR4, pCMV-UL4, pCMV-ETIF and pSV-IR2; either individually (C) or in combination with the IEP (D). Luciferase activity was measured at 48-72 hours post-transfection. Error bars show standard deviation of triplicate assays.
Characterization of the UL4 protein
To begin to characterize the UL4 protein, a rabbit polyclonal anti-UL4P specific antibody was generated (Materials and Methods). To verify the specificity of the anti-UL4P antibody, RK13 cells were transfected with plasmids that express UL4P, UL4 fused to either the carboxy- or amino-terminus of the green fluorescent protein (GFP), or GFP alone or cells were infected with RacL11 EHV-1. Cell lysates were harvested and subjected to western blot analysis using the OC95 anti-UL4P antibody and a mouse monoclonal anti-GFP antibody. In the transfected cells, the anti-GFP antibody detected bands corresponding to GFP (26 kDa; Fig. 3A lane 2), the GFP-UL4 fusion protein (46 kDa; Fig. 3A lane 7), and the UL4-GFP fusion protein (46 kDa; Fig. 3A lanes 5 and 6). This anti-GFP antibody also detected GFP (Fig. 3A lane 8) in lysates of cells infected with the BAC-derived RacL11 virus that expresses the GFP gene inserted during the generation of the BAC (Rudolph et al., 2002). The anti-UL4P antibody detected a 23 kDa protein from cells transfected with UL4 expression plasmids (Fig. 3B lanes 3 and 4) as well as from infected cell lysates (Fig. 3B lane 8). Additionally, the anti-UL4P antibody detected the UL4-GFP fusion protein (Fig. 3B lanes 5 and 6) and the GFP-UL4 fusion protein (Fig. 3B lane 7). No bands were detected with the anti-UL4P antibody in mock transfected cells (Fig. 3B lane 1) or cells transfected with the GFP expression plasmid (Fig. 3B lane 2). Thus, the OC95 antibody was highly specific for the UL4P and demonstrated very little cross reactivity with cellular proteins.
Fig. 3.

UL4 protein synthesis and absence of the UL4 protein in purified EHV-1 virions. Reactivity and specificity of the OC95 anti-UL4 protein polyclonal antiserum were verified by western blot analyses of lysates of RK13 cells transfected with various expression constructs and lysates of cells infected with BAC-derived (bd) RacL11 EHV-1. Lysates prepared at 48 hours post-transfection and at 24 hours post-infection were subjected to western blot analysis and reacted with either (A) a mouse monoclonal antibody to GFP or (B) purified OC95 anti-UL4P antibody. (C) Western blot analyses of lysates of EHV-1 infected RK13 cells with the purified anti-UL4P antibody. (D) Western blot analysis of purified virions with antibodies specific for the EHV-1 UL31 DNA-binding protein, glycoprotein D, or UL4P.
A time course experiment was completed to examine UL4 protein translation. Western blot analysis of lysates of infected RK13 cells with the purified rabbit anti-UL4P antibody revealed that the 23 kDa protein seemed to accumulate after 4 hpi (Fig. 3C). To determine whether the UL4 protein is a component of the EHV-1 virion, purified virus particles were prepared and subjected to western blot analyses. Nitrocellulose membranes were reacted with primary antibodies specific for the UL4P, glycoprotein D (gD), an envelope glycoprotein essential for EHV-1 replication (Csellner et al., 2000; Flowers and O’Callaghan, 1992; Frampton et al., 2005), and the EHV-1 nonstructural UL31 protein, the major DNA binding protein (Lewis et al., 1995). As shown in Figure 3D, the preparation of virions was free of cellular protein contamination as demonstrated by the lack of the nonstructural UL31 protein in the virion preparation, whereas this protein was present in large amounts in lysates of infected cells. As a positive control, the anti-gD antibody reacted with viral protein present in both infected cell lysates and purified virions. In contrast, the anti-UL4P antibody reacted strongly only with infected cell lysates and failed to react with proteins in the purified virion preparation, indicating that the UL4P is not a component of the EHV-1 virion.
Utilizing the monospecific anti-UL4P antibody, immunofluorescence assays were completed to determine the cellular localization of the UL4 protein in RK13 cells transfected with plasmids expressing either the UL4 gene or the UL4-GFP fusion gene, or cells infected with EHV-1. Expression of the UL4 protein (Fig. 4A) or the UL4-GFP fusion protein (Fig. 4B) in the absence of other viral proteins resulted in a broad distribution of the UL4P within both the cytoplasm and nucleus of these cells. A similar fluorescent staining pattern was observed in cells infected with EHV-1 (Fig. 4C), suggesting that the 23 kDa UL4 protein localizes within both the nucleus and cytoplasm during infection. To verify the localization of the UL4 protein in both cellular compartments, RK13 cells were infected with EHV-1, and cell lysates were separated into cytoplasmic and nuclear fractions as described in the Materials and Methods. Western blot analysis of the proteins isolated from these cellular fractions confirmed that the UL4 protein was located within both the cytoplasm and nucleus (data not shown).
Fig. 4.

Localization of the UL4 protein in transfected and EHV-1 infected cells by immunofluorescence with purified anti-UL4P antibody. RK13 cells were transfected with either the (A) pCMV-UL4 or the (B) pUL4-GFP expression plasmid, or (C) were infected with RacL11 EHV-1. Cells were fixed with 4% paraformaldehyde at 48 hours post-transfection or at 12 hours post-infection and permeabilized with 0.2% Triton X100 in PBS. Panels A and C: purified anti-UL4P antibody was used as the primary antibody and an Alexa Fluor 488 goat anti-rabbit IgG was the secondary antibody. Panel B shows fluorescence due to GFP expression. Coverslips were mounted with a DAPI solution to stain nuclei (top photograph of each panel). Arrows in the upper panels indicate the nuclei of the cells of interest stained in the lower panels.
UL4P is an inhibitor of gene expression and antagonizes IEP trans-activation of EHV-1 promoters
Although known to be conserved perfectly within the genome of defective interfering particles (Baumann et al., 1984, 1986; Ebner et al., 2008; Ebner and O’Callaghan, 2006), no function has been reported for UL4. Herpes simplex virus 1 (HSV-1) UL55, the UL4P homolog, was demonstrated to inhibit transient gene expression when co-transfected into cells along with plasmids expressing the immediate early gene α27 as well as alpha genes α0 and α4 (Block et al., 1991). Additionally, Yamada and coworkers (1998) characterized the UL55 protein of HSV-2 as a potential accessory protein in virion assembly and maturation and found it to co-localize with capsid protein ICP35 at the periphery of infected cell nuclei. To determine whether UL4P possesses any regulatory activity, luciferase reporter assays were performed. The reporter plasmid expressed the firefly luciferase gene under the control of various EHV-1 promoters, including the immediate early (IE), early promoters EICP0, thymidine kinase (TK), and IR2, and late promoters ETIF and glycoprotein K (gK). Rabbit kidney cells were transfected with the various reporter plasmids along with the effector plasmids pSV-IE and/or pCMV-UL4, and luciferase activity was measured at 48-72 hours post-transfection. For the IE promoter, luciferase activity was inhibited by 77% in the presence of the UL4 protein (Fig. 5A). In a separate reporter assay employing the chloramphenicol acetyltransferase (CAT) gene under the control of the EHV-1 IE promoter, UL4 protein expression inhibited CAT activity by 87% (data not shown). UL4P expression decreased luciferase activity for the early EHV-1 TK and IR2 promoters 95% and 85%, respectively (Fig. 5C and D). In addition, luciferase activity driven by EHV-1 late promoters was negatively affected by the UL4P, such that in the case of the ETIF and gK promoters the inhibition reached levels of 78% and 98%, respectively (Fig. 5E and F). Conversely, when cells were transfected with the IE effector plasmid, luciferase activity was greatly increased for the early promoters and the ETIF promoter; whereas, the late gK promoter and the IE promoter were inhibited by expression of the IE protein, confirming our previous reports (Caughman et al., 1985; Harty and O’Callaghan, 1991; Kim et al., 1999; Smith et al., 1992, 1994). When the UL4 and IE expression plasmids were co-transfected into cells, the ability of the IEP to trans-activate the early EICP0, TK, IR2, and late ETIF promoters was diminished (Fig. 5B to E). IEP trans-activation of these four promoters was inhibited by 45% to 70%. The IEP has been shown to inhibit its own promoter (Harty and O’Callaghan, 1991; Smith et al., 1994), and the coexpression of the UL4 and IE proteins further abrogated the luciferase activity driven by the IE promoter (Fig. 5A). These data suggest that the UL4 protein plays an inhibitory role in governing the expression of EHV-1 genes.
Fig. 5.

Luciferase assays examining the effect of the UL4 protein on luciferase expression driven by EHV-1 promoters of all gene classes. RK13 cells in 24-well plates were transfected with 0.5 pmol of the expression plasmids pCMV-UL4, pSV-IE, or both plasmids, along with 1 pmol of the luciferase reporter plasmid under the control of the EHV-1 (A) immediate early promoter; or early promoters (B) EICP0, (C) thymidine kinase (TK), and (D) IR2, or late promoters (E) ETIF and (F) glycoprotein K (gK). Luciferase activity was assayed at 48 hours post-transfection as described in Materials and Methods.
Given that the UL4P appeared to function across all gene classes, we next wanted to establish if the negative effect on reporter expression was specific for EHV-1 genes. In a similar reporter assay, luciferase activity was measured from reporter plasmids that used the simian vacuolating virus 40 (SV40) large T antigen promoter and the human cytomegalovirus (CMV) IE promoter. In the presence of UL4P, luciferase activity was decreased 90% from these heterologous viral promoters as well (Fig. 6A), indicating that UL4P inhibition is not virus specific. The inhibitory effect of the UL4P was not specific for the firefly luciferase reporter gene as similar levels of inhibition were obtained in assays that employed the renilla gene as the reporter (data not shown). Lastly, a number of cellular promoters was tested to ascertain whether expression of the UL4 protein affects non-viral gene expression. To this end, expression of the UL4 protein was found to inhibit luciferase activity driven by the GAPDH, eIF4E, IFNβ, and HPRT cellular promoters by 44%, 86%, 75%, and 78%, respectively (Fig. 6B). These decreases in luciferase activity were not attributable to cytotoxicity in the presence of the UL4 protein, as demonstrated by similar levels of cell viability in cells expressing the UL4P compared to non-transfected cells (data not shown). Taken together, the data presented in Figures 5 and 6 indicate that the UL4 protein negatively affects gene expression, not only from viral promoters but cellular promoters as well.
Fig. 6.

Luciferase assays examining the effect of the UL4 protein on luciferase expression driven by heterologous viral promoters or cellular promoters. (A). Luciferase activity driven by the SV40 large T antigen promoter or the CMV IE promoter as compared to activity controlled by the EHV-1 IE promoter. (B). Luciferase activity driven by cellular promoters glyceraldehyde phosphate dehydrogenase (GAPDH), eukaryotic initiation factor 4E (eIF4E), interferon β (IFNβ), or hypoxanthine-guanine phosphoribosyltransferase (HPRT).
UL4 is not required for EHV-1 lytic replication
After observing the inhibitory activity of the UL4 protein against a broad range of viral and cellular promoters, we wanted to examine if UL4 plays an essential role in EHV-1 lytic infection. It was hypothesized that deletion of the UL4 gene would increase the ability of the virus to replicate due to the absence of the additional inhibitory activity of the UL4P. To begin to address this hypothesis, a UL4-null EHV-1 was generated utilizing the galK bacterial artificial chromosome (BAC) technology (Rudolph et al., 2002; Warming et al., 2005). Initially, the UL4 gene was replaced by the galK gene to create a UL4-deleted virus. The galK gene was then replaced with a mutant form of UL4 that contained a STOP codon at position 18 within the amino acid sequence (UL4aa18stop) or a wild-type UL4 to generate a revertant virus (ΔUL4R). Once the mutant and revertant BACs were generated, the gross genomic structure was examined by BamH I restriction enzyme digestion. Figure 7A demonstrates that the process used for generating the mutant and revertant BACs did not result in any spurious recombination events or alterations in the overall genomic structure, as evidenced by similar digestion patterns of the parental RacL11, ΔUL4R, and UL4aa18stop BACs. The second BamH I fragment in the ΔUL4 (galK+) BAC migrated in agarose gels more slowly than the other BACs, indicative of the additional base pairs within the galK gene compared to the UL4 gene (Fig. 7A; lane 2). Southern blot analysis for the presence of the UL4 gene within the various BACs revealed that the UL4 sequence was absent from the ΔUL4 (galK+) BAC, while the UL4-specific probe hybridized to DNA sequences within the parent, mutant, and revertant BACs (Fig. 7B). Furthermore, PCR analysis and DNA sequencing verified that the BACs were correct (data not shown).
Fig. 7.

Generation and verification of the UL4aa18stop EHV-1 and ΔUL4R revertant EHV-1 by galK BAC technology. Homologous recombination was used to delete the UL4 gene as described in Materials and Methods. Briefly, PCR was utilized to append UL4 DNA flanking sequences to the galK gene. SW106 E. coli harboring the EHV-1 BAC were transformed with the resultant PCR product and plated on positive selection plates. To generate the revertant BAC, wild-type UL4 sequences were transformed into bacteria containing the mutant BAC and plated on negative selection plates with 2-deoxygalactose. The mutant and revertant BAC genomes were analyzed and verified by (A) BamH I restriction enzyme digestion and (B) Southern blot analysis. (C) RK13 cells were transfected with each of the three BAC genomes to generate wild-type, UL4aa18stop, and ΔUL4R viruses. RK13 cells were then infected with each virus, and at 9 hours post-infection lysates were prepared and subjected to western blot analysis with the purified anti-UL4P antibody.
To determine whether UL4 was essential for EHV-1 replication, the BACs were transfected into permissive RK13 cells. As shown in Fig. 8A, each BAC resulted in the formation of plaques in transfected cells, indicating that UL4 is dispensable for EHV-1 lytic replication. However, it was observed that the plaques formed by the UL4aa18stop virus were smaller in size compared to those of the parent and revertant viruses, being approximately one-third the size of wild-type and revertant plaques (Fig. 8B). To verify that the stop mutant virus did not produce UL4 protein, lysates from cells infected with the wild-type, mutant, and revertant viruses were subjected to western blot analysis. As expected, the UL4 protein was detected in cells infected with the wild-type RacL11 and ΔUL4R viruses, but not in cells infected with the UL4aa18stop virus (Fig. 7C). These data indicated that the UL4 protein is not essential for EHV-1 replication, but the smaller plaque size of the UL4aa18stop virus may indicate a deficiency in its ability to replicate or spread efficiently.
Fig. 8.

Morphology of plaques produced on RK13 cells transfected with the parent, UL4aa18stop, and ΔUL4R BACs. (A) RK13 cells were transfected with the various BACs, overlaid with medium containing 1.5% methylcellulose, and examined after 5 days for plaque formation by staining with 0.5% methylene blue dye. (B) Plaque size was analyzed with the ImageJ software (Materials and Methods). 30 plaques produced by each virus were measured and the Student’s-t test was used for statistical analysis.
Single-step growth kinetics experiments performed to address this possibility revealed that the UL4aa18stop mutant replicated with similar kinetics and to equivalent levels as the wild-type and revertant viruses that express UL4 (Fig. 9A). Similar virus titers were obtained from the extracellular supernatant of RK13 cells infected with all three viruses, indicating that impairment of virion release is not responsible for the smaller plaque phenotype. Finally, cell tropism was examined to determine whether UL4P plays a role in expanding the host range of EHV-1, as shown for the EHV-1 early IR4 regulatory gene (Breitenbach et al., 2009) as well as for the IR4 homologue EICP22 of HSV-1 (Poffenberger et al., 1993; Post and Roizman, 1981). The UL4aa18stop virus was able to replicate in cells of mouse, rabbit, equine, monkey, and human origin (Fig. 9B). Virus titers in cells infected with all three viruses were similar, with exceptions of mouse L-M and human HeLa cells, for which an approximately one log reduction in maximal virus titer was observed for the UL4aa18stop mutant virus, indicating that UL4 does not contribute to the broad host range of EHV-1 (O’Callaghan and Osterrieder, 2008). However, upon detailed statistical analysis of the viral titers from multiple experiments, we found that the values were significantly different between wild-type and the UL4aa18stop EHV-1 for all cell types. Although the levels of maximal virus production were statistically different, the biological relevance is not likely to be consequential.
Fig. 9.

Growth kinetics and cell tropism of the wild-type EHV-1, UL4aa18stop EHV-1 and ΔUL4R EHV-1. (A) RK13 cells in 60-mm dishes were infected with the wild-type, mutant, and revertant viruses at a MOI of 5. Virus was harvested from the extracellular supernatant as well as from freeze-thawed cells at the indicated times post-infection and enumerated by plaque assay. (B) The ability of the wild-type, mutant, and revertant viruses to replicate in various cell types was determined by infecting mouse L-M, rabbit RK13, equine NBL-6, African green monkey Vero, and human HeLa cells at a MOI of 5. Maximal viral titers at 72 hours post-infection were determined by plaque assay on RK13 cells.
The UL4 gene is not a virulence factor in the CBA mouse model of EHV-1 pathogenesis
Although no striking differences were observed between the UL4-mutant virus and the parental EHV-1 in their abilities to infect and replicate in cell culture, we wanted to determine whether the UL4 gene plays a role in EHV-1 pathogenesis. Utilizing the well characterized CBA mouse model of EHV-1 infection (Colle et al., 1996; Frampton et al., 2004, 2002; Matsumura et al., 1996; O’Callaghan and Osterrieder, 2008; Smith et al., 2000), groups of 12 female mice were intranasally inoculated with wild-type, UL4aa18stop, or ΔUL4R viruses as well as with sterile medium for a control group, and monitored for signs of morbidity and mortality over the course of 9 days. As expected, mice in the sterile medium group demonstrated no signs of infection and exhibited increases in body weight (Fig. 10A). On the other hand, all mice infected with each of the three EHV-1 displayed ruffled fur and a huddling phenotype, along with significant decreases in overall body weight (Fig. 10A). EHV-1 lacking UL4 gene expression caused decreases in body weight similar to that of the wild-type highly pathogenic EHV-1, suggesting that UL4 is not required for EHV-1 pathogenesis in the mouse. Furthermore, mortality rates were similar for mice infected with either the wild-type virus or the UL4aa18stop EHV-1 (Fig. 10B). In cell culture, there was an approximate one log decrease in the ability of the UL4aa18stop mutant virus to replicate in mouse L-M cells compared to the parental virus (Fig. 9B). To determine whether the reduced ability of the UL4-mutant virus to replicate in mouse cells in culture was likewise reflected in vivo, infected mice were sacrificed at days 2, 3, and 4 post-infection, and virus was titered from infected lungs. As shown in Fig. 10C, the UL4aa18stop virus replicated as efficiently as the wild-type EHV-1 in murine lungs. Taken together, the results from these animal experiments indicate that the UL4 gene is not a determinant of virulence for EHV-1.
Fig. 10.

Comparison of the pathogenic properties of wild-type, UL4-mutant and revertant EHV-1 in the CBA mouse model. Groups (n=12 mice) of 6 week old female CBA mice were mock infected with sterile medium or were infected intranasally with 1.25×106 pfu of wild-type RacL11, UL4aa18stop, or ΔUL4R EHV-1. Mice were monitored daily for clinical signs of infection, gain or loss in body weight, and morbidity. (A) Changes in body weight; (B) Percent survival; (C) Virus lung titer. Whole lungs were harvested from mice sacrificed on days 2, 3 and 4 post-infection, and virus was titered by plaque assay.
Viral transcripts of all gene classes are increased in the absence of UL4 protein synthesis
We previously observed that the UL4P was capable of inhibiting luciferase gene expression driven by representative EHV-1 promoters from all three gene classes (Fig. 5). To extend these findings, the effect of UL4P on the synthesis of EHV-1 transcripts was addressed. Cells were infected with wild-type virus or the UL4aa18stop EHV-1 and examined for the effect of the absence of the UL4 protein synthesis on the level of selected IE, E, and L transcripts. RNA was isolated and analyzed by northern blot assays at 4 hpi for IE, 8 hpi for early TK, and 12 hpi for late gK gene expression. As shown in Fig. 11, the levels of the transcripts for each of the three viral genes were greater in the absence of the UL4 protein as compared to those of WT infected cells. Densitometry measurements revealed that the levels of the IE, TK, and gK transcripts were increased a minimum of 20% in the absence of the UL4P, and in repeated experiments these increases were as great as 42%. These data indicate that the absence of the UL4P allows for increased viral transcription at all times during infection, and that the UL4P is indeed an inhibitor of EHV-1 gene expression.
Fig. 11.
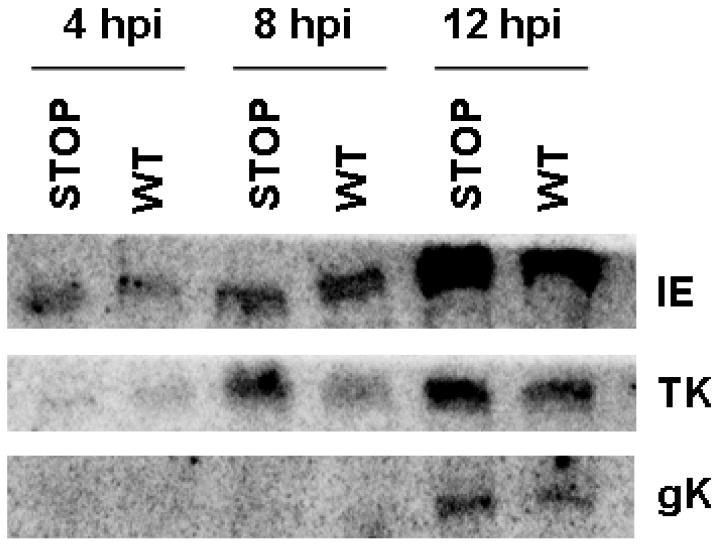
Northern blot analysis examining gene expression in cells infected with wild-type (WT) or STOP-mutant EHV-1. RK13 cells were infected with wild-type EHV-1 or the UL4aa18stop EHV-1, and RNA was isolated at 4, 8, and 12 hours post-infection. Northern blot analysis was used to determine transcript levels for the immediate early (IE) gene, the early thymidine kinase (TK) gene, and the late glycoprotein K (gK) gene.
DISCUSSION
The EHV-1 UL4 gene, conserved within the genome of defective interfering particles, has no attributed function during lytic replication or DIP-mediated persistent infection (Baumann et al., 1984, 1986; Ebner et al., 2008; Ebner and O’Callaghan, 2006). In this report, we presented findings suggesting that this early gene encodes an inhibitory protein that negatively affects viral and cellular gene expression. The expression of the UL4 protein was capable of inhibiting gene expression driven by promoters from all EHV-1 gene classes by greater than 75%. These data appear very similar to the results from experiments concerning the IR2 protein, a negative regulatory protein that was demonstrated to down-regulate all EHV-1 gene promoters tested to date (Kim et al., 2006). Comparing the broad negative regulatory activity of the IR2P and UL4P suggests that UL4P may serve as an auxiliary inhibitory protein to govern EHV-1 gene expression during lytic infection. However, this inhibitory function appears to differ from that reported for the HSV-1 UL55 protein (Block et al., 1991), which shares only 32% amino acid identity with UL4P. In that report, the authors concluded that the inhibition of transient gene expression relied on the presence of other alpha genes expressed in conjunction with the UL55 protein, such that the UL55 protein alone was not sufficient to negatively affect viral gene expression. Conversely, in the case of the UL4 protein, EHV-1 gene expression was significantly inhibited when only the UL4P was expressed. Although these homologs may have an inhibitory function in common, our findings suggest that their mechanism of action is different. However, we cannot rule out the possibility that in the prior study involving HSV-1 UL55, alpha genes were required solely for the efficient expression of the UL55 gene. In that case, the alpha genes per se contributed no inhibitory function, and thus it is conceivable that UL4 and UL55 proteins function through a similar mechanism. We are interested in exploring how the UL4 protein functions to inhibit gene expression, and current work is focused on elucidating a mechanism. Due to the broad inhibitory activity of the UL4P, it appears that the mechanism is not gene-specific as supported by the fact that the UL4P does not bind to specific promoter DNA sequences (data not shown). We have considered the possibility that the UL4 protein interacts with another protein involved in gene expression. GST-pulldown assays are being utilized to examine protein-protein interactions that may occur between UL4P and other viral and/or cellular proteins. In preliminary studies, the UL4 protein did not interact with the IEP; however, it appears that the UL4P functions at the level of gene transcription via an interaction with general transcription factors (unpublished data).
To determine whether the inhibitory function of UL4P was important for viral replication, a mutant virus (UL4aa18stop) was generated. Surprisingly, we found that the replication kinetics, cell tropism, and pathogenesis of an EHV-1 lacking UL4 expression were similar to those of the parental and UL4 revertant viruses (Figs. 9 and 10). These findings were contrary to our hypothesis that removing UL4 gene expression would allow enhanced growth of the mutant virus compared to wild-type EHV-1. As UL4 is not a diploid gene, serial passage of the UL4aa18stop EHV-1 was not expected to result in its reversion to a wild-type phenotype. Indeed, the UL4aa18stop virus failed to produce the UL4 protein upon serial passage in cell culture (data not shown). The varicella-zoster virus (VZV) ORF3 protein is a UL4P counterpart that has 37% identity in amino acid sequence. Similar to these findings with the UL4-mutant EHV-1, it was shown that deletion of VZV ORF3 did not affect virus replication in vivo or in vitro (Zhang et al., 2007). One significant difference between the UL4aa18stop EHV-1 and the parental virus was a reduction in plaque size (Fig. 8). One possible explanation for the smaller plaques is that the virus has a defect in the ability to egress from infected cells, but that possibility was not supported by the observation that the extracellular virus titers were identical for RK13 cells infected with the parental virus and the UL4aa18stop virus.
During DIP-mediated persistent infection, it is believed that the highly expressed IR4/UL5 hybrid protein is responsible for mediating the interference of standard virus replication, as demonstrated by the observation that recombinant DI particles deleted of the UL3 and UL4 genes and engineered to express only the hybrid protein retained DIP interference activity (Ebner et al., 2008). However, it remains unclear what actually triggers the establishment of persistent infection and how high levels of DIP maintain persistent infection. Here, we presented data that demonstrate that the UL4 protein negatively affects all EHV-1 gene classes. As this gene is conserved perfectly within the DIP genome and expressed at very high levels in persistently infected cells (Gray et al., 1989), it is possible the UL4 protein could greatly impair the expression of standard viral genes. This possibility is supported, in part, by the finding that EHV-1 gene expression is tempered in cells infected with the wild-type virus that expresses UL4P, as compared to gene expression in cells infected with the UL4aa18stop mutant EHV-1. This broad inhibitory activity may facilitate the establishment and/or maintenance of DIP-mediated persistent infection. As attractive as this may be, the studies to determine the role UL4 plays during persistency will require a better understanding of the function(s) mediated by this early gene product.
These studies are the first to characterize UL4 as a member of the early class within the EHV-1 gene program. The UL4 protein localizes within both the nucleus and cytoplasm of infected cells, but is not a component of mature virus particles. The luciferase reporter assays demonstrated that the UL4P is an inhibitory protein, a finding that was supported by the results of the viral gene expression studies using the UL4aa18stop EHV-1. Thus, the UL4 protein joins the IR2P as the second EHV-1 early gene product that possess predominantly negative activity. Current studies are addressing how this protein mediates this inhibitory outcome in the case of EHV-1 genes and if this mechanism applies to genes of heterologous viruses and host cells.
MATERIALS AND METHODS
Cell culture and virus propagation
Mouse L-M, rabbit RK13, equine NBL-6, African green monkey Vero and human HeLa cells were grown in Dulbecco’s minimum essential medium (DMEM) supplemented with either 5% or 10% fetal bovine serum at 37°C in a 5% CO2 incubator. The pathogenic EHV-1 strain RacL11 was propagated in equine NBL-6 cells, while the attenuated KyA strain was grown in mouse L-M cells. After growing a high-titer stock of KyA virus, the supernatant virus was collected and clarified by removing cells by centrifugation. Highly purified virions were prepared as previously described in detail (O’Callaghan and Randall, 1976; Perdue et al., 1974).
Plasmids
Manipulation and cloning of the expression plasmids were performed according to the procedures described by Sambrook et al. (1989). pGEX-UL4, which expresses the UL4 gene as a GST fusion protein, was generated by PCR amplifying the UL4 gene from the RacL11 BAC (Rudolph et al., 2002) to include the restriction enzyme sites EcoR I and Sal I with forward primer: 5’- CATGAATTCCCATGCTGCCGGCAAACCGCGCAGAACAC-3’ and reverse primer: 5’-CTAGGTCGACTTATCGTTTATTTTCTCGCTGGCGCTCTTTGGCCGA-3’. The PCR product and the pGEX-4T-2 plasmid were digested with the appropriate enzymes and ligated. pGFP-UL4 expresses a GFP-UL4 fusion protein and was generated by PCR using forward primer: 5’-CATGTGTACAAGATGCTGCCGGCAAACCGCGCAGAA-3’ and reverse primer: 5’-CATTGCGGCCGCTTATCGTTTATTTTCTCGCTG-3’ to append restriction enzyme sites BsrG I and Not I, respectively, to the UL4 gene. The digested insert was cloned into the pEGFP-N1 plasmid that contains the enhanced GFP gene under the control of the HCMV IE promoter. The pUL4-GFP plasmid expresses an amino-terminal UL4-GFP fusion protein. PCR was used to attach EcoR I and BamH I enzyme sites to the UL4 gene using forward primer: 5’-AATTGAATTCCGCCACCATGCTGCCGGCAAACCGCGCAGAAC-3’ and reverse primer: 5’-CATGGGATCCCGTTTATTTTCTCGCTGGCGCTCTTTG-3’, respectively. The digested fragment was again inserted into the pEGFP-N1 plasmid. To generate pCMV-UL4, an expression plasmid that expresses the UL4 gene under the control of the HCMV IE promoter, forward primer: 5’- AATTGAATTCCGCCACCATGCTGCCGGCAAACCGCGCAGAAC-3’ and reverse primer: 5’- CATTGCGGCCGCTTATCGTTTATTTTCTCGCTG-3’ were used to add the EcoR I and Not I sites to the UL4 gene, respectively. The digested insert was then cloned into the pEGFP-N1 plasmid to replace the GFP gene. pUL4-Luc, a reporter plasmid that contains the firefly luciferase gene driven by the UL4 promoter, was created using PCR to attach Kpn I and Bgl II restriction sites flanking the UL4 promoter region. Forward primer: 5’-CATGGTACCCCAACGCAAACAGTTGGCACCGTG-3’ and reverse primer: 5’-CATAGATCTCAGGCTGGGAATTTGCTCGACTGAAG-3’ were used. The resulting 1.5 kb enzyme fragment encompassing the UL4 promoter with TATA box was inserted into the pGL3-Basic plasmid. The remaining expression and reporter plasmids were generated elsewhere (Bowles et al., 2000; Holden et al., 1995; Kim and O’Callaghan, 2001; Smith et al., 1992; Zhao et al., 1995).
Expression and purification of GST-UL4 fusion protein
The induction of GST fusion protein synthesis and its purification have been described previously (Albrecht et al., 2004; Jang et al., 2001). To prepare a purified UL4 protein lacking the GST moiety, GST-UL4 was treated with a thrombin cleavage capture kit (Novagen, Madison, WI) per the manufacturer’s instructions, and the GST portion was removed using GST-bind resin beads (Novagen). The purified protein was concentrated using Amicon Ultra centrifugation filter devices with a size exclusion of 10 and 30 kilodaltons (Millipore, Billerica, MA).
Generation of anti-UL4 protein polyclonal antibody
Antibody generation has been described elsewhere (Albrecht et al., 2004). Briefly, two New Zealand White rabbits (approximately 4 kg per rabbit) were immunized with either the GST-UL4 fusion protein (OC94) or the purified UL4 protein (OC95). The primary inoculum was emulsified in Freund’s complete adjuvant, and after 4-8 weeks, booster immunizations emulsified in incomplete Freund’s adjuvant were administered every 14 days for a total of five booster injections for each rabbit. Before each booster injection, small quantities of serum were drawn from each rabbit to test antibody titers before the final bleed was performed. The anti-UL4P antibody was purified using protein A agarose beads (Pierce, Rockford, IL) according to the manufacturer’s directions. The working dilution in western blot assays was 1:5,000 or greater.
Immunofluorescence microscopy and cell fractionation
RK13 cells were seeded onto No. 1.5 glass coverslips (Fischer Scientific, Pittsburgh, PA) in the bottom of a 6-well plate. The cells were either infected with wild-type RacL11 EHV-1 or transfected with the expression plasmids pCMV-UL4 or pUL4-GFP. Infected cells were fixed at 12 hours post-infection, while transfected cells were fixed at 48 hours post-transfection. Cells were fixed with a 4% paraformaldehyde solution in PBS, and cellular membranes were permeabilized with 0.2% Triton X100 in PBS. After washing the cells with PBS, 10% goat serum in PBS was used to block non-specific antibody interactions. Anti-UL4P OC95 antiserum was used as the primary antibody at a dilution of 1:100 in PBS/0.1% Tween-20 and the secondary antibody was Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, Carlsbad, CA) at 1:100 in PBS/0.1% Tween-20. Unbound antibody was removed by washing, and the coverslips were removed from the 6-well plates and placed on glass slides. SlowFade Gold antifade reagent with DAPI (Invitrogen) was used as the mounting solution (arrows indicate nuclei in Fig. 4). Clear nail polish sealed the edges of the coverslips, and the slides were viewed on a Nikon Eclipse TE300 inverted fluorescent microscope (Melville, NY). Cells were separated into cytoplasmic and nuclear fractions by the following procedure, modified from Muramatsu et al. (1963). Cells were washed with cold PBS, then scraped and pelletted in a microfuge tube. Resuspend pellets in ice-cold Buffer A (10mM HEPES, pH 7.9; 1.5mM MgCl2; 10mM KCl; 0.5mM DTT) and keep on ice for 5 minutes. Cells were disrupted by repeated passage through a 26 gauge needle. Disrupted cells were centrifuged at 300 g for 5 minutes at 4° C to pellet the nuclei. The supernatant was saved as the cytoplasmic fraction. The nuclear pellet was resuspended in sucrose buffer A (0.25M sucrose, 10mM MgCl2), layered over a cushion of sucrose buffer B (0.88M sucrose, 0.5mM MgCl2), and centrifuged at 3000 g for 10 minutes at 4° C. Nuclear pellets were disrupted with RIPA buffer (50mM Tris, pH 7.5; 150mM NaCl; 1% NP-40; 0.5% deoxycholate) and centrifuged at 3000 g for 10 minutes at 4° C to pellet insoluble debris.
Luciferase reporter assays
RK13 cells were seeded into 24-well plates and used the subsequent day at a confluency of 80%. Cells were transfected using lipofectin (Invitrogen) and Opti-MEM medium (Gibco, BRL, Carlsbad, CA) as detailed elsewhere (Ahn et al., 2007). Briefly, 1 pmol of the various reporter plasmids and 0.5 pmol of the expression plasmids were co-transfected into RK13 cells. Eight hours later, regular growth medium was added, and luciferase activity was assayed at 48-72 hours post-transfection utilizing a luciferase assay kit (Promega, Madison, WI) and a POLARstar OPTIMA plate reader (BMG LABTECH Inc., Cary, NC) following the manufacturer’s instructions.
Generation and confirmation of UL4-null EHV-1
A RacL11 BAC deleted for the UL4 gene was generated through galK BAC recombineering (Ahn et al., 2010; Warming et al., 2005). To replace the UL4 gene, PCR amplification (forward primer: 5’-TTATCGTTTATTTTCTCGCTGGCGCTCTTTGGCCGAGGTTATTCCCCTAGCCTGTTGACAATT AATCATCGGCA-3’ and reverse primer: 5’-ATGCTGCCGGCAAACCGCGCAGAACACTCATCTGATGCAGAGCCGCGGGATCAGCACTGTC CTGCTCCTT-3’) was used to append UL4 flanking sequences to the ends of the galK selection gene, and the subsequent PCR product was transformed into SW106 E. coli harboring the RacL11 BAC. Bacteria were plated onto positive selection agar plates containing galactose, and colonies were screened by PCR (5’- CAGACCCAGAGCTCCACGCACCGTCC-3’/5’-GCAGATCTTGCTCCCAGACCTGACC-3’ and 5’-CCCTCTTCTCGAACACGCCGATGAAAAAGGCG-3’/5’-GGCAGATACCTGCAGCCTTGTATCGGCC-3’) for the correct junction sequences. The galK marker was replaced with a mutant form of the UL4 gene that contains a STOP codon at position 18 (underlined; UL4aa18stop) or a wild-type form of the UL4 gene to generate a UL4 revertant BAC (ΔUL4R). PCR was used to create the desired DNA fragments that were transformed into the E. coli harboring the galK inserted RacL11 BAC (5’-TTATCGTTTATTTTCTCGCTGGCGC-3’/5’-ATGCTGCCGGCAAACCGCGCAGAACACTCATCTGATGCAGAGCCGCGGGATTAAATAGGCT CCCACGGGAGGA-3’ and 5’-TTATCGTTTATTTTCTCGCTGGCGCTCTTTGGCCGAGGTTATTCCCCTAG-3’/5’-ATGCTGCCGGCAAACCGCGCAGAACACTCATCTGATGCAGAGCCGCGGGA-3’). Transformed bacteria were plated on negative selection plates containing glycerol and 2-deoxygalactose. Resulting colonies were again screened by PCR for the junction sequences as well as DNA sequence analysis. The desired BACs were further verified by BamH I digestion and Southern blot analysis. To reconstitute the mutant and revertant RacL11 BACs as standard virus, the BAC sequences were replaced with viral DNA sequences for the EUs4 gene by transfecting RK13 cells with both BAC and a plasmid containing the EUs4 sequences. Plaques lacking GFP expression were plaque purified for three rounds and then propagated to high titer on equine NBL-6 cells.
Southern, northern and western blot analyses
Mutant and revertant RacL11 BACs were subjected to BamH I digestion and then electrophoretically separated on a 0.6% agarose gel. Digested DNA was transferred onto a positively-charged nylon membrane (Ambion, Austin, TX) using a semi-dry electroblotter (Bio-Rad Laboratories, Hercules, CA). The transferred DNA was hybridized with a fragment of the UL4 gene (PCR product forward primer: 5’-GGCCTGGGCAGAGTTGGCTGCCTGCC-3’ and reverse primer: 5’-GCAGATCTTGCTCCCAGACCTGACC-3’) end-labeled with [γ-32P]ATP (New England Nuclear Corporation, Boston, MA) by T4 polynucleotide kinase (Promega) in ULTRAhyb ultrasensitive hybridization buffer (Ambion). Free radio-labeled probe was washed away with 2x SSC/0.1% SDS followed by 0.1x SSC/0.1% SDS. A final wash with 2x SSC was completed before wrapping the membrane in plastic and exposing to a phosphor screen and scanning on the molecular imager FX system (Bio-Rad Laboratories). Northern blot analysis was performed by isolating total RNA from RK13 cells infected with RacL11 EHV-1 at the indicated times using the RNA-Bee RNA isolation reagent (AMS Biotechnology (Europe) Ltd, Abingdon, U.K.) per the manufacturer’s procedure (Chomczynski and Sacchi, 1987). RNA samples were separated on a 6% denaturing urea-polyacrylamide gel. The above procedures were followed for transferring and probing the RNA on the membranes. The probe used was a short fragment of the UL4 DNA (5’-TTATCGTTTATTTTCTCGCTGGCGCTCTTTGGCCGAGGTTATTCCCCTAG-3’). Western blot analyses were performed using protein lysates from RK13 cells infected with virus or transfected with expression plasmids. The protein samples were separated on 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes using the semi-dry electroblotter. Membranes were blocked with 1-5% dry milk in PBS/0.3% Tween-20 (PBST). Membranes were rinsed with PBST and incubated with EHV-1 specific primary antibodies, as indicated, followed by goat anti-rabbit or anti-mouse secondary antibody conjugated to alkaline phosphatase (Sigma, Saint Louis, MO). Protein-antibody complexes were visualized using the AP color reagent (Bio-Rad Laboratories) following the manufacturer’s instructions.
Use of metabolic inhibitors
The procedure for virus infection with the use of metabolic inhibitors is described elsewhere (Gray et al., 1987). To detect immediate early transcripts, cells were incubated in the presence of the protein synthesis inhibitor cycloheximide (CHX, 100 μg/mL; Sigma) for 30 minutes prior to infection with EHV-1. Cells were infected with wild-type EHV-1 (MOI of 10) and maintained in the presence of CHX for 4 hours, at which time RNA was isolated. To distinguish early from late transcripts, cells were treated with phosphonoacetic acid (PAA, 100 μg/mL; Sigma) for 30 minutes prior to infection and then for 8 hours after infection when RNA was isolated. The RNA was subjected to northern blot analysis as detailed above.
Growth kinetics, cell tropism and plaque morphology
Cells were seeded into 60 mm dishes to 80% confluency and infected at an MOI of 5. Virus was harvested from the cells and supernatant at the indicated times post-infection and serially diluted to perform plaque assays on RK13 monolayers. Infected monolayers were incubated with medium containing 1.5% methylcellulose, and plaques were enumerated after three days by fixing with 10% formalin and staining with 0.5% methylene blue (Perdue et al., 1974). Plaque morphology was examined using the ImageJ software (NIH, http://rsbweb.nih.gov/ij/).
Animal experiments and statistical analysis
Animal experiments were conducted as published previously (Osterrieder et al., 1996; von Einem et al., 2004). Groups (n=12) of 6 week-old female CBA mice were intranasally infected with 1.25×106 pfu of wild-type RacL11, UL4aa18stop, or ΔUL4R EHV-1 or mock-infected with sterile medium. Mice were weighed before infection and every day post-infection for 9 days to monitor changes in body weight. Mice were sacrificed at days 2, 3, and 4 post-infection, and whole lungs were harvested. Whole lungs were also isolated from animals that succumbed to virus infection. Lung tissue was disrupted using silica beads and BeadBeater (BioSpec Products, Inc., Bartlesville, OK), and virus was titered as described above. Statistical analysis was performed using the two-tailed Student’s-t test to compare changes in body weight as well as mortality rates.
Gene expression in cells infected with wild-type and UL4aa18stop EHV-1
RK13 cells were infected with wild-type or UL4aa18stop EHV-1 to determine whether gene expression differs in the absence of the UL4P. RNA was isolated at 4, 8, and 12 hpi and examined by northern blot analysis using probes specific for the IE, TK and gK genes.
Acknowledgments
We thank Mrs. Suzanne Zavecz for excellent technical assistance and the members of our laboratory for their helpful suggestions. These investigations were supported by research grant AI-22001 from the National Institute of Allergy and Infectious Diseases and grant P20-RR018724 from the National Center for Research Resources of the National Institutes of Heath.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BC, Breitenbach JE, Kim SK, O’Callaghan DJ. The equine herpesvirus-1 IR3 gene that lies antisense to the sole immediate-early (IE) gene is trans-activated by the IE protein, and is poorly expressed to a protein. Virology. 2007;363:15–25. doi: 10.1016/j.virol.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn BC, Zhang Y, O’Callaghan DJ. The equine herpesvirus-1 (EHV-1) IR3 transcript downregulates expression of the IE gene and the absence of IR3 gene expression alters EHV-1 biological properties and virulence. Virology. 2010;402:327–37. doi: 10.1016/j.virol.2010.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht RA, Kim SK, Zhang Y, Zhao Y, O’Callaghan DJ. The equine herpesvirus 1 EICP27 protein enhances gene expression via an interaction with TATA box-binding protein. Virology. 2004;324:311–26. doi: 10.1016/j.virol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Allen GP, Bryans JT. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog Vet Microbiol Immunol. 1986;2:78–144. [PubMed] [Google Scholar]
- Baumann RP, Dauenhauer SA, Caughman GB, Staczek J, O’Callaghan DJ. Structure and genetic complexity of the genomes of herpesvirus defective-interfering particles associated with oncogenic transformation and persistent infection. J Virol. 1984;50:13–21. doi: 10.1128/jvi.50.1.13-21.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann RP, Staczek J, O’Callaghan DJ. Cloning and fine mapping the DNA of equine herpesvirus type one defective interfering particles. Virology. 1986;153:188–200. doi: 10.1016/0042-6822(86)90022-x. [DOI] [PubMed] [Google Scholar]
- Block T, Jordan R, Farkas DH, Hughes RG., Jr Inhibition of transient gene expression with plasmids encoding herpes simplex virus type 1 UL55 and alpha genes. J Gen Virol. 1991;72:131–41. doi: 10.1099/0022-1317-72-1-131. [DOI] [PubMed] [Google Scholar]
- Bowles DE, Holden VR, Zhao Y, O’Callaghan DJ. The ICP0 protein of equine herpesvirus 1 is an early protein that independently transactivates expression of all classes of viral promoters. J Virol. 1997;71:4904–14. doi: 10.1128/jvi.71.7.4904-4914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles DE, Kim SK, O’Callaghan DJ. Characterization of the trans-activation properties of equine herpesvirus 1 EICP0 protein. J Virol. 2000;74:1200–8. doi: 10.1128/jvi.74.3.1200-1208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbach JE, Ebner PD, O’Callaghan DJ. The IR4 auxiliary regulatory protein expands the in vitro host range of equine herpesvirus 1 and is essential for pathogenesis in the murine model. Virology. 2009;383:188–94. doi: 10.1016/j.virol.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DE, Kemp MC, Perdue ML, Randall CC, Gentry GA. Equine herpesvirus in vivo: cyclic production of a DNA density variant with repetitive sequences. Virology. 1976;69:737–50. doi: 10.1016/0042-6822(76)90502-x. [DOI] [PubMed] [Google Scholar]
- Caughman GB, Staczek J, O’Callaghan DJ. Equine herpesvirus type 1 infected cell polypeptides: evidence for immediate early/early/late regulation of viral gene expression. Virology. 1985;145:49–61. doi: 10.1016/0042-6822(85)90200-4. [DOI] [PubMed] [Google Scholar]
- Chen M, Garko-Buczynski KA, Zhang Y, O’Callaghan DJ. The defective interfering particles of equine herpesvirus 1 encode an ICP22/ICP27 hybrid protein that alters viral gene regulation. Virus Res. 1999;59:149–64. doi: 10.1016/s0168-1702(98)00128-2. [DOI] [PubMed] [Google Scholar]
- Chen M, Harty RN, Zhao Y, Holden VR, O’Callaghan DJ. Expression of an equine herpesvirus 1 ICP22/ICP27 hybrid protein encoded by defective interfering particles associated with persistent infection. J Virol. 1996;70:313–20. doi: 10.1128/jvi.70.1.313-320.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Colle CF, 3rd, Tarbet EB, Grafton WD, Jennings SR, O’Callaghan DJ. Equine herpesvirus-1 strain KyA, a candidate vaccine strain, reduces viral titers in mice challenged with a pathogenic strain, RacL. Virus Res. 1996;43:111–24. doi: 10.1016/0168-1702(96)01324-x. [DOI] [PubMed] [Google Scholar]
- Csellner H, Walker C, Wellington JE, McLure LE, Love DN, Whalley JM. EHV-1 glycoprotein D (EHV-1 gD) is required for virus entry and cell-cell fusion, and an EHV-1 gD deletion mutant induces a protective immune response in mice. Arch Virol. 2000;145:2371–85. doi: 10.1007/s007050070027. [DOI] [PubMed] [Google Scholar]
- Dauenhauer SA, Robinson RA, O’Callaghan DJ. Chronic production of defective-interfering particles by hamster embryo cultures of herpesvirus persistently infected and oncogenically transformed cells. J Gen Virol. 1982;60:1–14. doi: 10.1099/0022-1317-60-1-1. [DOI] [PubMed] [Google Scholar]
- Ebner PD, Kim SK, O’Callaghan DJ. Biological and genotypic properties of defective interfering particles of equine herpesvirus 1 that mediate persistent infection. Virology. 2008;381:98–105. doi: 10.1016/j.virol.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner PD, O’Callaghan DJ. Genetic complexity of EHV-1 defective interfering particles and identification of novel IR4/UL5 hybrid proteins produced during persistent infection. Virus Genes. 2006;32:313–20. doi: 10.1007/s11262-005-6916-y. [DOI] [PubMed] [Google Scholar]
- Elliott GD, O’Hare P. Equine herpesvirus 1 gene 12, the functional homologue of herpes simplex virus VP16, transactivates via octamer sequences in the equine herpesvirus IE gene promoter. Virology. 1995;213:258–62. doi: 10.1006/viro.1995.1568. [DOI] [PubMed] [Google Scholar]
- Flowers CC, O’Callaghan DJ. Equine herpesvirus 1 glycoprotein D: mapping of the transcript and a neutralization epitope. J Virol. 1992;66:6451–60. doi: 10.1128/jvi.66.11.6451-6460.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton AR, Jr, Goins WF, Cohen JB, von Einem J, Osterrieder N, O’Callaghan DJ, Glorioso JC. Equine herpesvirus 1 utilizes a novel herpesvirus entry receptor. J Virol. 2005;79:3169–73. doi: 10.1128/JVI.79.5.3169-3173.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton AR, Jr, Smith PM, Zhang Y, Grafton WD, Matsumura T, Osterrieder N, O’Callaghan DJ. Meningoencephalitis in mice infected with an equine herpesvirus 1 strain KyA recombinant expressing glycoprotein I and glycoprotein E. Virus Genes. 2004;29:9–17. doi: 10.1023/B:VIRU.0000032785.19420.14. [DOI] [PubMed] [Google Scholar]
- Frampton AR, Jr, Smith PM, Zhang Y, Matsumura T, Osterrieder N, O’Callaghan DJ. Contribution of gene products encoded within the unique short segment of equine herpesvirus 1 to virulence in a murine model. Virus Res. 2002;90:287–301. doi: 10.1016/s0168-1702(02)00245-9. [DOI] [PubMed] [Google Scholar]
- Gray WL, Baumann RP, Robertson AT, Caughman GB, O’Callaghan DJ, Staczek J. Regulation of equine herpesvirus type 1 gene expression: characterization of immediate early, early, and late transcription. Virology. 1987;158:79–87. doi: 10.1016/0042-6822(87)90240-6. [DOI] [PubMed] [Google Scholar]
- Gray WL, Yalamanchili R, Raengsakulrach B, Baumann RP, Staczek J, O’Callaghan DJ. Viral transcripts in cells infected with defective interfering particles of equine herpesvirus type 1. Virology. 1989;172:1–10. doi: 10.1016/0042-6822(89)90101-3. [DOI] [PubMed] [Google Scholar]
- Grundy FJ, Baumann RP, O’Callaghan DJ. DNA sequence and comparative analyses of the equine herpesvirus type 1 immediate early gene. Virology. 1989;172:223–36. doi: 10.1016/0042-6822(89)90124-4. [DOI] [PubMed] [Google Scholar]
- Harty RN, Holden VR, O’Callaghan DJ. Transcriptional and translational analyses of the UL2 gene of equine herpesvirus 1: a homolog of UL55 of herpes simplex virus type 1 that is maintained in the genome of defective interfering particles. J Virol. 1993;67:2255–65. doi: 10.1128/jvi.67.4.2255-2265.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, O’Callaghan DJ. An early gene maps within and is 3’ coterminal with the immediate-early gene of equine herpesvirus 1. J Virol. 1991;65:3829–38. doi: 10.1128/jvi.65.7.3829-3838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden VR, Harty RN, Yalamanchili RR, O’Callaghan DJ. The IR3 gene of equine herpesvirus type 1: a unique gene regulated by sequences within the intron of the immediate-early gene. DNA Seq. 1992;3:143–52. doi: 10.3109/10425179209034010. [DOI] [PubMed] [Google Scholar]
- Holden VR, Zhao Y, Thompson Y, Caughman GB, Smith RH, O’Callaghan DJ. Characterization of the regulatory function of the ICP22 protein of equine herpesvirus type 1. Virology. 1995;210:273–82. doi: 10.1006/viro.1995.1344. [DOI] [PubMed] [Google Scholar]
- Huang AS, Baltimore D. Defective viral particles and viral disease processes. Nature. 1970;226:325–7. doi: 10.1038/226325a0. [DOI] [PubMed] [Google Scholar]
- Jang HK, Albrecht RA, Buczynski KA, Kim SK, Derbigny WA, O’Callaghan DJ. Mapping the sequences that mediate interaction of the equine herpesvirus 1 immediate-early protein and human TFIIB. J Virol. 2001;75:10219–30. doi: 10.1128/JVI.75.21.10219-10230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Ahn BC, Albrecht RA, O’Callaghan DJ. The unique IR2 protein of equine herpesvirus 1 negatively regulates viral gene expression. J Virol. 2006;80:5041–9. doi: 10.1128/JVI.80.10.5041-5049.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Bowles DE, O’Callaghan DJ. The gamma2 late glycoprotein K promoter of equine herpesvirus 1 is differentially regulated by the IE and EICP0 proteins. Virology. 1999;256:173–9. doi: 10.1006/viro.1999.9608. [DOI] [PubMed] [Google Scholar]
- Kim SK, Holden VR, O’Callaghan DJ. The ICP22 protein of equine herpesvirus 1 cooperates with the IE protein to regulate viral gene expression. J Virol. 1997;71:1004–12. doi: 10.1128/jvi.71.2.1004-1012.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, O’Callaghan DJ. Molecular characterizations of the equine herpesvirus 1 ETIF promoter region and translation initiation site. Virology. 2001;286:237–47. doi: 10.1006/viro.2001.0988. [DOI] [PubMed] [Google Scholar]
- Lewis JB, Thompson YG, Caughman GB. Transcriptional control of the equine herpesvirus 1 immediate early gene. Virology. 1993;197:788–92. doi: 10.1006/viro.1993.1658. [DOI] [PubMed] [Google Scholar]
- Lewis JB, Thompson YG, Feng X, Holden VR, O’Callaghan DJ, Caughman GB. Structural and antigenic identification of the ORF12 protein (alpha TIF) of equine herpesvirus 1. Virology. 1997;230:369–75. doi: 10.1006/viro.1997.8477. [DOI] [PubMed] [Google Scholar]
- Lewis JB, Thompson YG, Jenkins AC, Caughman GB. Characterization and localization of the equine herpesvirus 1 major DNA binding protein. Virology. 1995;207:380–91. doi: 10.1006/viro.1995.1097. [DOI] [PubMed] [Google Scholar]
- Matsumura T, O’Callaghan DJ, Kondo T, Kamada M. Lack of virulence of the murine fibroblast adapted strain, Kentucky A (KyA), of equine herpesvirus type 1 (EHV-1) in young horses. Vet Microbiol. 1996;48:353–65. doi: 10.1016/0378-1135(09)59999-3. [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC, Keil GM, Fuchs W. Molecular Biology of Animal Herpesviruses. In: Mettenleiter TC, Sobrino F, editors. Animal Viruses: Molecular Biology. Caister Academic Press; Norfolk, UK: 2008. pp. 375–455. [Google Scholar]
- Misra V, Bratanich AC, Carpenter D, O’Hare P. Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the BHV alpha gene trans-inducing factor. J Virol. 1994;68:4898–909. doi: 10.1128/jvi.68.8.4898-4909.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriuchi H, Moriuchi M, Cohen JI. Proteins and cis-acting elements associated with transactivation of the varicella-zoster virus (VZV) immediate-early gene 62 promoter by VZV open reading frame 10 protein. J Virol. 1995;69:4693–701. doi: 10.1128/jvi.69.8.4693-4701.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Smetana K, Busch H. Quantitative aspects of isolation of nucleoli of the Walker carcinosarcoma and liver of the rat. Cancer Res. 1963;23:510–8. [PubMed] [Google Scholar]
- O’Callaghan DJ, Osterrieder N. Herpesviruses of Horses. In: Mahy BWJ, VanRegenmortel MHV, editors. Encyclopedia of Virology. Third. Vol. 2. Elsevier Ltd; Oxford: 2008. pp. 411–420. [Google Scholar]
- O’Callaghan DJ, Randall CC. Molecular anatomy of herpesviruses: recent studies. Prog Med Virol. 1976;22:152–210. [PubMed] [Google Scholar]
- Osterrieder N, Holden VR, Brandmuller C, Neubauer A, Kaaden OR, O’Callaghan DJ. The equine herpesvirus 1 IR6 protein is nonessential for virus growth in vitro and modified by serial virus passage in cell culture. Virology. 1996;217:442–51. doi: 10.1006/viro.1996.0138. [DOI] [PubMed] [Google Scholar]
- Perdue ML, Kemp MC, Randall CC, O’Callaghan DJ. Studies of the molecular anatomy of the L-M cell strain of equine herpes virus type 1: proteins of the nucleocapsid and intact virion. Virology. 1974;59:201–16. doi: 10.1016/0042-6822(74)90216-5. [DOI] [PubMed] [Google Scholar]
- Poffenberger KL, Raichlen PE, Herman RC. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes. 1993;7:171–86. doi: 10.1007/BF01702397. [DOI] [PubMed] [Google Scholar]
- Post LE, Roizman B. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell. 1981;25:227–32. doi: 10.1016/0092-8674(81)90247-6. [DOI] [PubMed] [Google Scholar]
- Purewal AS, Allsopp R, Riggio M, Telford EA, Azam S, Davison AJ, Edington N. Equid herpesviruses 1 and 4 encode functional homologs of the herpes simplex virus type 1 virion transactivator protein, VP16. Virology. 1994;198:385–9. doi: 10.1006/viro.1994.1047. [DOI] [PubMed] [Google Scholar]
- Rudolph J, O’Callaghan DJ, Osterrieder N. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and racL11 as bacterial artificial chromosomes (BAC) J Vet Med B Infect Dis Vet Public Health. 2002;49:31–6. doi: 10.1046/j.1439-0450.2002.00534.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, F EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Springs Harbor Laboratory Press; Cold Springs Harbor, NY: 1989. [Google Scholar]
- Smith PM, Kahan SM, Rorex CB, von Einem J, Osterrieder N, O’Callaghan DJ. Expression of the full-length form of gp2 of equine herpesvirus 1 (EHV-1) completely restores respiratory virulence to the attenuated EHV-1 strain KyA in CBA mice. J Virol. 2005;79:5105–15. doi: 10.1128/JVI.79.8.5105-5115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Zhang Y, Grafton WD, Jennings SR, O’Callaghan DJ. Severe murine lung immunopathology elicited by the pathogenic equine herpesvirus 1 strain RacL11 correlates with early production of macrophage inflammatory proteins 1alpha, 1beta, and 2 and tumor necrosis factor alpha. J Virol. 2000;74:10034–40. doi: 10.1128/jvi.74.21.10034-10040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Caughman GB, O’Callaghan DJ. Characterization of the regulatory functions of the equine herpesvirus 1 immediate-early gene product. J Virol. 1992;66:936–45. doi: 10.1128/jvi.66.2.936-945.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Zhao Y, O’Callaghan DJ. The equine herpesvirus type 1 immediate-early gene product contains an acidic transcriptional activation domain. Virology. 1994;202:760–70. doi: 10.1006/viro.1994.1398. [DOI] [PubMed] [Google Scholar]
- von Einem J, Schumacher D, O’Callaghan DJ, Osterrieder N. The alpha-TIF (VP16) homologue (ETIF) of equine herpesvirus 1 is essential for secondary envelopment and virus egress. J Virol. 2006;80:2609–20. doi: 10.1128/JVI.80.6.2609-2620.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Einem J, Wellington J, Whalley JM, Osterrieder K, O’Callaghan DJ, Osterrieder N. The truncated form of glycoprotein gp2 of equine herpesvirus 1 (EHV-1) vaccine strain KyA is not functionally equivalent to full-length gp2 encoded by EHV-1 wild-type strain RacL11. J Virol. 2004;78:3003–13. doi: 10.1128/JVI.78.6.3003-3013.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Jiang YM, Oshima S, Daikoku T, Yamashita Y, Tsurumi T, Nishiyama Y. Characterization of the UL55 gene product of herpes simplex virus type 2. J Gen Virol. 1998;79:1989–95. doi: 10.1099/0022-1317-79-8-1989. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rowe J, Wang W, Sommer M, Arvin A, Moffat J, Zhu H. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J Virol. 2007;81:9024–33. doi: 10.1128/JVI.02666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Holden VR, Smith RH, O’Callaghan DJ. Regulatory function of the equine herpesvirus 1 ICP27 gene product. J Virol. 1995;69:2786–93. doi: 10.1128/jvi.69.5.2786-2793.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]