


Abstract
Histone modifications are fundamental to chromatin structure and transcriptional regulation, and are recognized by a limited number of protein folds. Among these folds are PHD fingers, which are present in most chromatin modification complexes. To date, about 15 PHD finger domains have been structurally characterized, whereas hundreds of different sequences have been identified. Consequently, an important open problem is to predict structural features of a PHD finger knowing only its sequence. Here, we classify PHD fingers into different groups based on the analysis of residue–residue co-evolution in their sequences. We measure the degree to which fixing the amino acid type at one position modifies the frequencies of amino acids at other positions. We then detect those position/amino acid combinations, or ‘conditions’, which have the strongest impact on other sequence positions. Clustering these strong conditions yields four families, providing informative labels for PHD finger sequences. Existing experimental results, as well as docking calculations performed here, reveal that these families indeed show discrepancies at the functional level. Our method should facilitate the functional characterization of new PHD fingers, as well as other protein families, solely based on sequence information.
INTRODUCTION
Databases of protein sequences have grown rapidly in recent years. These data, whether restricted to a single family of genes or proteins, a single organism or to a functional group of proteins, enable the discovery of common structural or functional features within protein groups. One such application is the detection of protein ‘functional’ residues (1–3).
The in silico detection of functional residues often uses multiple sequence alignments. Among the detection algorithms for ranking positions within an alignment, several are based on scores derived from information-theoretic tools, including mutual information and relative entropy. In many cases, such scores are used to determine which residues in a given family share evolutionary or sequence variation patterns, yielding groups of residues which are likely to be involved in similar structures or communication pathways (4,5). Another assumption of residue–residue correlated evolution is that, indeed, residues that co-evolve are ‘in contact’, i.e. in a non-bonding chemical interaction (6), although a strict correlation between residue–residue contact and co-evolution remains to be established.
Some in silico methods are also specifically dedicated to the detection of features which are specific to sub-families (7,8). Indeed, the detection of sites that show diversity at the scale of the whole protein family but are conserved within sub-groups, enables one to go beyond the original view that conservation means function, i.e. that residues which evolve more slowly than others are more likely to be functional (9). For example, the Evolutionary Trace Method was designed for this purpose and can highlight conserved binding surfaces in proteins (10). The detection of such sites could also be achieved by combining conservation and physical–chemical properties of residues (11). Another motivation for these methods is rooted in the conjecture that residues that possess a large number of co-evolutionary relationships, or that are strongly evolutionarily correlated with many other residues, are likely to play a critical role in protein function, or are involved in active sites (1).
Here we concentrate on certain zinc-binding protein domains, the PHD fingers (12), which are short domains for which various functions and binding partners have been identified thus far, and to which numerous studies have been dedicated over the last 5 years. These domains are involved in histone recognition (13), and the initial consensus proposal was that these domains specifically recognized histones bearing a methylated lysine residue on their N-terminal end (14,15). More recent results suggest a broader diversity of substrates for these domains (16,17). We therefore analyze a large set of sequences in order to highlight what similarities and differences exist between PHD fingers at the sequence level. Specifically, we demonstrate how a computational analysis of multiple sequences for PHD fingers can detect sub-families, which putatively share similar functions or selectivities towards different substrates.
We classify PHD fingers relying on the measure of correlated evolution between their residues. An information-based method is introduced in order to detect which residues in the alignment are critical for discriminating among PHD fingers based solely on their sequences. The aim of the study is to pinpoint the alignment positions or the subset of residue types at these positions which are the most critical for distinguishing among PHD finger sequences. The measure used for comparing protein residues is relative entropy, or Kullback–Leibler (KL) divergence. Relative entropy is a measure of the similarity between two probability distributions, which has proven effective in various protein sequence analyses, both theoretical (7,18) and directly applied (19). The proposed labeling of PHD finger sequences is derived by using relative entropy to compare the amino acid distributions at a given position with and without fixing the amino acid at another position. More specifically, we compare the frequency of amino acids at one position within the full population with the frequency at the same position for the sub-population determined by fixing the amino acid at another position, which we refer to as a ‘condition.’ Each condition provides a ‘tag’ for labeling a PHD finger sequence, each sequence possibly having multiple tags. We then detect those conditions which have the strongest impact on other positions and these conditions are clustered into disjoint families.
The results are validated in three ways. First, comparison with existing experimental results reveals that some of these groups of tags are clearly critical in defining substrate selectivity. Second, a structural study is performed on two PHD fingers. Prediction of a structure for these two domains, along with the docking of Histone 3 peptides, further demonstrates the coherence of our grouping with respect to preferred substrates for each of these families and the corresponding utility of our prediction. Finally, a validation of the method is performed on two additional protein families, as well as a comparison to other existing methods.
MATERIALS AND METHODS
Protein sequence alignment
An alignment of PHD fingers was downloaded from the PFAM database (20) (www.pfam.org) as of January 2010. The alignment was first filtered using CD-HIT (21) for 90% maximum identity, then manually edited so as to align all zinc-binding residues, and finally re-filtered at 70% maximum identity [a value in the range suggested by Buslje et al. (22)]. This produced an alignment of 926 PHD finger sequences (Supplementary Data S1).
The alignment positions were numbered according to the most extended sequences present, with the final position at a count of 133. Conserved zinc-binding residues corresponded to positions 14 (Cys1), 20 (Cys2), 49 (Cys3), 62 (Cys4), 69 (His), 73 (Cys5), 114 (Cys6) and 119 (Cys7). A positioning of residues in the alignment relative to one of these eight zinc-ligand positions will often be used in the text. Such relative numbering often does not correspond to the actual alignment numbering, where all sequences present in the alignment are considered, but rather only to a majority of the sequences.
Alignment positions and conditions
For sequence analysis to be performed in a statistically robust manner, enough residues need be present at each analyzed position. We thus applied a threshold to the number of gaps: all positions at which <30% of sequences had gaps, and which were not zinc ligands (as strictly conserved), were considered for analysis. This produced 39 candidate observation positions. Our analysis of residue pairwise evolution is based on a condition, meaning a fixed amino acid type for a given position. Conditions were used to define alignment subsets and are summarized as ‘position: residue type’, e.g. 67:G. In order to reduce sample-size effects, we only considered conditions which were fulfilled in at least 50 protein sequences.
Calculation of conservation
Each observation position was described by a vector of length 21 representing the frequency of all amino acid types, including gaps, at that position. We denote the frequency in the whole set of sequences of amino acid (or gap) a at position i by pi(a). Conservation of alignment positions was measured by comparing distribution pi(a) with the frequency ui(a) of any amino acid a as observed in the Uniprot database (number of times the amino acid is present in the database over all amino acids from the database). The comparison was performed using relative entropy (also known as the KL divergence), a standard, information-theoretic measure for comparing two probability distributions, which has been used in multiple sequence analysis studies (3,19,23). The conservation value at alignment position i is then:
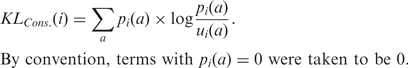 |
Residue–residue correlated evolution
The correlation in the evolution of two positions in the alignment was also calculated using relative entropy (see above). For each observation position i, the strength of its coupling to condition ‘j:b’ (position j is amino acid b) was measured by comparing the conditional distribution obtained for position i when only considering sequences that meet condition ‘j:b’, 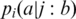 , with the unconditional distribution. [Conditional amino acid distributions have been proposed earlier by the group of Ranganathan in order to analyze sequence alignments (4)]. The comparison between the conditional and the unconditional distributions was done by calculating the relative entropy between these distributions over all amino acid types (including gaps):
, with the unconditional distribution. [Conditional amino acid distributions have been proposed earlier by the group of Ranganathan in order to analyze sequence alignments (4)]. The comparison between the conditional and the unconditional distributions was done by calculating the relative entropy between these distributions over all amino acid types (including gaps):
| (1) |
It should be emphasized that Equation (1) does not measure the disparity between the amino acid distributions at two different positions, but rather the coupling of a conditional and an unconditional amino acid distribution at a fixed position. The larger the value of KLi ( j:b), the larger is the influence of fixing the amino acid at position j to be b on the frequencies at position i. Relative entropy was preferred to other coupling measures, such as the weighted L1-distance: 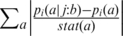 , where stat(a) represents the frequency of residue a in a reference database, e.g. the Uniprot database. It was also preferred to mutual information:
, where stat(a) represents the frequency of residue a in a reference database, e.g. the Uniprot database. It was also preferred to mutual information:
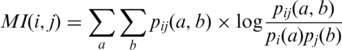 |
(2) |
Here, pij (a;b) is the relative frequency of simultaneously observing amino acid a at position i and amino acid b at position j, which provides only one value for each pair i,j of positions. Mutual information averages out some of the effects we wish to capture. This can be seen in the following formula: 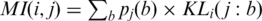 . Moreover, there is no apparent way to use mutual information to compare a distribution with a conditional distribution, as can be done with KL, because mutual information is defined in terms of the joint probability of two random variables. Still, for the sake of comparison, the results obtained with mutual information for pairs of positions are described in the last part of the ‘Results’ section.
. Moreover, there is no apparent way to use mutual information to compare a distribution with a conditional distribution, as can be done with KL, because mutual information is defined in terms of the joint probability of two random variables. Still, for the sake of comparison, the results obtained with mutual information for pairs of positions are described in the last part of the ‘Results’ section.
Second protein data set: inclusion of N-terminal domains
A second, smaller protein alignment was considered, where sequences extended further N-terminal to Cys1 than in the initial PFAM alignment. This alignment was created by editing over 200 sequences for PHD fingers as present in the UniProt database (www.Uniprot.org), and filtering the resulting sequences with extended N-terminal sequences to a maximum conservation rate of 70%, as above. The CD-Hit filtered alignment included 122 sequences, and was used for relative entropy analysis, with consideration of all positions with <30% gaps and of conditions present in at least 20 sequences (this smaller value being chosen due to a smaller size of the alignment).
Most significant conditions
In order to determine which of the analyzed conditions had the strongest overall influence on the alignment, the conditions with the most significant coupling values were first identified and then clustered so as to group together conditions that had similar effects at similar positions. The relative entropies for all couplings of an observation position i and a condition j:b were ordered and only those conditions which presented at least one coupling among the highest 1% of the relative entropy values were retained. A permutation test was run in order to assign a statistical significance to each of the retained conditions [see e.g. refs (24,25)]. The test was performed by randomly permuting the amino acids at position i among the sequences (or shuffling), thereby keeping the overall distribution at this position constant, and without modifying the corresponding amino acids at position j, where the condition was applied. The aim is to render the distributions of amino acids at positions i and j statistically independent, or to artificially ‘uncouple’ them, by canceling the evolutionary relationship between residues belonging to a same sequence [see ref. (26) for limitations on the uncoupling thus obtained]. Therefore measuring relative entropy KLi (j:b) on this shuffled distribution provides a value for the situation when there is a vanishing effect of condition j:b on position i. The test was repeated 1000 times for each of the possible condition and position pairs. Each of the retained couplings had a P-value smaller than 10−3 with respect to random. Whereas all coupling values in the first 5% of relative entropy values had a P-value smaller than 2.5 × 10−3, we observed that using the larger set of conditions produced redundant information with respect to protein sequences. Consequently, in order to consider only the conditions with high information content, the smaller set was used. Moreover, it was verified that the calculated relative entropy values were not affected by the size of the alignment used (Supplementary Figure S2). Finally, a comparison of the frequencies of significant conditions with respect to the most frequent amino acid types at the corresponding alignment positions also showed that our selection of conditions was not biased towards the highest amino acid frequencies (Supplementary Figure S3).
The retained conditions, i.e. the ‘first-percentile conditions’, were then compared with each other on the basis of the disparity of the amino acid distributions each of them produced at all observation positions. The disparity distance between conditions j:b and k:c was calculated according to the following formula:
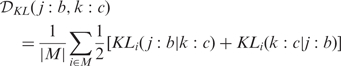 |
(3) |
which resulted in a ‘distance matrix’ between all first-percentile conditions. Here M corresponds to the first decile of the values of symmetrized relative entropy over all observation positions (thus |M| = 4), which produced a stabilized version of the maximum value, and proved more discriminating than averaging over all positions. The first decile was chosen after considering the distribution of the symmetrized relative entropy values.
The L1-distance was also used, for comparison, as a measure for the disparity between the conditions:
| (4) |
with M being defined as above. Disparity values for the first-percentile conditions as obtained using Equation (3) ranged from 0.1 (conditions 42:N with 70:Q) to 2.18 (condition 68:Q with 77:P), with an average of 1.12. Finally, these conditions were clustered by directly applying ‘affinity propagation’ (27) to the distance matrix, with the affinity clustering parameter p set identical for all conditions. The most central conditions for each cluster (the centroid), along with all members of each cluster, are provided. Similar results were obtained with spectral clustering. Affinity propagation was preferred as providing more reproducible results. The clustering which was obtained using Equation (4) instead of Equation (3) for the calculation of disparities is provided as Supplementary Data (Supplementary Table S4).
The resulting families of conditions were used to assign a membership to protein sequences: each protein was assigned to the family for which the number of conditions from that family it fulfilled was highest. It should be noted that, in most cases, conditions from a single family were strongly predominant, thus making the assignment straightforward. Half of the sequences satisfied conditions from a single family, and the respective numbers of sequences included in each family were 113 for family I, 373 for family II, 105 for family III and 159 for family IV.
Structure prediction
The 3D structures for the PHD fingers of the Transcription intermediary protein 1 α (TIF1A) and of bromodomain and PHD-finger protein 1 (BRPF1) were predicted using version 9v5 of Modeller (28). For TIF1A, the structures of the PHD fingers of BHC80, AIRE (first PHD finger) and CHD4 (second PHD finger) were used as templates (PDB entries: 2PUY, 2KFT, 1MM2). For BRPF1, the templates were protein PHD-finger 22 from mouse, the PHD finger from metal-response element-binding transcription factor 2 and PHD3 of JARID1A, with respective PDB entries 1WEV, 2YT5 and 3GL6. Docking to the predicted structures was performed using FlexX (BioSolveIT GmbH, Sankt Augustin, Germany), with no ‘access scaling’ for calculations, and the single best scoring solution conserved. Images were prepared using PyMol (29).
Additional protein data sets
Two further protein alignments were submitted to our condition-based clustering analysis. Their sequence alignments were obtained from the PFAM database, with respective entry PF00385 and PF01553, and filtered by CD-Hit at a maximum sequence conservation rate of 70%. The Chromatin Organization Modifier domain (chromodomain) data set contained 801 sequences after filtering. It produced 45 observation positions and had 24 distinct conditions involved in the first percentile of relative entropy values, with a threshold of 40 on the number of times a condition was met (50 was used for the PHD finger alignment, which included 926 sequences).
The acyltransferase data set contained 2059 sequences after filtering. It produced 119 observation positions and had 98 distinct conditions in the first percentile of relative entropy values. A threshold of 100 was used for the minimal number of times a condition was fulfilled.
RESULTS
The purpose of this study was to analyze a multiple sequence alignment of PHD fingers in order to classify them into different groups sharing functional properties. Nearly 1000 PHD fingers were analyzed computationally. Calculations were solely based on alignment properties, and did not involve any substitution or similarity matrix or any prior knowledge about family memberships.
Residue conservation
The first analysis that was performed over the sequence alignment was the calculation of position conservation. The measure used was the relative entropy between the observed residue distribution at one position and the average residue distribution observed in the Uniprot database. This relative measure was preferred to a plain entropy calculation of the amino acid distribution because over-representation of ‘rare’ amino acids (Trp, Cys, His) at a given position is often more meaningful than over-representation of more common ones (Leu, Ala). Moreover, a recent methodological study by Wang and Samudrala showed that incorporation of background frequencies improved entropy-based conservation analysis (23).
The position that showed the strongest discrepancy with respect to the reference distribution was, unsurprisingly, position 111, which is conserved as a Trp in most PHD fingers (Figure 1 and Supplementary Figure S5). The presence of an aromatic residue at this position, two residues N-terminal to Cys6, is indeed part of the sequence signature of PHD fingers, with 67.3% of proteins having a Trp and 31.2% having Tyr or Phe at that position in our alignment. The other positions that diverged significantly from the reference distribution were position 52, which is located one residue C-terminal to Cys3 in most PHD fingers, positions 67 and 68, which are respectively two and one residue N-terminal to the zinc-binding His, positions 46 and 47, which are directly N-terminal to Cys3, and position 19, which is located N-terminal to Cys2. Most of these residues are fundamental for the PHD-finger fold; for example, positions 46, 47 and 111 form a hydrophobic core in most known PHD-finger structures, whereas position 67 (His-2), when a Trp, is involved in hydrophobic interactions with the lysine side-chain in LysMe3-binding PHD fingers (14,30,31).
Figure 1.

Alignment of PHD fingers from the N-terminal extended alignment. Five positions N-terminal to Cys1 are shown. Zinc ligand positions are shown as gray boxes. Sequences are grouped according to their family assignment by our method (Table 1), with first-percentile conditions satisfied indicated in bold, and conditions originating from a family different from the one the sequence was assigned to in bold and italics. Sequences named without a suffix are from human. Those terminated with AT, CA, SP and Y are respectively from Arabidopsis thaliana, Candida albicans, Schizosaccharomyces pombe and Saccharomyces cerevisiae. Many of these sequences were not present in the initial (non-extended) CD-Hit-filtered alignment, and were thus not used for the determination of the families of conditions, but were added here as being better described in the literature.
Whereas this residue conservation calculation delineates common features of PHD fingers, it does not enable one to distinguish among different classes of PHD fingers on grounds of structural or substrate preferences. We therefore turned to a more refined analysis of PHD finger sequences by considering pairs of alignment positions rather than single positions.
Measuring residues co-evolution: relative entropy calculations
We next performed a pairwise analysis of the sequence alignment, in which all alignment positions with <30% gaps were considered. As described in ‘Materials and Methods’ section, amino acid distributions at all of these positions were calculated in the full alignment; these are the ‘unconditional’ distributions. We then calculated amino acid distributions at these positions in the subset of alignments defined by fixing the residue type at each of the other positions (a condition). For each position, this provides a ‘conditional’ distribution for any given condition; see ‘Materials and Methods’ section for details. We then compared the conditional and unconditional distributions using relative entropy [Equation (1)], thus obtaining a value for each coupling of a position to a condition. It was checked, by performing relative entropy calculations on random subsets of the alignment of variable sizes, that there was no bias due to sample size in the relative entropy calculations (Supplementary Figure S2). All conditions which had at least one coupling among the first percent of coupling values were retained for further analysis.
The first-percentile conditions (see Supplementary Table S6 for a complete list) were then clustered: conditions with a similar impact on alignment positions according to a symmetrized relative entropy measure [Equation (3)] were placed in the same cluster, yielding four clusters. The resulting clusters of conditions are listed in Table 1. Within families, conditions were ranked by increasing average distance to the most central condition (the ‘centroid’) of the cluster, with low ranking signaling high similarity to the centroid in terms of the modification of residue frequencies. Comparing the conditions using the L1 measure [Equation (4)] produced identical families of conditions, except that the two most distant conditions from family II, 63:P and 48:C, segregated into a separate cluster (Supplementary Table S4). Before illustrating the relevance of the clustering with respect to the preference of different PHD fingers for differently-modified histone substrates, we now discuss the direct significance of this clustering at the sequence level. One should bear in mind that it is conditions, not sequences, which are clustered into disjoint families, before assigning family memberships to each protein sequence.
Table 1.
Families of conditions obtained on PHD fingers
| I | II | III | IV |
|---|---|---|---|
| 66:E | 77:P | 70:Q | 111:Y |
| 37:S | 79:L | 42:N | 70:G |
| 48:A | 41:P | 46:I | 43:F |
| 36:V | 67:G | 74:H | 68:Q |
| 48:G | 79:I | 68:V | 48:E |
| 59:D | 72:Y | 74:Y | 66:V |
| 70:T | |||
| 63:P | |||
| 48:C |
Families of conditions obtained by clustering the PHD finger alignment. The first condition indicated (first line below the family number) is the family centroid. Then, from top to bottom, all the conditions from the family are listed by decreasing similarity to the centroid (‘Materials and Methods’ section). Families are ranked by increasing core size (‘Materials and Methods’ section). Underlined positions are those that were predicted as ‘specificity determining’ by method SDR. Note that SDR only considers positions (e.g. 66) and not conditions (e.g. 66:E).
Clustering results: conditions
Four families of conditions were produced after clustering by affinity propagation, with six to nine conditions per family (Table 1). The four centroid conditions each corresponded to different alignment positions, with one position belonging to the short Cys4-His segment (position 66, or His-3), one being between His and Cys5 (position 70) and two belonging to the Cys5-Cys6 segment (77 and 111, or Cys6-3). It should be noted that positions that produced multiple significant conditions, such as positions 48 (Cys3-1), 67 (His-2), 70 (His+1) and 79 (Cys5+5) could either belong to a similar cluster of conditions, or to different ones. Thus, the two conditions that involve position 79 belong to cluster II (Table 1), whereas conditions that derived from position 48 were present in all the clusters, with two conditions in cluster I (48:G and A) and one in each of the other clusters. Conditions involving position 70 were similarly present in all but one cluster. The interest of the condition definition as opposed to position is again suggested by the absence of condition 48:F from our set of conditions, while other conditions occurring at position 48 are involved. This condition being fulfilled in proteins belonging to family I and in proteins belonging to family IV (Figure 1), it would prove little discriminating with respect to a family assignment.
Clusters of conditions can also be analyzed by considering pairs of conditions. Consider for instance positions 48 and 66. The centroid of family I is condition 66:E (Table 1); disparity distances between condition 66:E on the one hand and conditions 48:A and 48:G on the other hand are respectively 0.19 and 0.23 [Equation (3), see ‘Materials and Methods’ section for value ranges]. Condition 48:E, which is a member of family IV, has a disparity of 0.613 to condition 66:E, while condition 48:C has a disparity of 1.23 to it. These discrepancies are clearly reflected in the family assignment of each condition. This analysis thus supports the argument that extra information may be conveyed by considering conditions and not only positions in the correlated evolution calculations, as a position-only analysis would miss the details conveyed by the different amino acid types.
Clustering results: assigning a family to the protein sequences
The families of conditions produced by affinity clustering were used to assign a family membership to the protein sequences used in the analysis (‘Materials and Methods’ section). This assignment enabled us to highlight common features shared by distinct protein sequences that corresponded to a similar family. We will first analyze the different families at the sequence level, and then compare our assignment to a sequence clustering. We will also discuss the relevance of our family assignment with respect to protein structures in the ‘Discussion’ section.
Among the five position-unique conditions that define family I, two of them, 36:V and 37:S, correspond to an extra insert in many PHD fingers. For example, in a quarter of the proteins that simultaneously satisfy conditions 77:P and 79:L, and almost all (97%) of the proteins satisfying 63:P or 48:C, all being conditions from family II, both positions 36 and 37 were gaps. In other proteins, such as all INGs and YNGs (which were not all present in the alignment, due to filtering on sequences identities), all but the last position-unique conditions that define family I are satisfied with, in addition, condition 59:D fulfilled in ING3, 4 and 5, with 59 as a Glu in ING1 and 2 (Figure 1).
Sequences of proteins from family II have a strong bias away from the majority of the PHD fingers from our alignment at position 67. While this residue is a Trp in about half of the proteins studied, none of the proteins satisfying the two most central conditions of family II, conditions 77:P and 79:L, have a Trp at this position; instead, the most preferred residues, Gly, Ala or Ser, have small side-chains. This feature will be interesting when discussing a favored substrate for this family (‘Discussion’ section).
The validity of the clustering was confirmed when comparing it to a more traditional clustering of the PHD finger sequences, such as that obtained using an amino acid similarity matrix (Figure 2). One clear feature from Figure 2 is that most proteins from family II did cluster together when using such a matrix, except for the second PHD finger from ATX5 and the first PHD finger of MYST3. Family III appeared to be more spread out within the tree, which suggested that the conditions it involves induce a less global change in the sequences than those from the other families. Finally, the two sequences from family I we incorporated in the tree were close neighbors, and had as closest neighbors sequences from family IV. This fact is interesting in regard to the substrate preference we assign to each family (‘Discussion’ section).
Figure 2.

Family assignment for some PHD finger sequences and comparison with a sequence clustering. The tree was created by a comparison of sequences using BLOSUM62 average distances. The name of each protein was colored according to its family assignment (Table 1): red for family I, cyan for family II, blue for family III and orange for family IV. For protein names and suffixes, see Figure 1.
Extending the alignment: introduction of N-terminal motifs
Sequence alignment and structural results suggest the critical importance of the sequence fragment N-terminal to Cys1. Nonetheless, this fragment is absent from most databases, including the one we used for our analysis, the PFAM database. The first PHD finger of AIRE (AIRE 1) has been shown to interact with unmodified histones mainly through its Asn295 and Asp297 residues (32), as well as Glu298 (17), which, respectively, correspond to positions Cys1-7, Cys1-4 and Cys1-3 in an alignment where N-terminal domains were included (Figure 1). The importance of these positions was also confirmed by site-directed mutagenesis, as mutations of Asp297 to Ala (33) and of Asn295 to Ala (17) in AIRE1 abolished interactions with histones. A similar result was obtained on the equivalent position, Asp489, in protein BHC80 (16). Moreover, mutation of the Asp at position Cys1-7 to Ala in Dnmt3l abolished interactions with Histone 3 (34).
We therefore created and analyzed a new sequence alignment, where the N-terminal region of PHD fingers was present. A total of 200 PHD finger sequences were manually edited, and clustered on a maximum sequence identity of 70% using program CD-Hit, which produced 122 sequences. This new, N-terminal extended alignment, was submitted to the same residue co-evolution analysis as the previous one. Nine conditions had at least one coupling within the first percentile of coupling values. Among these conditions, two corresponded to the newly introduced N-terminal region, Cys1-3 (Glu298 in AIRE 1, Figure 1) with two significant couplings and Cys1-7 (Asn295 in AIRE 1) with one significant coupling (Table 2).
Table 2.
Number of couplings in the first percentile of coupling values for the N-terminal extended alignment of PHD fingers, as grouped by conditions
| Condition | Number of couplings |
|---|---|
| 111 (Cys6-3):Y | 6 |
| 67 (His-2):A | 3 |
| 48 (Cys3-1):Q | 2 |
| Cys1-3:D | 2 |
| 63:D | 1 |
| 75 (Cys5+2):G | 1 |
| 67:W | 1 |
| Cys1-7:D | 1 |
| 46 (Cys3-3):M | 1 |
Position Cys1-3 from AIRE 1 hydrogen-binds Lys9 from Histone 3, reduces 6-fold the association constant between these two peptides when mutated to Ala (17), and its NMR shift in CHD4 is affected by the presence of unmodified or modified Histone 3 (35). These results support the relevance of our analysis as well as the potential to classify unmodified histone-specific PHD fingers according to a residue located N-terminal to Cys1. The use of this sequence region could also help classify the few protein sequences for which no family assignment was provided by our algorithm, such as Pygopus 1 (36) or isoform RE-IIBP of WHSC1/MMSET (37). Other significant conditions in this alignment, as shown in Table 2, agree with the calculations performed on the previous, N-terminal deprived, alignment. Discrepancies might originate in the smaller size of this second sequence set, which could imply a less exhaustive coverage of the existing PHD finger sequences.
New putative interactions for PHD fingers
The importance of the N-terminal domains of PHD fingers in histone recognition, as discussed above, has been experimentally observed for position Cys1-2 in Pygopus [Tyr339 in mouse, Tyr or Phe in other species (36)] and Transcription Factor IID (38) (corresponding residue: Trp868). Moreover, the classification we propose in Table 1 suggests that a key feature shared by both the PHD fingers in family II and in family III is their interaction with unmodified histones, a recently observed property of PHD fingers; see e.g. refs (16,17) and below. We completed our analysis from a structural perspective, and predicted structures for the PHD fingers of BRPF1, a member of the MOZ acetyltransferase complex (39) which satisfies all the position-unique conditions from family III (Table 1), and that of TIF1A (also called TRIM24), which belongs to family II, with four conditions fulfilled (Figure 1).
BRPF1 is interesting with respect to histone recognition, as it bears two domains capable of interacting with histones, a bromodomain and a PHD finger. The structure of its PHD finger was predicted using templates that were selected according to the significant conditions that define family III (Table 1 and Figure 1). Comparison of the predicted structure to complexed PHD finger structures (e.g. ING2 or ING4) suggests that Asp214 replaces the hydrophobic residue (Tyr198 for ING4) at the end of the substrate-binding socket, thus disfavoring the presence of methyl groups on lysine 4. Docking of Histone 3 to the predicted structure confirmed the possibility of a binding of the ammonium group of Lys4 by Asp214, with salt bridges with the side chain of Asp222 for Arg2 and of Glu253 for Arg8. Multiple interactions between the protein and the backbone atoms of Histone 3 are also present (Figure 3 and Supplementary Table S7).
Figure 3.

Docking of Histone 3 to the predicted structure for the PHD finger of BRPF1. BRPF1 is shown as ribbons, with carbon atoms in green, and side-chains with interactions to peptide as sticks. The Histone 3 peptide is shown as balls and sticks with carbon atoms in orange. Interactions (hydrogen bonds and hydrophobic contacts) between the peptide and TIF1A are shown as yellow dotted lines.
The PHD finger of TIF1A has extended sequence similarities with the PHD fingers of AIRE, BHC80 and CHD4 (Figure 2) at sites involving significant conditions. Positions 19 (Val), 42 (Gly), 46 (Leu), 77 (Pro) and 79 (Leu) are strictly conserved over these four proteins. Moreover, positions 41 (Gly), 48 (Cys) and 63 (Pro) are identical in AIRE, CHD4 and TIF1A. This latter domain thus satisfies four of the eight position-unique conditions that define family II (Table 1). A structure was calculated for this domain using CHD4 (second PHD finger), AIRE (first PHD finger) and BHC80 as templates. The overall fold showed the conserved β/β/α arrangement of PHD fingers, with an additional α-helix in the C-terminal, as observed e.g. in ING2 (Figure 4). The possible binding mode of Histone 3 to this domain was first analyzed by superimposing the predicted structure to that of ING2 and ING4, which suggested that a negatively-charged residue (Cys1-7 or Cys1-8 in Figure 2) could be present at the end of the socket into which Lys4-Me3 binds, and that Leu838 (Cys3-3) from TIF1A could develop hydrophobic interactions with the alkyl groups of Lys4-Me3. Docking of unmodified Histone 3 to the predicted structure of TIF1A showed multiple favorable interactions (Figure 4). Both the side chains of Lys4 and Arg2 showed stabilizing interactions with the side chain of Asp823, while Arg8 developed an interaction with Asn825 through its Nε atom. Moreover, multiple interactions take place between the backbone of the histone terminal and the backbone atoms of residues 224–227 from the PHD finger (Supplementary Table S7).
Figure 4.

Docking of Histone 3 to the PHD finger of TIF1A. TIF1A is shown as ribbons, with carbon atoms in green and side-chain with interactions to peptide as sticks. The Histone 3 peptide is shown as balls and sticks with carbon atoms in cyan. Other details are as in Figure 3.
For these two predicted structures, further docking calculations were performed using Lys4-Me3, Lys9-Ac and Lys14-Ac modified Histone 3 as ligands, with no favorable interaction involving these modified residues observed. These new structural and docking studies, in addition to confirming the attribution of unmodified Histone 3 as a preferred substrate for two of our families of conditions, as observed experimentally with members AIRE 1, CHD4 2 and BHC80 for family II, further confirm the importance of the N-terminal region of PHD-fingers in their interaction with histone peptides. The incorporation of the region N-terminal to Cys1 in databases and their inclusion in structural studies would therefore be helpful in enriching our understanding of the function of PHD fingers.
Comparison with mutual information and other methods
Published methods for the determination of important residues use mostly alignment positions and not conditions as we do. In order to compare our results with existing methods, we therefore used only the position at which our important conditions were applied, thus losing a major part of the information obtained through our analysis.
Our relative entropy analysis was first compared to a mutual information analysis [Equation (2)], as this measure has been used in multiple sequence alignment studies [see e.g. (26) and other references in the ‘Introduction’ section]. Mutual information (MI) values obtained from the PHD fingers alignment were first submitted to a comparison to random, using a sequence shuffling algorithm (see ‘Materials and Methods’ section). Still, these values could not be segregated using their P-values, as MI values between all pairs of observation positions were highly significantly different from random (P < 10−4). This provided a first indication of the higher relevance of the relative entropy analysis, as an analysis of statistical significance did distinguish among coupling values, with multiple couplings having P-values superior to 2.5 × 10−3 among the first decile of coupling values. MI values were thus divided into two groups with respect to their ranking. A comparison of the set of pairs of positions having the highest MI values (first 5%, i.e. 37 pairs) with an identical number of ‘condition:position’ pairs resulting from the highest coupling values [Equation (1)] is shown in Table 3. Though similar pairs of positions are detected by the two methods in multiple cases (positions in bold), some important pairs are absent from the MI analysis. Position pairs 42 and 70 (His+1) on the one hand, and 66 and 68 on the other hand, do not have high MI values. However, they were each involved in high-value couplings both as a condition (first column of Table 3) and as an observation position (Couplings column), and proved important for discriminating the sequences into different families (Figure 1 and Table 1, family III and IV). Moreover, high MI values were observed for residues 86 to 90 (Table 3), and yet these positions do not seem to be discriminating between the various PHD finger sequences (Figure 1), even though position 88 was also highlighted by method SDR (see below).
Table 3.
Comparison of the results obtained with MI and coupling based on relative entropy
| First position | Mutual information | Couplings |
|---|---|---|
| 15 (Cys1+1) | 46, 48, 66, 67, 77, 79 | |
| 35 | 36, 37, 42, 48, 63, 70 | |
| 36 | 35, 37, 41, 42, 46, 48, 63, 67 | 66 |
| 37 | 35, 36, 48, 63 | 41 |
| 42 | 35, 36 | 70, 74 |
| 43 | 70 | |
| 46 | 15, 48, 67 | |
| 48 (Cys3-1) | 15, 35, 36, 63, 67, 70 | 37 |
| 66 | 15 | 68 |
| 67 (His-2) | 15, 36, 46, 48, 79 | |
| 68 (His-1) | 74 | 66, 67, 70, 111, 115 |
| 70 (His+1) | 35, 43, 48, 79 | 42, 46, 74 |
| 74 (Cys5+1) | 68, 77 | 48, 66, 70, 79, 115 |
| 75 (Cys5+2) | 77, 79 | |
| 77 | 15, 74, 75, 79 | |
| 79 | 15, 70, 75, 77, 80 | |
| 86 | 87 | |
| 87 | 88 | |
| 88 | 87, 89, 90 |
Pairs of positions are obtained by matching an entry in the first column with one of the other two columns. The column ‘mutual information’ contains all positions with mutual information with ‘first position’ in the first 5% of mutual information values. The ‘couplings’ column contain the observation position that were highly coupled (37 highest couplings, in order to match the number of position pairs used for mutual information) to a condition occurring at the ‘first position’. Positions in bold were present both among the highest couplings as observation positions and in the first 5% of mutual information values. As coupling are not symmetrical with respect to positions, mutual information pairs were indicated twice [e.g. both at line 48 and 70 for the pair (48,70)].
The PHD finger family was also analyzed with method SDR (http://paradox.harvard.edu/sdr), an algorithm which was recently proposed by Donald and Shaknovich for the determination of ‘specificity-determining residues’ (40). Ten alignment positions were determined as such by method SDR: positions 42, 48, 52, 67, 68, 70, 74, 77, 88 and 111 (Table 1, underlined positions). Similarly to MI, this algorithm does not output amino acid types but only alignment positions. A comparison was also performed with the ‘specificity’ residues proposed by the proteinkeys server (www.proteinkeys.org), which corresponded to positions 42, 48, 66, 67 (His-2) and 70 (His+1).
Calculations with these different methods first confirmed the importance of positions 48 (Cys3-1), 68 (His-1) and 74 (Cys5+1) as positions for significant sequence tags (Table 3). In addition, both SDR and proteinkeys suggested a role for position 42 in defining specificity, while this position defines the second most central condition of family III. Position 66 (His-3), which is involved in two first-percentile conditions, was neither evidenced by MI nor by SDR calculations. This might be due to the fact that, apart from these two conditions, the other amino acid values may produce no strong effect on the alignment, which would prevent this position from being detected by position-based calculations. Overall, this comparison supports our detection of the sequence conditions that are the most effective at classifying PHD fingers.
A further validation of our method is provided by the consideration of two additional protein families that recognize histones, the Chromatin Organization Modifier (chromo-) domain and the acyltransferase domain. These families were submitted to the same computational process as the PHD finger alignment, from a download from the PFAM database to a clustering of alignment conditions (‘Materials and Methods’ section). The resulting clusters obtained for the chromodomains are summarized below, while those for the acetyltransferase domain are provided as Supplementary Data. These results were contrasted with the position predictions available online for the SDR algorithm and the proteinkeys server (www.proteinkeys.org).
For the chromodomain family, the 24 first-percentile conditions are clustered into four families. Two of the four positions that were involved in cluster centroids were also predicted as specificity-determining by method SDR (Supplementary Table S8, underlined). Moreover, three of the five conditions from family I, which includes e.g. over 50 proteins from rice (with all conditions satisfied by Uniprot entry Q01JF9, see Figure 5) as well as some from maize, were detected by that method. Note that Polyprotein from maize (A5JSC4) satisfies three conditions from family I and one from family III (106:K), which makes it a member of family I. Three of the positions that are involved in our cluster-defining conditions were also predicted as specificity-determining by the proteinkeys server (Supplementary Table S8, gray boxes), among which the centroid of family IV. The relevance of the clustering can be seen for family II, with the first chromodomain from fission yeast HRP3 satisfying five of its conditions, HRP1 its four most central conditions and PKL from Arabidopsis thaliana that satisfies four of its conditions (Figure 5). Position 113, which produced one condition for family II, is involved in the docking of Lys9-trimethylated Histone 3 by Chp1, where it corresponds to Tyr47 (41). Overall, twenty-one proteins fulfill the four most central conditions for this family. Family III includes for example the DNA-methyltransferase from Arabidopsis, CMT3, with five conditions fulfilled, or the cytosine-specific methyltransferase from mustard (Figure 5). Family IV includes fewer proteins, among which elongation factor 3 or mRNA export factor elf1 (Q5KQ02, Figure 5). These results confirm that our method is not valid only on PHD finger domains, but can be applied to other protein families.
Figure 5.

Sample alignment for proteins from the chromodomain data set. The sequences are ordered by family assignment. Bold residues correspond to first-percentile conditions (Supplementary Table S8) and the box to a residue strictly conserved over the protein sequences shown. Some columns were removed before positions 51, 59, 128 and 158. Suffix ZM indicates a protein from maize, OZ a protein from rice, MD one from Marine diatom, MT one from mustard and CN one from Cryptococcus neoformans. SP is as indicated in Figure 1.
DISCUSSION
Correlated evolution as a tool for detecting important positions or residues
The present study relies on the generally accepted assumption that if two protein residues ‘evolve’ simultaneously, then they possess either a structural or a functional relationship, e.g. they belong to the same communication pathway or the same binding surface. Analysis of correlated evolution in proteins was first proposed as a tool for predicting 3D contacts between residues using only protein sequences (6), in order to address the fundamental biochemical issue of sequence-structure relationship. Still, there is no clear agreement today whether co-evolution is a satisfactory tool for the prediction of contacts. The group of Ranganathan successfully applied correlated evolution analysis to the detection of allostery communication pathways in various protein families (4,5). More recently, multiple studies have used this concept for the determination of residues critical for function (1,42).
Our study, aside from being the first to analyze sequences of PHD fingers from a global perspective, with more than 900 distinct sequences considered, also introduces a novel method of analysis, namely the identification of ‘statistically significant conditions’, i.e. a position and residue-type pair within the protein family studied, where significance is measured by the degree of influence of a condition on the amino acid distribution at other sites. We have attempted to demonstrate the gain in information obtained by considering conditions rather than positions alone. Indeed, multiple conditions involving a given position may have distinct effects or scores, as for example discussed above when comparing condition 66:E respectively to condition 48:A and condition 48:C. One interpretation of this observation is that, in some proteins, a given position may play an important role, whereas, in others, the same position may correspond to a more ‘functionally silent’ region, e.g. due to local structure differences or neighboring insertions or deletions, and therefore carry a smaller evolutionary stress. In this respect, one of the possible outcomes of our study is the use of weighted alignments for PHD fingers, where conservation of either of our significant conditions would be assigned a more important weight than other conservations. Such an alignment would then directly group sequences according to the residue identities we believe are the most biologically meaningful.
Prediction of substrate selectivities among PHD fingers from their sequences
Relying on our family classification, some trends relating PHD finger sequences to their most favored substrate can be found among available experimental results. A preferred substrate for proteins from family I, which corresponds to the PHD fingers of ING proteins, is Histone 3 trimethylated on Lys4. The docking of this modified histone through a hydrophobic cage, which involves conserved Met at position 46, Trp at position 67 and Tyr at position Cys1-2 (Figure 1), has indeed been described in multiple studies (31,43). PHD fingers that satisfy conditions from family II are likely to have Histone 3 unmodified at Lys4 as a preferred substrate, as e.g. observed for the first PHD finger of AIRE (17) or that of BHC80 (16). These proteins are also likely to have a secondary interaction with Lys9, as an effect of its modification state on the affinity of Histone 3 for AIRE and CHD4 has been reported (17,35). Some proteins that harbor a bromodomain in addition to their PHD finger belong to family III, such as BRPF1 and BRPF3. It is therefore likely that their PHD fingers either recognize unmodified, as suggest by our docking study, or acetylated histones [see Figure 1b in (16)]. Two recent reports show that two proteins in family IV, namely PHD fingers 2 and 8, which fulfill conditions 111:Y, 70:G, 43:F and 48:E, bind to trimethylated Lys4 from Histone 3, suggesting that the entire family may do so as well (44,45).
Though PHD fingers are often referred to as ‘methylated-lysine binding domains’, in our analysis multiple domains that recognize non-modified Histone 3 could be assigned to a single family. Indeed, the PHD fingers of AIRE (first finger), CHD4 (second finger) and BHC80 (also named PF21A, see Figure 2) belong to family II. Moreover, most proteins from this family have a small amino acid at position His-2 (‘Results’ section), while the Trp that is present at this position in all trimethylated Lys4-binding PHD-fingers characterized to date is involved in the formation of the hydrophobic substrate-recognition socket (14,43). The fact that the conditions that define family II could be highlighted in our calculations, even while maintaining at least 50 occurrences for each condition analyzed, and that conditions 77:P and 79:L, its most central ones, are simultaneously fulfilled in 180 proteins in our alignment, suggest that the binding of PHD fingers to unmodified Histone 3 holds for a significant fraction of PHD fingers.
Regarding the PHD fingers of AIRE, no binding to histones has been described for its second PHD finger (33). As it fulfills condition 77:P from family II and condition 74:H from family III (Figure 1), no strict family assignment can be made for it. One more definite criterion regarding its sequence is the absence of an aromatic residue at position 111 (Cys6-2). We propose that the presence of a non-aromatic residue at this position (that of a Leu being moreover unique in our data set) implies that this domain should not be classified as a PHD finger.
The clustering of alignment conditions into different families was also analyzed at the structural level. Since, for example, we propose that both family I and IV have trimethylated lysine as a most favored substrate, one could wonder whether 3D structures support both the similarity of the substrate and the meaningfulness of their grouping into two families instead of one. We therefore superimposed the structure of the PHD finger of ING4 (family I) to that of the PHD finger of PHF8 (family IV, Figure 6), which was recently described (44). While residues at positions Cys1-1 and 46, which are respectively conserved as Tyr and Met, occupy nearly identical positions in the two proteins, three conditions from family IV evidence major differences with family I: 48:E, 70: G and 43:F. Position 48, a glycine (residue 211) in ING4, is located close to the guanidinium group of Arg2 from Histone 3. In PHF8, where this residue is a glutamine, a condition from family IV, a similar orientation of Arg2 is prevented by this more bulky residue (bottom of the image, Histone peptides respectively in limegreen and orange). Condition 43:F and 70:G, which are also fulfilled by PHF8, reveal the necessity of co-evolution of these two positions: in PHF8, the aromatic ring of Phe at position 43 (residue 19 in the crystal structure) points towards the Gly at position 70; in the superimposed structures, the Phe at 70 from ING4 occupies the same volume as 43:F from PHF8, while the corresponding residue at position 43 for ING4, a Glu, has a different orientation. Moreover, the two structures evidence a displacement of Thr3 from Histone 3, which could be due to the different free space provided by residues at positions 43 and 70. These overall differences, as well as the additional recognition of Lys9 by the bromodomain of PHF8 (44), contribute to justifying a different orientation of the bound Histone-derived peptide to these two proteins. A similar analysis was performed for the PHD fingers of AIRE 1 and BRPF1, which respectively belong to family II and III. This comparison also highlighted the importance of condition at position 48 in affecting the interaction with Histone, as well as 63:P and 77:P in altering the overall fold of the PHD finger (Supplementary Figure S11). Interestingly, in both Figure 6 and Supplementary Figure S11, the strongest discrimination with respect to histone docking does not appear to involve Lys4, but more likely Arg2.
Figure 6.

Superposition of the three dimensional structure of ING4 on that of PHF8 (respective PDB entries: 2VNF and 3KV4). The PHD fingers are respectively shown as cyan and pale yellow ribbons, with docked Lys4-trimethylated peptides with carbon atoms in limegreen and orange, respectively. Conditions for ING4 (family I) are shown with side chains as blue ball-and-sticks models, those for PHF8 (family IV) as yellow sticks, except for Gly which were shown with their Cα atom as a sphere. The zinc ions from PHF8 are displayed as gray spheres. Additional highlighted residues are shown as sticks using the color of their corresponding peptide.
Finally, the classification into four families opens new possibilities in the consideration of interactions to simultaneous histone modifications i.e. going beyond attributing the modification state of a single histone residue to a given group of PHD fingers. Recent work highlights a tight correlation between histone tagging at different positions, such as the mass spectroscopy study by Vermeulen, Mulder et al. that identifies a correlation between the modifications of Lys4 and Lys9 from Histone 3 (46), the study by Kirmizis et al. (47) which shows that methylation of Arg2 from Histone 3 inhibits methylation of Lys4 from the same protein, and the work on AIRE 1 which reveals an influence of the modifications at Arg2 and Lys9 on the binding association of the protein to Histone 3 unmodified at Lys4 (17). More data involving simultaneous histone modifications are indeed needed to refine the substrate preferences we propose here.
Relevance and perspectives
A new method that applies statistical and information-theoretic tools to identify critical protein residues was described and applied to PHD fingers, a family of histone-interacting protein domains (13). Twenty-seven position-residue type pairs were used to divide this family into four distinct subfamilies (Table 1). The importance of these pairs was confirmed by comparison to existing structural and functional results. The classification we propose should enable easier sequence-based attribution of substrate-binding selectivity for PHD fingers. In addition, as no criterion specific to this family was applied in the course of study, this method should also be relevant to any family of proteins, provided a sufficiently large alignment of protein sequences be available. One modification currently under study which could extend this method to smaller alignments is the consideration of a reduced amino acid alphabet (48) instead of all possible residue types. Moreover, additional structure predictions and docking calculations are under way that may provide promising directions for the further development of our method.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The work of P.S. and D.G. was partially supported by NIH-NCRR Grant UL1 RR 025005. P.S. also acknowledges the MPI for Molecular Genetics (Berlin, Germany) for funding the initial steps of this project. Funding for open access charge: above Grant.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr Joel Bader, Dr Nicolas Biais and Dr Laurent Younès for insightful comments.
REFERENCES
- 1.Chakrabarti S, Lanczycki CJ. Analysis and prediction of functionally important sites in proteins. Protein Sci. 2007;16:4–13. doi: 10.1110/ps.062506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panchenko AR, Kondrashov F, Bryant S. Prediction of functional sites by analysis of sequence and structure conservation. Protein Sci. 2004;13:884–892. doi: 10.1110/ps.03465504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- 4.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 5.Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell. 2004;116:417–429. doi: 10.1016/s0092-8674(04)00119-9. [DOI] [PubMed] [Google Scholar]
- 6.Gobel U, Sander C, Schneider R, Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 7.Capra JA, Singh M. Characterization and prediction of residues determining protein functional specificity. Bioinformatics. 2008;24:1473–1480. doi: 10.1093/bioinformatics/btn214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hannenhalli SS, Russell RB. Analysis and prediction of functional sub-types from protein sequence alignments. J. Mol. Biol. 2000;303:61–76. doi: 10.1006/jmbi.2000.4036. [DOI] [PubMed] [Google Scholar]
- 9.Hughes AL. Near neutrality. Ann. NY Acad. Sci. 2008;1133:162–179. doi: 10.1196/annals.1438.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 11.Chakrabarti S, Bryant SH, Panchenko AR. Functional specificity lies within the properties and evolutionary changes of amino acids. J. Mol. Biol. 2007;373:801–810. doi: 10.1016/j.jmb.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bienz M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem. Sci. 2006;31:35–40. doi: 10.1016/j.tibs.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Taverna S, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–723. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chignola F, Gaetani M, Rebane A, Org T, Mollica L, Zucchelli C, Spitaleri A, Mannella V, Peterson P, Musco G. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 2009;37:2951–2961. doi: 10.1093/nar/gkp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sterner B, Singh R, Berger B. Predicting and annotating catalytic residues: an information theoretic approach. J. Comp. Biol. 2007;14:1058–1073. doi: 10.1089/cmb.2007.0042. [DOI] [PubMed] [Google Scholar]
- 19.Kapoor K, Rehan M, Kaushiki A, Pasrija R, Lynn AM, Prasad R. Rational mutational analysis of a multidrug MFS transporter CaMdr1p of Candida albicans by employing a membrane environment based computational approach. PLoS Comp. Biol. 2009;5:e1000624. doi: 10.1371/journal.pcbi.1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer ELL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 22.Buslje CM, Santos J, Delfino JM, Nielsen M. Correction for phylogeny, small number of observations and data redundancy improves the identification of coevolving amino acid pairs using mutual information. Bioinformatics. 2009;25:1125–1131. doi: 10.1093/bioinformatics/btp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Samudrala R. Incorporating background frequency improves entropy-based residue conservation measures. BMC Bioinformatics. 2006;7:385. doi: 10.1186/1471-2105-7-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahony S, Auron PE, Benos PV. Inferring protein-DNA dependencies using motif alignments and mutual information. Bioinformatics. 2007;23:i297–i304. doi: 10.1093/bioinformatics/btm215. [DOI] [PubMed] [Google Scholar]
- 25.Weil P, Hoffgaard F, Hamacher K. Estimating sufficient statistics in co-evolutionary analysis by mutual information. Comp. Biol. Chem. 2009;33:440–444. doi: 10.1016/j.compbiolchem.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Martin LC, Gloor GB, Dunn SD, Wahl LM. Using information theory to search for co-evolving residues in proteins. Bioinformatics. 2005;21:4116–4124. doi: 10.1093/bioinformatics/bti671. [DOI] [PubMed] [Google Scholar]
- 27.Frey BJ, Dueck D. Clustering by passing messages between data points. Science. 2007;315:972–977. doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- 28.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 29.DeLano WL. DeLano Scientific. CA: Palo Alto; 2002. [Google Scholar]
- 30.Martin DG, Baetz K, Shi X, Walter KL, MacDonald VE, Wlodarski MJ, Gozani O, Hieter P, Howe L. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell Biol. 2006;26:7871–7879. doi: 10.1128/MCB.00573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peña PV, Hom RA, Hung T, Lin H, Kuo AJ, Wong RPC, Subach OM, Champagne KS, Zhao R, Verkhusha VV, et al. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J. Mol. Biol. 2008;380:303–312. doi: 10.1016/j.jmb.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Org T, Chignola F, Hetényi C, Gaetani M, Rebane A, Liiv I, Maran U, Mollica L, Bottomley MJ, Musco G, et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008;9:370–376. doi: 10.1038/embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh AS, Kuo AJ, Park SY, Cheung P, Abramson J, Bua D, Carney D, Shoelson SE, Gozani O, Kingston RE, et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc. Natl Acad. Sci. USA. 2008;105:15878–15883. doi: 10.1073/pnas.0808470105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ooi SKT, Qiu C, Bernstein E, Li KQ, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musselman C, Mansfield R, Garske A, Davrazou F, Kwan A, Oliver S, O'Leary H, Denu J, Mackay J, Kutateladze T. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem. J. 2009;423:179–187. doi: 10.1042/BJ20090870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiedler M, Sanchez-Barrena MJ, Nekrasov M, Mieszczanek J, Rybin V, Müller J, Evans P, Bienz M. Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol. Cell. 2008;30:507–518. doi: 10.1016/j.molcel.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JY, Kee HJ, Choe NW, Kim SM, Eom GH, Bqek HJ, Kook H, Kook H, Seo SB. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol. Cell Biol. 2008;28:2023–2034. doi: 10.1128/MCB.02130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Ingen H, van Schaik FMA, Wienk H, Ballering J, Rehmann H, Dechesne AC, Kruijzer JAW, Liskamp RMJ, Timmers HTM, Boelens R. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure. 2008;16:1245–1256. doi: 10.1016/j.str.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 39.Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, Tahmasebi S, Cayrou C, Doyon Y, Goh SL, et al. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 2008;28:6828–6843. doi: 10.1128/MCB.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donald JE, Shakhnovich EI. SDR: a database of predicted specificity-determining residues in proteins. Nucleic Acids Res. 2009;37:D191–D194. doi: 10.1093/nar/gkn716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schalch T, Job G, Noffsinger VJ, Shanker S, Kuscu C, Joshua-Tor L, Partridge JF. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell. 2009;34:36–46. doi: 10.1016/j.molcel.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sankararaman S, Kolaczkowski B, Sjölander K. INTREPID: a web server for prediction of functionally important residues by evolutionary analysis. Nucleic Acids Res. 2009;37:W390–W395. doi: 10.1093/nar/gkp339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taverna S, Ilin S, Rogers R, Tanny J, Lavender H, Li H, Baker L, Boyle J, Blair L, Chait B, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010;17:38–44. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen H, Li J, Song T, Lu M, Kan PY, Lee MG, Sha B, Shi X. Recognition of histone H3K4 trimethylation by the PHD finger of PHF2 modulates histone demethylation. J. Biol. Chem. 2010;285:9322–9326. doi: 10.1074/jbc.C109.097667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vermeulen M, Mulder K, Denissov S, Pijnappel W, van Schaik F, Varier R, Baltissen M, Stunnenberg H, Mann M, Timmers H. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 47.Kirmizis A, Santos-Rosa H, Penkett C, Singer M, Vermeulen M, Mann M, Bähler J, Green R, Kouzarides T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007;449:928–932. doi: 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melo F, Marti-Renom MA. Accuracy of sequence alignment and fold assessment using reduced amino acid alphabets. Proteins Struct. Funct. Bioinf. 2006;63:986–995. doi: 10.1002/prot.20881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.