


Abstract
Reverse gyrase is the only topoisomerase that can introduce positive supercoils into DNA in an ATP-dependent process. It has a modular structure and harnesses a helicase-like domain to support a topoisomerase activity, thereby creating the unique function of positive DNA supercoiling. The isolated topoisomerase domain can relax negatively supercoiled DNA, an activity that is suppressed in reverse gyrase. The isolated helicase-like domain is a nucleotide-dependent switch that is attenuated by the topoisomerase domain. Inter-domain communication thus appears central for the functional cooperation of the two domains. The latch, an insertion into the helicase-like domain, has been suggested as an important element in coordinating their activities. Here, we have dissected the influence of the latch on nucleotide and DNA binding to the helicase-like domain, and on DNA supercoiling by reverse gyrase. We find that the latch is required for positive DNA supercoiling. It is crucial for the cooperativity of DNA and nucleotide binding to the helicase-like domain. The latch contributes to DNA binding, and affects the preference of reverse gyrase for ssDNA. Thus, the latch coordinates the individual domain activities by modulating the helicase-like domain, and by communicating changes in the nucleotide state to the topoisomerase domain.
INTRODUCTION
DNA topoisomerases catalyze the inter-conversion of DNA topoisomers in processes such as DNA replication, recombination and repair (1). Reverse gyrase, first identified in the hyperthermophilic archaeon Sulfolobus acidocaldarius (2), is the only topoisomerase that can introduce positive supercoils into DNA in an ATP-dependent process. Reverse gyrases are unique to thermophiles and hyperthermophiles, and presumably protect their DNA at high temperatures via DNA chaperone and renaturase activities (3,4). In general, reverse gyrases have a modular structure, comprised of an N-terminal helicase-like domain covalently linked to a C-terminal topoisomerase I domain (Figure 1) (5). The helicase-like domain consists of two RecA-like subdomains (H1 and H2) that harbor all signature motifs of superfamily (SF) two helicases, though with altered sequences (6). The H2 domain is interrupted by an insertion called the ‘latch’ (H3), which structurally resembles the RNA-binding region of the transcription termination factor Rho (7,8). The C-terminal topoisomerase domain is homologous to prokaryotic type IA DNA topoisomerases, and consists of four subdomains (T1–T4) equivalent to domains I-IV of Escherichia coli topoisomerase I (9). The isolated topoisomerase domain can relax negatively supercoiled DNA in vitro (10). By contrast, neither the isolated helicase-like domain nor reverse gyrase exhibits any nucleic acid unwinding activity (10,11). The helicase-like domain is a nucleotide-dependent switch that exhibits a strong preference for ssDNA in the nucleotide-free and ADP state, but binds ssDNA and dsDNA with similar affinities in the ATP state (11). Reverse gyrase, in contrast, preferentially interacts with ssDNA in all states, and it has been shown that positive supercoiling is more efficient with a plasmid containing single-stranded regions (4). Furthermore, the DNA-stimulated ATPase activity of the helicase-like domain of Thermotoga maritima reverse gyrase is 10-fold higher than that of full-length reverse gyrase, pointing toward a suppression of the activity of the helicase-like domain by the topoisomerase domain (11). Harnessing a helicase-like domain to support a topoisomerase activity creates the unique function of positive DNA supercoiling and thus generates an enzyme that is clearly more than the sum of its parts. Undoubtedly, inter-domain communication is central for the combined activities of the helicase-like and topoisomerase domains in the supercoiling reaction.
Figure 1.

Reverse gyrase structure and constructs used. (A) Structure of reverse gyrase from A. fulgidus (PDB-ID 1gku) (6). The helicase-like domain is shown in dark blue (H1, H2) and orange (H3, latch, residues 389–459), and the topoisomerase domain (T1-T4) in cyan. The figure was created with Pymol (www.pymol.org). (B) Constructs used in this study. rgyr: reverse gyrase, rgyr_Δlatch: reverse gyrase lacking the latch region H3, rgyr_hel: helicase-like domain comprising H1, H2 and H3 (latch), rgyr_hel_Δlatch: helicase-like domain comprising H1 and H2.
Based on biochemical data and structural information, a strand-passage mechanism, similar to DNA relaxation by type IA topoisomerases, has been proposed for positive supercoiling of DNA by reverse gyrase (6). The salient features of this putative mechanism are a conformational change in the helicase-like domain that brings domains H1 and H2 closer together, and a movement of the latch domain away from the topoisomerase domain that allows the lid (T3) to swing up. The latch has thus been suggested as an important element in inter-domain communication, and was shown to inhibit the DNA relaxation activity of the topoisomerase domain of Archaeoglobus fulgidus reverse gyrase (12). Moreover, mutants lacking the latch exhibited altered DNA-dependent ATPase activity, indicating a role of the latch in coupling ATP hydrolysis to the supercoiling reaction. By contrast, the latch was not required for the positive supercoiling activity of the enzyme (13).
Here, we have dissected the influence of the latch on nucleotide and DNA binding to the helicase-like domain, and on DNA supercoiling by reverse gyrase. The latch can be deleted without affecting the structure of the H2 domain. We find that the latch contributes to DNA binding, and inhibits nucleotide binding, but not ATP hydrolysis. The latch is crucial for the cooperativity of DNA and nucleotide binding to the helicase-like domain, and affects the preference of reverse gyrase for ssDNA. Most importantly, the latch is required for positive DNA supercoiling. Our data demonstrate that the latch modulates the properties of the helicase-like domain, and communicates changes in the nucleotide state to the topoisomerase domain, Thereby, it coordinates and couples individual domain activities to allow for positive supercoiling of DNA.
MATERIALS AND METHODS
Cloning, protein production and purification
Reverse gyrase lacking the latch region (aa 389–459) was constructed by inverse polymerase chain reaction (PCR) using pET28a with the reverse gyrase gene inserted into the NcoI and XhoI sites (14) as a template, and primers containing a BamH1 restriction site and the region downstream from the codon for aa 459 (forward primer), or upstream from the codon for aa 389 (reverse primer). The amplified DNA was restricted with BamH1, and circularized by ligation, resulting in pET28a with the gene for reverse gyrase lacking the latch domain (rgyr_Δlatch). The helicase-like domain lacking the latch (rgyr_hel_Δlatch) was constructed by overlap extension PCR with pET28 containing the coding region for aa 59–541 as a template. The generated fragment was amplified using primers containing NcoI and XhoI restriction sites, and ligated into the NcoI and XhoI sites of pETM28. The region coding for the latch (aa 388–459) was amplified using primers containing NcoI and XhoI restriction sites and ligated into the NcoI and XhoI sites of pETM30 (G. Stier, EMBL Heidelberg). Reverse gyrase (rgyr) and the rgyr_Δlatch were produced in E. coli BL21(DE3) Star RIL and were purified as described (11). Rgyr_hel, rgyr_hel_Δlatch and the isolated latch domains were produced in E. coli Rosetta(DE3). Rgyr_hel and rgyr_hel_Δlatch were purified as described (11). Cells overproducing the latch as a GST fusion were disrupted in a Microfluidizer in 50 mM Tris/HCl, pH 7.5, 1 M NaCl, 10 mM MgCl2, 10 mM Zn(OAc)2, 2 mM β-mercaptoethanol, and the crude extract was cleared by centrifugation. All purification steps were performed at room temperature. The NaCl concentration of the supernatant was adjusted to 0.2 M, and it was applied to a Glutathione sepharose column equilibrated in 50 mM Tris/HCl, pH 7.5, 0.2 M NaCl, 10 mM MgCl2, 10 mM Zn(OAc)2, 2 mM β-mercaptoethanol. The GST fusion protein was eluted with the same buffer containing 20 mM reduced glutathione, and cleaved overnight with TEV protease. Remaining fusion protein and GST were removed by purification via size-exclusion chromatography on a calibrated S75 column in 50 mM Tris/HCl, pH 7.5, 0.2 M NaCl, 10 mM MgCl2, 10 mM Zn(OAc)2, 2 mM β-mercaptoethanol. As the latch does not contain tryptophans, the protein concentration was determined with the Bradford method.
Protein crystallization, data collection and structure determination
Diffraction quality crystals for rgyr_hel_Δlatch were obtained at 25°C from the PEG/ion screen (Hampton). 20 mg ml−1 protein was mixed 1:1 (v/v) with reservoir consisting of 0.2 M magnesium formate, 20% PEG 3350. Crystals were cryo-protected with paraffin oil, mounted in an arbitrary orientation and data were collected at SLS beamline X06DA on a MAR-CCD detector. Data were indexed in a primitive orthorhombic lattice, integrated with XDS (15), scaled with MOSFLM (16) and corrected for anisotropy with XPREP (Bruker). Data statistics are collected in Table 1. Systematic absences identified space group P212121. A Matthews coefficient of 2.8 Å3/Da corresponding to 56% solvent content indicated two rgyr_hel_Δlatch molecules per asymmetric unit.
Table 1.
Data collection, phasing and refinement statistics
| Data set | 3OIY |
|---|---|
| Data collection | 132.7–2.35 |
| Resolution range (Å)a | (2.44–2.35) |
| Space group | P212121 |
| Cell dimensions (Å) | a = 59.6, b = 126.5, c = 132.7 |
| Unique reflections | 42 547 (4453) |
| Multiplicity | 7.1 (7.4) |
| Completeness (%) | 99.6 (100) |
| Rsym (%)b | 9.9 (65.9) |
| Average I/σ(I) | 9.7 (1.3) |
| Refinement | 44.4–2.35 |
| Resolution range (Å) | (2.41–2.35) |
| Rcryst (%)c | 20.8 (39.4) |
| Rfree (%)c | 25.1 (41.5) |
| # of residues/waters | 807/99 |
| Phase/coordinate errors (°/Å)d | 28.3/0.38 |
| rmsd bonds/angles (Å/ °) | 0.012/1.56 |
| Ramachandran plot (%)e | 96.8/3.1/0.1 |
aValues in parentheses correspond to the highest resolution shell.
bRsym = 100·ΣhΣi|Ii(h) – <I(h)>|/ΣhΣiIi(h), where Ii(h) is the i-th measurement of reflection h and <I(h)> is the average value of the reflection intensity.
cRcryst=Σ|Fo| – |Fc|/Σ|Fo|, where Fo and Fc are the structure factor amplitudes from the data and the model, respectively; Rfree is Rcryst with 5 % of test set structure factors.
dBased on maximum likelihood.
eCalculated using COOT (19); numbers reflect the percentage amino acid residues of the core, allowed and disallowed regions, respectively.
The structure was determined by molecular replacement using the truncated (termini and loops were removed, the sequence of conserved residues was adjusted to match T. maritima reverse gyrase) H1 and H2 domains of A. fulgidus reverse gyrase as separate search models with PHASER (16). Both H2 domains, but only one H1 domain, were found. Refinement of this model with BUSTER (17) resulted in spurious electron density for the second H1 domain, which could be placed manually. Further refinement using PHENIX (18) and rebuilding in COOT (19) enabled tracing of the connections between the H1 and H2 domains to generate a model of two complete helicase modules. Refinement statistics are summarized in Table 1. The coordinates and structure factors have been deposited in the Protein Data Bank (accession code 3oiy).
Nucleotides and nucleic acid substrates
Nucleotides were purchased from Jena Biosciences. Oligonucleotides for ssDNA or dsDNA substrates were purchased from Purimex. The 60 b ssDNA was 5′-(Fl)- AAG CCA AGC TTC TAG AGT CAG CCC GTG ATA TTC ATT ACT TCT TAT CCT AGG ATC CCC GTT-3′. The 60-bp dsDNA substrate was formed by annealing with the complementary strand. Negatively supercoiled pUC18 was purified from transformed E. coli XL1-Blue cells.
Fluorescence equilibrium titrations
Dissociation constants of enzyme/nucleotide complexes at 37°C were determined in fluorescence equilibrium titrations with a Fluoromax-3 fluorimeter (Jobin Yvon) using the fluorescent ADP analog mantADP (20). In titrations of 1 µM mantADP in 50 mM Tris/HCl, pH 7.5, 0.15 M NaCl, 10 mM MgCl2, 100 µM Zn(OAc)2, 2 mM β-mercaptoethanol, binding was monitored via Förster resonance energy transfer (FRET) from tryptophan to the mant group. Tryptophan fluorescence was excited at 295 nm (3 nm bandwidth), and mant fluorescence was detected at 440 nm (3 nm bandwidth). The Kd value was determined using the solution of the quadratic equation describing a 1:1 complex formation [Equation (1)].
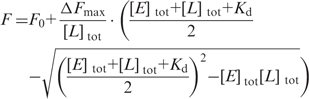 |
(1) |
where F0 is the fluorescence of free mantADP, ΔFmax is the amplitude, [E]tot is the total enzyme concentration and [L]tot is the total ligand concentration.
DNA binding was measured in fluorescence anisotropy titrations of DNA labeled with fluorescein at the 5′-end in 50 mM Tris/HCl, pH 7.5, 0.15 M NaCl, 10 mM MgCl2, 100 µM Zn(OAc)2, 2 mM β-mercaptoethanol, and analyzed as described (11).
Steady-state ATPase activity
The steady-state ATPase activity was measured in a coupled ATPase assay via the decrease in A340 due to oxidation of NADH to NAD+ (21) as described (11). Assay conditions were 50 mM Tris/HCl, pH 7.5, 0.15 M NaCl, 10 mM MgCl2, 2 mM β-mercaptoethanol, 0.4 mM phosphoenolpyruvate and 0.2 mM NADH, 23 µg ml−1 lactate dehydrogenase, 37 µg ml−1 pyruvate kinase. Initial velocities v (in μM ATP s−1) were calculated from the absorbance change ΔA340/Δt with ε340, NADH = 6220 M−1 cm−1, and converted to kcat. Data were analyzed according to the Michaelis–Menten equation.
Topoisomerase activity
Relaxation and supercoiling activities were measured in 50 mM Tris/HCl, pH 7.5, 0.15 M NaCl, 10 mM MgCl2, 100 μM Zn(OAc)2, 2 mM β-mercaptoethanol, 10% (w/v) polyethylene glycol 8000 and 15 nM negatively supercoiled pUC18 plasmid as a substrate. Reactions were started by adding 2 mM ATP, ADP, ATPγS or ADPNP to 1 μM reverse gyrase at 75°C, and samples taken at different time points were analyzed on a 1.2% (w/v) agarose gel run at 11 V cm−1. To separate positively and negatively supercoiled species, two-dimensional electrophoresis in 2% agarose gels was performed in the presence of 10 μg ml−1 chloroquine in the second dimension.
RESULTS
The latch interferes with nucleotide binding, but contributes to DNA binding to the helicase-like domain
We have previously shown that the helicase-like domain, rgyr_hel, is a nucleotide-dependent switch that binds DNA and ATP cooperatively (11). In the ATP state of rgyr_hel, the affinities for ssDNA and dsDNA are comparable, whereas in the ADP state, ssDNA is bound preferentially. In the context of full-length rgyr, the differences in affinities for ssDNA and dsDNA are reduced, as is the cooperativity between ATP and DNA binding.
To dissect the role of the latch region in positive DNA supercoiling, we constructed a latch deletion mutant of T. maritima reverse gyrase in which residues 389–459 of the helicase-like domain are replaced by a glycine (Figure 1, rgyr_hel_Δlatch). Residues 387 and 460 are prolines in T. maritima reverse gyrase (Pro352 and Pro424 in A. fulgidus numbering) that are conserved among reverse gyrases. In the structure of A. fulgidus reverse gyrase, Pro352 and Pro424 mark the beginning and end, respectively, of β-strands leading into and out of the latch domain. These β-strands are part of a β-sheet that connects the latch domain to H2. The Cα-atoms of the prolines are 0.66 nm apart, a distance that can be covered by two amino acids. To confirm that the replacement of the latch domain by a glycine does not alter the overall structure of reverse gyrase, we determined the crystal structure of rgyr_hel_Δlatch to 2.35-Å resolution (Table 1, Figure 2). The H2 domains of T. maritima and A. fulgidus reverse gyrases superpose with an rmsd of 1.6 Å over 181 residues, proving that the deletion of the latch domain does not significantly alter the structure of the H2 domain (Figure 2A). Importantly, the glycine residue replacing the latch domain is well defined by electron density (Figure 2B). The region around the conserved proline residues is similar in both structures with a Cα–Cα distance of 0.57 nm for Pro387 and Pro460. Thus, the deletion of the latch region does not alter the RecA fold of H2, and, consequently, should not have any undesired effects on the overall structure of reverse gyrase.
Figure 2.

Structure of H2 in rgyr_hel_Δlatch. (A) Superposition of the H2 domain in rgyr_hel_Δlatch (T. maritima, gray) and H2/H3 of A. fulgidus reverse gyrase. H2 is depicted in dark blue, the latch domain (H3) in orange. (B) Close-up of the deletion region. P387, S388 and P460, as well as the introduced glycine are shown in stick representation; S388 and the glycine are highlighted in cyan. The σA-weighted 2mFo–DFc electron density in this region is contoured at the 1σ level and depicted as a gray mesh. The figure was created with Pymol (www.pymol.org).
We also measured far-ultraviolet (UV) circular dichroism (CD) spectra of rgyr and rgyr_Δlatch at 37°C, 50°C and 70°C (Supplementary Figure S1). The CD spectra indicate that the two proteins are thermostable and have similar folds in this temperature range. Both enzymes are active topoisomerases at 75°C (see below). Overall, we thus have no indication for large structural changes in rgyr_Δlatch.
As the helicase-like domain is responsible for ATP binding and hydrolysis, we first analyzed the effect of the latch on adenine nucleotide binding (Figure 3). We have previously established that mantADP is a suitable analog to monitor adenine nucleotide binding to rgyr and rgyr_hel (11,14). Deletion of the latch in rgyr led to a 7-fold increase in affinity for mantADP, and a 2-fold increase in the affinity for ADPNP (Table 2). In the context of the isolated helicase-like domain, deletion of the latch led to a 1.6-fold increase in mantADP affinity, and a 2.7-fold in (mant)ADPNP affinity. Thus, the latch insertion in H2 affects adenine nucleotide binding.
Figure 3.

Nucleotide binding. (A) Titration of 1 µM mantADP with rgyr (filled squares) and rgyr_Δlatch (open squares). The Kd values are 1.1 ± 0.3 µM (rgyr) and 0.16 ± 0.02 µM (rgyr_Δlatch). (B) Titration of 1 µM mantADP with rgyr_hel (filled circles) and rgyr_hel_Δlatch (open circles). The Kd values are 4.4 ± 0.2 µM (rgyr_hel) and 2.7 ± 0.1 µM (rgyr_hel_Δlatch). Binding was detected via energy transfer from tryptophans to the mant group. For Kd values in the presence of DNA, see Table 2.
Table 2.
Nucleotide binding
| mADP | ||
| rgyraKd (µM) | rgyr_Δlatch Kd (µM) | |
| – | 1.1 ± 0.3 | 0.16 ± 0.02 |
| ssDNA | 2.2 ± 0.4 | 0.67 ± 0.05 |
| dsDNA | 0.5 ± 0.2 | 0.13 ± 0.03 |
| rgyr_hel Kd (µM) | rgyr_hel_Δlatch Kd (µM) | |
| – | 4.4 ± 0.2 | 2.7 ± 0.1 |
| ssDNA | 10 ± 1 | 2.6 ± 0.7 |
| dsDNA | 1.7 ± 0.4 | 0.9 ± 0.4 |
| ADPNP | ||
| rgyr Kd (µM) | rgyr_Δlatch Kd (µM) | |
| – | 10 ± 0.8 | 4.7 ± 0.8 |
| ssDNA | 12 ± 3 | 12 ± 2 |
| dsDNA | 8 ± 1 | 8 ± 1 |
| rgyr_helbKd (µM) | rgyr_hel_ΔlatchbKd (µM) | |
| – | 36 ± 8 | 13 ± 4 |
| ssDNA | 12 ± 2 | 16 ± 8 |
| dsDNA | 1.9 ± 0.4 | 11 ± 4 |
aData from ref. (11) .bTitrations were performed with mADPNP.
To dissect contributions of the latch domain to DNA binding, anisotropy titrations of fluorescently labeled ssDNA and dsDNA were performed (Figure 4). Ideally, DNA binding studies should be performed with a reverse gyrase mutant in which the catalytic tyrosine involved in DNA cleavage is replaced by a phenylalanine to prevent covalent binding of the DNA. However, the DNA-cleavage-deficient rgyr_Δlatch_Y851F mutant was not soluble. Therefore, we performed comparative DNA-binding studies with rgyr_Δlatch and wild-type reverse gyrase that also contained the catalytic tyrosine (Table 3) to dissect the influence of the latch. The effects of the latch deletion on DNA binding to rgyr were small, with a 2-fold reduction of ssDNA affinity (Kd 17 nM for rgyr versus 33 nM for rgyr_Δlatch), and a 1.3-fold reduction of dsDNA affinity (74 nM versus 96 nM). These differences in Kd values correspond to a small energetic contribution of the latch to DNA binding by <2 kJ/mol. For rgyr_hel, the effects of the latch deletion on DNA binding were more pronounced. The ssDNA affinity was decreased 2.1-fold (Kd 0.20 µM for rgyr_hel versus 0.42 µM for rgyr_hel_Δlatch), and the dsDNA affinity was decreased 5.4-fold (3.9 µM versus 21 µM), corresponding to small but significant energetic contributions of the latch to DNA binding by 2–4 kJ/mol. To test directly for DNA binding to the latch, we produced the isolated latch domain, and determined its affinities for ssDNA and dsDNA (Figure 4E). The isolated latch binds both ssDNA and dsDNA with low affinities (Kd 64 ± 29 µM for ssDNA, 78 ± 39 µM for dsDNA), confirming that it contributes to DNA binding. Although differences in the helicase-like domain reflect contributions of the latch to DNA binding to the helicase-like domain only (and suggest that the latch is involved in DNA binding to this site), Kd values for the full-length enzyme also contain contributions from the (nucleotide-independent) DNA-binding sites in the topoisomerase domain, masking the influence of the latch and rendering a dissection of effects difficult.
Figure 4.

DNA binding. (A) Titration of 10 nM ssDNA with rgyr (filled squares) and rgyr_Δlatch (open squares). (B) Titration of 25 nM ssDNA with rgyr_hel (filled circles) and rgyr_hel_Δlatch (open circles). (C) Titration of 10 nM dsDNA with rgyr (filled squares) and rgyr_Δlatch (open squares). (D) Titration of 25 nM dsDNA with rgyr_hel (filled circles) and rgyr_hel_Δlatch (open circles). (E) Titration of 25 nM ssDNA (red) or dsDNA (blue) with the isolated latch domain. Binding curves for ssDNA are shown in red, binding curves for dsDNA are depicted in blue. For Kd values from the depicted data and Kd values in different nucleotide states, see Table 3. (F) Preference for ssDNA in rgyr and rgyr_Δlatch (upper panel) and rgyr_hel and rgyr_hel_Δlatch (lower panel) in different nucleotide states. (G) Thermodynamic cycle for ADPNP (N) and dsDNA (D) binding to rgyr_hel (rgh). The Kd values in µM are indicated. Numbers in the center refer to the coupling factor (n-fold decrease in Kd when the first ligand is already bound). The coupling factor for binding of ADPNP and dsDNA to rgyr_hel is ∼20. (H) Thermodynamic cycle for ADPNP (N) and dsDNA (D) binding to rgyr_hel_Δlatch (rghΔ). The coupling factor for ADPNP and dsDNA binding is ∼1, indicating that coupling is lost upon latch deletion.
Table 3.
DNA binding
| rgyr |
rgyr_Δlatch |
|||
| Kd (µM) | rel. | Kd (µM) | rel. | |
| ssDNA | ||||
| – | 0.017 + 0.004 | 1 | 0.033 ± 0.077 | 1 |
| ADP state | 0.013 ± 0.003 | 0.8 | 0.025 ± 0.005 | 0.8 |
| ATP state (ADPNP) | 0.015 ± 0.006 | 1.2 | 0.040 ± 0.010 | 1.2 |
| dsDNA | ||||
| – | 0.074 ± 0.016 | 4.3 | 0.096 ± 0.019 | 2.9 |
| ADP state | 0.11 ± 0.02 | 6.5 | 0.058 ± 0.010 | 1.8 |
| ATP state (ADPNP) | 0.055 ± 0.015 | 3.2 | 0.104 ± 0.015 | 3.2 |
| rgyr_hela |
rgyr_hel_Δlatch |
|||
| Kd (µM) | rel. | Kd (µM) | rel. | |
| ssDNA | ||||
| – | 0.20 ± 0.01 | 1 | 0.42 ± 0.07 | 1 |
| ADP state | 0.28 ± 0.01 | 1.4 | 2.6 ± 0.3 | 6.2 |
| ATP state (ADPNP) | 0.46 ± 0.03 | 2.3 | 1.1 ± 0.1 | 2.6 |
| dsDNA | ||||
| – | 3.9 ± 0.6 | 20 | 21 ± 5 | 50 |
| ADP state | 3.7 ± 0.5 | 19 | 24 ± 5 | 57 |
| ATP state (ADPNP) | 0.19 ± 0.03 | 1 | 36 ± 8 | 86 |
aData from ref. (11).
Deletion of the latch diminishes cooperativity in the helicase-like domain
We next characterized the effect of the latch on ATP hydrolysis, and stimulation of ATP hydrolysis by DNA in steady-state ATPase assays (Figure 5, Table 4). The kcat values for the intrinsic ATPase activities of rgyr and rgyr_Δlatch are small, with 30 × 10−3 s−1 and 18 × 10−3 s−1, respectively, indicating that the latch is not involved in ATP hydrolysis. The KM value for ATP is slightly reduced in rgyr_Δlatch (44 µM for rgyr, 26 µM for rgyr_Δlatch). In the context of the isolated helicase-like domain, the analogous deletion of the latch also does not affect the intrinsic ATPase activity much (kcat = 30 × 10−3 s−1, KM,ATP = 77 µM for rgyr_hel, and kcat = 20 × 10−3 s−1, KM,ATP = 67 µM for rgyr_hel_Δlatch). The small reduction in KM,ATP upon deletion of the latch is in-line with the interference of the latch with nucleotide binding (see above, Table 2). The effect is more pronounced for rgyr than for rgyr_hel. The similar kcat values demonstrate that the latch does not play a role in basal ATP hydrolysis within the helicase-like domain of reverse gyrase.
Figure 5.

ATPase activity. Steady-state ATPase activity as a function of the ATP concentration. (A) rgyr, (B) rgyr_Δlatch, (C) rgyr_hel, (D) rgyr_hel_Δlatch. Black: no DNA, red: ssDNA, blue: dsDNA. Data for rgyr are depicted with squares, data for rgyr_hel with circles. Data for wild-type proteins are depicted with filled symbols, data for latch deletions with open symbols. For kcat and KM values, see Tables 4 and 5.
Table 4.
Steady state ATPase parameters (ATP dependence)
| rgyra |
rgyr_Δlatch |
|||
| KM,ATP (µM) | kcat (10−3 s−1) | KM,ATP (µM) | kcat (10−3 s−1) | |
| – | 44 ± 6 | 20 ± 0.8 | 26.5 ± 3.5 | 18 ± 0.5 |
| ssDNA | 12 ± 1.5 | 108 ± 2.1 | 33.3 ± 5.5 | 101 ± 4 |
| dsDNA | 16 ± 1.5 | 117 ± 1.6 | 18.3 ± 4.1 | 131 ± 6 |
| pUC18 | 15.6 ± 0.9 | 199 ± 1.6 | 42.1 ± 2.3 | 164 ± 3 |
| rgyr_hela |
rgyr_hel_Δlatch |
|||
| KM,ATP (µM) | kcat (10−3 s−1) | KM,ATP (µM) | kcat (10−3 s−1) | |
| – | 77 ± 23 | 30 ± 2 | 67 ± 18 | 20 ± 2 |
| ssDNA | 4.8 ± 1 | 992 ± 12 | 27 ± 2 | 660 ± 10 |
| dsDNA | 11 ± 4 | 1435 ± 57 | 30 ± 4 | 850 ± 20 |
| pUC18 | 7.4 ± 1.1 | 818 ± 10 | n.d. | n.d. |
aData from ref. (11).
In the presence of ssDNA, dsDNA or pUC18 plasmid, the kcat values for ATP hydrolysis by reverse gyrase are increased 5–10-fold (Table 4, ref. 11). Similar to wild-type reverse gyrase, ATP hydrolysis by rgyr_Δlatch was stimulated 6–9-fold by DNA (Table 4), excluding a role of the latch for stimulation of the ATPase activity by DNA. At 75°C, similar effects of ssDNA and plasmid DNA on ATP hydrolysis by rgyr and rgyr_Δlatch were observed (Supplementary Figure S2).
Rgyr binds ATP and DNA cooperatively, reflected in a 2.8–3.7-fold decrease of the KM,ATP in the presence of DNA (Table 4, ref. 11). By contrast, decreases in KM,ATP values in the presence of ssDNA, dsDNA or pUC18 plasmid are only 0.7–1.3-fold for rgyr_Δlatch (Table 4), pointing toward a complete loss in cooperativity upon deletion of the latch region. A similar effect is observed with rgyr_hel, where the cooperativity of DNA and ATP binding leads to a large, 16- and 7-fold decrease of the KM,ATP in the presence of ssDNA and dsDNA, respectively. Deletion of the latch reduces the cooperativity, and the decrease is only 2.5- and 2.2-fold. Thus, the latch is crucial for coupling ATP and DNA binding within the helicase-like domain of reverse gyrase.
Measuring the steady-state ATPase activity as a function of the DNA concentration allows for the determination of apparent KM values for DNA. These values provide a relative measure for DNA affinity to the predominant states populated under steady-state conditions. For reverse gyrase, the ATP state should be the predominant form as ATP hydrolysis is the rate-limiting step in the nucleotide cycle of reverse gyrase (14) (in the absence of DNA). In contrast to Kd values above that describe overall DNA binding to all sites, the KM values only report on DNA-binding sites on reverse gyrase that are coupled to ATP hydrolysis. The KM,DNA values for rgyr are 0.45 µM (ssDNA), and 2.2 µM (dsDNA), respectively (Table 5, ref. 11). The corresponding KM,DNA values for rgyr_Δlatch are 0.27 µM (ssDNA), and 0.12 µM (dsDNA), corresponding to a slight increase in affinity for ssDNA (1.7-fold), and a significant increase for dsDNA (18-fold). The affinity for pUC18 plasmid was also increased by deleting the latch (Table 5). These data suggest an inhibitory effect of the latch on (ds)DNA binding to the ATP state of rgyr. The KM,DNA values for rgyr contain contributions from DNA binding to the helicase-like domain and to the topoisomerase domain, if these are coupled to ATP hydrolysis. We therefore dissected the contribution of the latch to DNA binding by performing the same experiments with rgyr_hel. Interestingly, the effects observed with rgyr are not paralleled by rgyr_hel (Table 5). Here, the KM,DNA values are increased 3.6-fold for ssDNA and 1.6-fold for dsDNA upon latch deletion, in-line with a contribution of the latch to DNA binding to the helicase-like domain in the ATP state. Overall, these findings are thus consistent with contributions of the latch to ssDNA and dsDNA binding to the helicase-like domain of reverse gyrase, in agreement with the findings from anisotropy titrations of DNA (see above). The decrease in KM,DNA of rgyr_Δlatch that is not observed for rgyr_hel_Δlatch suggests that the latch (negatively) coordinates dsDNA binding within the topoisomerase domain to ATP hydrolysis by the helicase-like domain.
Table 5.
Steady state ATPase parameters (DNA dependence)
| rgyra |
rgyr_Δlatch |
|||
| KM,DNA (µM) | kcat (10−3 s−1) | KM,DNA (µM) | kcat (10−3 s−1) | |
| – | n.a. | 20 ± 0.8 | n.a. | 18 ± 0.5 |
| ssDNA | 0.45 ± 0.06 | 160 ± 7 | 0.271 ± 0.024 | 108 ± 2 |
| dsDNA | 2.2 ± 0.30 | 148 ± 9 | 0.124 ± 0.039 | 98 ± 8 |
| pUC18 | No sat. | No sat. | 0.020 ± 0.013 | 104 ± 14 |
| rgyr_hela |
rgyr_hel_Δlatch |
|||
| KM,DNA (µM) | kcat (10−3 s−1) | KM,DNA (µM) | kcat (10−3 s−1) | |
| – | n.a. | 30 ± 2 | n.a. | 20 ± 2 |
| ssDNA | 0.07 ± 0.04 | 1160 ± 188 | 0.25 ± 0.03 | 780 ± 20 |
| dsDNA | 0.18 ± 0.03 | 1450 ± 64 | 0.29 ± 0.06 | 780 ± 40 |
| pUC18 | 0.046 ± 0.006 | 673 ± 21 | n.d. | n.d. |
aData from ref. (11).
The effect of the latch on coupling nucleotide and DNA binding
The loss of cooperativity between ATP and DNA binding upon deletion of the latch region suggests a role of the latch for coupling DNA binding to the nucleotide state. We next determined the effect of the latch on nucleotide-dependent DNA binding (Table 3). In the nucleotide-free state, the latch contributes to ssDNA and dsDNA binding to the helicase-like domain of rgyr (see above, 1.3–2-fold loss in affinity by deleting the latch in rgyr, 2.1–5.4-fold in rgyr_hel). In the ADP state, deletion of the latch leads to a modest (2-fold) decrease in affinity for ssDNA, and an equally modest 2-fold increase in affinity of rgyr for dsDNA. These values suggest a contribution of the latch to ssDNA binding, but interference with dsDNA binding, in the ADP state. In the ADPNP state, in contrast, deletion of the latch causes a 2.7-fold and 1.9-fold loss in ssDNA and dsDNA affinity, in-line with a positive contribution of the latch to ssDNA and dsDNA binding to reverse gyrase.
Similar to the KM,DNA values, Kd values for rgyr/DNA complexes contain contributions from (nucleotide-dependent) DNA binding to the helicase-like domain and (nucleotide-independent) DNA binding to the topoisomerase domain, as well as possible coupling effects. To dissect the contribution of the latch to DNA binding within the helicase-like domain, we therefore also performed titrations with rgyr_hel. Here, the deletion of the latch consistently leads to a decrease in both ssDNA and dsDNA affinity in all nucleotide states (Table 3). In the ADP state, this loss in affinity is 9-fold for ssDNA and 7-fold for dsDNA; in the ADPNP state it is 2.4-fold for ssDNA and 190-fold for dsDNA. These values demonstrate that the latch contributes to ssDNA and dsDNA binding to the helicase-like domain in all nucleotide states. The largest contribution of the latch to DNA binding is observed for dsDNA in the ADPNP state.
As a reciprocal approach to monitoring DNA binding in different nucleotide states, we also measured nucleotide binding to different DNA-bound forms of rgyr and rgyr_hel (Table 2). Consistent with the observation in the absence of DNA, the affinity of rgyr and rgyr_hel for mantADP was increased in all DNA-bound states upon deletion of the latch, with more pronounced effects for rgyr (3–7-fold) than for rgyr_hel (2–4-fold). For rgyr, the increase in nucleotide affinity was most pronounced in the DNA-free state, whereas for rgyr_hel it was most pronounced in the ssDNA-bound state. The increased affinity of the latch deletions for (mant)ADPNP was alleviated in the presence of DNA, with identical nucleotide affinities for rgyr and rgyr_Δlatch, and similar values for rgyr_hel and rgyr_hel_Δlatch. The only exception is mantADPNP binding to rgyr_hel_Δlatch in the presence of dsDNA. Here, the latch deletion decreases the nucleotide affinity 5.8-fold. In combination with the nucleotide-dependent DNA binding studies (Table 3), thermodynamic cycles of DNA and nucleotide binding can be constructed (Figure 4). Coupling of overall DNA binding and nucleotide binding is small in reverse gyrase, and only small effects of the latch are observed. Coupling is larger for rgyr_hel, however, and here the effect of the latch becomes apparent. In rgyr_hel, ADPNP and dsDNA binding are thermodynamically coupled, with a coupling factor of 20 (Figure 4G). Upon deletion of the latch, this coupling is lost completely (Figure 4H). Some coupling is also present for ADPNP and ssDNA binding to rgyr_hel, which is also lost upon deletion of the latch. Overall, the data are thus consistent with a major effect of the latch on coupling (ds)DNA binding and ADPNP binding to the helicase-like domain.
The latch contributes to the discrimination between ssDNA and dsDNA
Reverse gyrase exhibits a clear (4–8-fold) preference for ssDNA over dsDNA in the nucleotide-free, the ADP and the ATP-state (Figure 4F, Table 3, ref. 11). Deletion of the latch reduces the preference for ssDNA significantly, and leads to a similar (2–3-fold) small preference for ssDNA in the nucleotide-free, the ADP- and the ADPNP state. In rgyr_hel, the latch deletion leads to a further increase of the preference for ssDNA in the nucleotide-free and the ADPNP state, but slightly reduces the preference in the ADP state (Figure 4F, Table 3). The preference of rgyr_hel_Δlatch for ssDNA in the ADPNP state is in stark contrast to similar affinities of rgyr_hel for ssDNA and dsDNA in the ADPNP state (11), strongly suggesting a role of the latch in this switch in DNA affinities upon ATP binding (switch from nucleotide-free to ADPNP state), and upon ATP hydrolysis/product release (switch from ADPNP to ADP state). This switch is suppressed in context of rgyr (11), and the effect of the latch can thus only be studied with the isolated helicase-like domain. Overall, our results point toward a role of the latch in coupling nucleotide and DNA-binding sites within the helicase-like domain, and in coordinating DNA binding within the topoisomerase domain with ATP hydrolysis in the helicase-like domain.
The latch is required for positive supercoiling of DNA by T. maritima reverse gyrase
Finally, we investigated the effect of the latch on the topoisomerase activities of reverse gyrase (Figure 6, Supplementary Figure S3). Similar to the wild-type enzyme, rgyr_Δlatch does not show significant relaxation activity in the absence of nucleotides, even at a large excess of enzyme (Supplementary Figure S3). In the presence of ATP and ATPγS, rgyr_Δlatch relaxes negatively supercoiled DNA, but is not capable of introducing positive supercoils as the wild-type enzyme (14). The relaxation activity of rgyr_Δlatch is also observed in the presence of ADP and ADPNP, indicating that it does not depend on ATP hydrolysis. Consequently, the latch region is required for the positive supercoiling reaction. Interestingly, this observation sets T. maritima reverse gyrase apart from the A. fulgidus homolog, which does not seem to require the latch for the positive supercoiling reaction (13).
Figure 6.

Topoisomerase activity. (A) Topoisomerase activity of rgyr and rgyr_Δlatch. (B) Topoisomerase activity of rgyr with ATP, ATPγS and ADPNP. Rgyr introduces positive supercoils in the presence of ATP and ATPγS, but only relaxes DNA in the presence of ADPNP. (C) Topoisomerase activity of rgyr_Δlatch with ATP, ATPγS and ADPNP. Rgyr_Δlatch relaxes DNA in the presence of ATP and ATPγS, and exhibits weak relaxation activity in the presence of ADPNP. In (A), reaction products from incubation of rgyr or rgyr_Δlatch with negatively supercoiled DNA were separated via one-dimensional gel electrophoresis. When two lanes are depicted for the same experimental conditions, the left lane corresponds to 1 h incubation time, the right lane to 2 h. In (B) and (C), reaction products were separated via two-dimensional gel electrophoresis (‘Material and Methods’ section). The species in the left arm of the arch represent negatively supercoiled DNA, the right arm of the arch represent positively supercoiled DNA. For analysis via one-dimensional gel electrophoresis in the presence of chloroquine, see Supplementary Figure S3.
DISCUSSION
The latch modulates the properties of the helicase-like domain in reverse gyrase
Studies on reverse gyrase from A. fulgidus have suggested that the latch region is involved in communication between the helicase-like and the topoisomerase domain and coordination of their activities, even though it was not required for positive supercoiling (13). Here, we have dissected the contributions of the latch to nucleotide binding and hydrolysis, DNA binding, and cooperativity in the isolated helicase-like domain of T. maritima reverse gyrase, as well as in the full-length enzyme. The latch affects nucleotide binding to the helicase-like domain, but does not alter the intrinsic or the DNA-stimulated ATPase activities. Coupling between DNA binding and ATP binding and hydrolysis in the helicase-like domain is lost when the latch is deleted, demonstrating that the latch is crucial for cooperativity within the helicase-like domain of reverse gyrase.
The helicase-like domain of reverse gyrase resembles DEAD box proteins. Members of this protein family within the SF2 helicases mediate ATP-dependent structural rearrangements of RNA, and typically exhibit cooperativity between nucleotide and DNA/RNA binding (22). This cooperativity has been rationalized from DEAD box protein structures that reveal a bipartite nucleic acid binding site that covers the surface of both RecA domains (corresponding to H1 and H2 in reverse gyrase) and a nucleotide binding site that is formed in the inter-domain cleft by residues from both domains (23). The similar spatial distribution of conserved motifs (6), mutational studies on the contributions of these motifs to supercoiling (24), as well as the existing cooperativity between DNA and ATP binding in the helicase-like domain of reverse gyrase altogether suggest similar concerted conformational changes upon ATP (and DNA) binding (11) in the helicase-like domain in reverse gyrase. The observation that the latch is involved in cooperativity of DNA and nucleotide binding suggests that it contributes to the DNA binding site within H2. On the other hand, the latch interferes with nucleotide binding. In the structure of A. fulgidus reverse gyrase with ADPNP bound to the helicase-like domain, the nucleotide only interacts with residues in H1, but not H2 (6), again supporting the notion that, similar to SF2 helicases, a closure of the cleft between H1 and H2 in response to nucleotide (and DNA) binding may lead to an increased nucleotide affinity. The interference of the latch with nucleotide binding to the helicase-like domain in the absence and presence of DNA suggests that the latch modulates the underlying conformational change, possibly by reducing the flexibility of the helicase-like domain. Consistent with the effect on the isolated helicase-like domain, inhibition of nucleotide binding by the latch is also observed in reverse gyrase. Here, binding of DNA and nucleotide is less cooperative, which may reflect an inhibition of the underlying conformational change in the helicase-like domain by the topoisomerase domain (11). In line with this scenario, the effect of the latch on nucleotide binding in reverse gyrase is less severe. In addition, it is alleviated in the presence of ssDNA and dsDNA. It is conceivable that nucleotide and DNA binding alleviate conformational restrictions caused by interactions between the helicase-like and the topoisomerase-like domains, consistent with a possible release of the latch from the topoisomerase domain in the presence of DNA (6), and a concomitant loss of conformational restrictions in the helicase-like domain.
The latch affects the discrimination between ssDNA and dsDNA
The DNA-binding site on the helicase-like domain exhibits a preference for ssDNA in the nucleotide-free and the ADP state, but switches to similar affinities for ssDNA and dsDNA in the ATP state. By contrast, in the absence of the latch the preference for ssDNA is high in the nucleotide-free and ATP state, but reduced for the ADP state, implicating the latch in the switching properties of the helicase-like domain. In line with the effect of the latch on cooperativity, it indeed contributes to DNA binding to the helicase-like domain in reverse gyrase, and it is capable of weak DNA binding on its own. The latch is structurally homologous to a region of the transcription termination factor Rho that is involved in RNA binding (7,8), but for A. fulgidus reverse gyrase no effect of the latch on DNA binding was observed (13). This may be due to the presence of various different DNA-binding sites within reverse gyrase that contribute to the overall DNA affinity, hiding contributions from the latch. These effects thus only become detectable in DNA-binding studies with the isolated helicase-like domain as we have performed here, and may have been missed in studies of the A. fulgidus enzyme. Even though the latch provides minor contributions to DNA binding, it is not required for high-affinity DNA binding to reverse gyrase. Its contribution may be relevant for transient contacts of the latch with DNA that have been proposed (6), consistent with a guiding effect of the latch for DNA during supercoiling and strand passage. Most importantly, the latch affects the discrimination of reverse gyrase between ssDNA and dsDNA. The strong preference of reverse gyrase for ssDNA in all nucleotide states (11) is thought to be important for sensing single-stranded regions in DNA at high temperatures (4). Upon deletion of the latch in reverse gyrase, this preference is reduced, and is now similar for the nucleotide-free, ATP and ADP states, consistent with the latch being involved in communicating the nucleotide state of the helicase-like domain to the DNA-binding sites on the topoisomerase domain.
The latch is required for positive supercoiling
It has been suggested previously that a conformational change bringing H1 and H2 close together may lead to a movement of the latch and to a release of the lid of the topoisomerase domain during positive supercoiling (6). In light of this scenario (that has not been proven experimentally as yet), a crucial role of the latch in positive supercoiling is expected. Interestingly, a deletion of the latch in A. fulgidus reverse gyrase did not abolish positive supercoiling, though the activity was significantly reduced (13). By contrast, we have shown here that the latch is absolutely required for positive supercoiling by T. maritima reverse gyrase. The latch deletion construct we used here does not contain the two-stranded β-sheet connecting the latch to the H2 domain. This region is absent in SF2 helicases, but was retained in the deletion construct of A. fulgidus reverse gyrase (13). On the other hand, the latch region shows less sequence conservation among reverse gyrases than the H1 and H2 domains, and may have different roles in different enzymes. It is thus unclear whether the different effects of the latch deletions in A. fulgidus and T. maritima reverse gyrase are due to different deletions studied, or reflect genuine differences between these enzymes.
Despite its requirement for positive DNA supercoiling by T. maritima reverse gyrase, the latch is not required for nucleotide-mediated relaxation of DNA in the presence of ATP, ATPγS and ADPNP, and is thus not necessary per se for strand passage in general, or for strand passage toward a higher linking number. The latch has been implicated in repressing relaxation of DNA by the topoisomerase domain (13) in the absence of nucleotides. However, deletion of the latch in T. maritima reverse gyrase does not lead to DNA relaxation in the absence of nucleotides, arguing against a general role of the latch in repressing this activity. Deletion of the latch leads to relaxation in the presence of all nucleotides tested, suggesting an important role of the latch in coupling ATP hydrolysis to strand passage towards positive DNA supercoiling. To dissect how DNA is guided toward positive DNA supercoiling, future studies will have to address DNA binding to individual sites on reverse gyrase, and their contributions to DNA binding at different stages of the supercoiling.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
VolkswagenStiftung (to D.K.); Swiss National Science Foundation (to D.K.); Marie Curie postdoctoral fellowship (to A.G.). Funding for open access charge: University of Basel.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank the staff at SLS beamlines PX-II and PX-III for support during data collection.
REFERENCES
- 1.Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 2.Kikuchi A, Asai K. Reverse gyrase–a topoisomerase which introduces positive superhelical turns into DNA. Nature. 1984;309:677–681. doi: 10.1038/309677a0. [DOI] [PubMed] [Google Scholar]
- 3.Kampmann M, Stock D. Reverse gyrase has heat-protective DNA chaperone activity independent of supercoiling. Nucleic Acids Res. 2004;32:3537–3545. doi: 10.1093/nar/gkh683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh TS, Plank JL. Reverse gyrase functions as a DNA renaturase: annealing of complementary single-stranded circles and positive supercoiling of a bubble substrate. J. Biol. Chem. 2006;281:5640–5647. doi: 10.1074/jbc.M513252200. [DOI] [PubMed] [Google Scholar]
- 5.Confalonieri F, Elie C, Nadal M, de La Tour C, Forterre P, Duguet M. Reverse gyrase: a helicase-like domain and a type I topoisomerase in the same polypeptide. Proc. Natl Acad. Sci. USA. 1993;90:4753–4757. doi: 10.1073/pnas.90.10.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez AC, Stock D. Crystal structure of reverse gyrase: insights into the positive supercoiling of DNA. EMBO J. 2002;21:418–426. doi: 10.1093/emboj/21.3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolan JW, Marshall NF, Richardson JP. Transcription termination factor rho has three distinct structural domains. J. Biol. Chem. 1990;265:5747–5754. [PubMed] [Google Scholar]
- 8.Dombroski AJ, Platt T. Structure of rho factor: an RNA-binding domain and a separate region with strong similarity to proven ATP-binding domains. Proc. Natl Acad. Sci. USA. 1988;85:2538–2542. doi: 10.1073/pnas.85.8.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lima CD, Wang JC, Mondragon A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature. 1994;367:138–146. doi: 10.1038/367138a0. [DOI] [PubMed] [Google Scholar]
- 10.Declais AC, Marsault J, Confalonieri F, de La Tour CB, Duguet M. Reverse gyrase, the two domains intimately cooperate to promote positive supercoiling. J. Biol. Chem. 2000;275:19498–19504. doi: 10.1074/jbc.M910091199. [DOI] [PubMed] [Google Scholar]
- 11.Del Toro Duany Y, Jungblut SP, Schmidt AS, Klostermeier D. The reverse gyrase helicase-like domain is a nucleotide-dependent switch that is attenuated by the topoisomerase domain. Nucleic Acids Res. 2008;36:5882–5895. doi: 10.1093/nar/gkn587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez AC. Investigating the role of the latch in the positive supercoiling mechanism of reverse gyrase. Biochemistry. 2003;42:5993–6004. doi: 10.1021/bi034188l. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez AC. Studies of a positive supercoiling machine. Nucleotide hydrolysis and a multifunctional “latch” in the mechanism of reverse gyrase. J. Biol. Chem. 2002;277:29865–29873. doi: 10.1074/jbc.M202853200. [DOI] [PubMed] [Google Scholar]
- 14.Jungblut SP, Klostermeier D. Adenosine 5′-O-(3-thio)triphosphate (ATPgammaS) promotes positive supercoiling of DNA by T. maritima. reverse gyrase. J. Mol. Biol. 2007;371:197–209. doi: 10.1016/j.jmb.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 15.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallog. 1993;26:795–800. [Google Scholar]
- 16.CCP4. The Collaborative Computational Project Number 4, suite programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 17.Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Cryst. 2004;D60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 18.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 19.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hiratsuka T. New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as substrates for various enzymes. Biochim. Biophys. Acta. 1983;742:496–508. doi: 10.1016/0167-4838(83)90267-4. [DOI] [PubMed] [Google Scholar]
- 21.Adam H. Methoden der enzymatischen Analyse. 1962. Bergmeyer, H.U.(Hrsg.), Verlag Chemie, Weinheim, pp. 573–577. [Google Scholar]
- 22.Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol. Chem. 2009;390:1237–1250. doi: 10.1515/BC.2009.135. [DOI] [PubMed] [Google Scholar]
- 23.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 24.Bouthier de la Tour C, Amrani L, Cossard R, Neuman K, Serre MC, Duguet M. Mutational analysis of the helicase-like domain of Thermotoga maritima reverse gyrase. J. Biol. Chem. 2008;283:27395–27402. doi: 10.1074/jbc.M800867200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.