

Abstract
In-stent restenosis is a clinical complication following coronary angioplasty caused by the implantation of the metal device in the atherosclerotic vessel. Histological examination has shown a clear contribution of both inflammatory and smooth muscle cells (SMCs) to the deposition of an excess of neointimal tissue. However, the sequence of events leading to clinically relevant restenosis is unknown. This paper aims to study the phenotype of SMCs when adhering on substrates and exposed to biochemical stimuli typical of the early phases of stent implantation. In particular, human SMC phenotype was studied when adhering on extracellular matrix-like material (collagen-rich gel), thrombus-like material (fibrin gel) and stent material (stainless steel) in the presence or absence of a platelet-derived growth factor (PDGF) stimulus. Cells on the collagen and fibrin-rich substrates maintained their contractile phenotype. By contrast, cells on stainless steel acquired a secretory phenotype with a proliferation rate 50 per cent higher than cells on the natural substrates. Cells on stainless steel also showed an increase in PDGF-BB receptor expression, thus explaining the increase in proliferation observed when cells were subject to PDGF-BB stimuli. The stainless steel substrate also promoted a different pattern of β1-integrin localization and an altered expression of hyaluronan (HA) synthase isoforms where the synthesis of high-molecular-weight HA seemed to be favoured. These findings highlighted the induction of a phenotypic pattern in SMC by the stainless steel substrate whereby the formation of a HA-rich neointimal tissue is enhanced.
Keywords: in-stent restenosis, biomaterials, smooth muscle cells, hyaluronan, platelet-derived growth factor
1. Introduction
In-stent restenosis (ISR) is a clinical complication consisting of the re-occlusion of the coronary artery lumen following stented angioplasty. In the case of the implantation of bare metal stents (e.g. stainless steel devices), ISR occurs in 20–30% of cases and its incidence can only be significantly reduced by the use of drug-eluting stents (DESs) [1]. However, the use of these medicated devices has been linked to the occurrence of late thrombosis and patient death by infarction [2,3]. Furthermore, the relatively higher costs of the DES limit their use to a specific category of patients who are at a risk of cardiovascular complications. These issues related to the use of DESs have prompted further research into the cause of ISR by non-medicated devices. It is envisaged that an in-depth understanding of the biochemical and cellular mechanisms by which these devices cause ISR may lead to the development of new and less expensive stents able to reduce the incidence of ISR without the use of DESs.
Thus far, clinical investigations have clearly shown that ISR has cellular and histological features that are distinct from those present in the neointima surrounding the atherosclerotic plaque [4]. In the case of ISR, the stent is mainly encased in an extracellular matrix (ECM) that, alongside collagen, is rich in hyaluronan (HA). HA is a glycosaminoglycan synthesized by three isoforms of HA synthase, HAS 1, 2 and 3, each responsible for the secretion of HA with different molecular weights that are able to promote different biochemical and cellular pathways. HA with a molecular weight larger than 2 million daltons is synthesized by HAS 1 and 2, and it has been reported to have both an anti-inflammatory and tissue remodelling properties, whereas HAS 3 synthesizes HA with a lower molecular weight that activates inflammatory cells and angiogenesis. Cells within this myxoid tissue have been shown to be positive to markers of expression of both inflammatory and smooth muscle cells (SMCs) [5]. The deposition of this type of tissue is always preceded by the earlier formation of a thrombus clot followed by the invasion of the stent area by the monocytes/macrophages [4]. Recently, the ability of stainless steel (St) to induce transdifferentiation of adhering mononuclear cells (i.e. monocytes/macrophages) into α actin-positive, spindle-like cells has been demonstrated [6]. These findings would suggest that monocytes/macrophages at the interface between the material and the tissue can transdifferentiate into a population of SMC-/myofibroblast-like cells. Furthermore, mononuclear cells adhering on a St surface acquire a post-inflammatory phenotype, able to secrete growth factors rather than pro-inflammatory cytokines [6,7]. In particular, two growth factors are released by the adhering mononuclear cell population upon contact with the St, platelet-derived growth factor-BB (PDGF-BB) and TGF-β1. These are widely recognized to be relevant to ISR owing to their ability to stimulate SMC proliferation [8–10]. It has been suggested that the combined surface activation of both inflammatory cells and SMCs play a major role in the thickening of the neointima [1,2,4].
The role played by SMCs in vascular complications has been widely investigated, with research focusing on mechanical stimuli, (ECM) protein adhesion and cell behaviour [11–14]. In particular, the SMCs ability to proliferate and produce HA has been closely examined. It is widely accepted that the combination of the host response following device implantation with the mechanical stimuli exerted by the stent and by the altered haemodynamics is able to stimulate SMC proliferation [13]. However, to our knowledge, no investigation has been performed to ascertain the effect of the substrates exposed during the early phases following stent implantation on SMC phenotype. This study hypothesizes that the damage of the endothelial layer of the arterial wall induced by the stent expansion exposes resident SMCs not only to the physiological extracellular matrix but also to the metal stent surface and to fibrin clots. The contact of the cells with these different surfaces can therefore lead to changes of their phenotype.
In this paper, SMC phenotype was assessed in the light of the cell contact with different substrates, in the presence and absence of a PDGF-BB stimulus. In particular, phenotypic characterization has been performed in vitro on SMCs adhering on (i) a typical stent material (i.e. St), (ii) a clot-like substrate (i.e. fibrin gel, Fib) such as that generated by the damage of the vessel wall upon stent expansion and (iii) an (ECM)-like substrate (i.e. collagen gel, ECM) that can be exposed after the loss of the endothelium in the traumatized vessel wall [5]. The SMCs adhering on these substrates were analysed for their morphology, viability and proliferation as well as for the expression of relevant markers such as the PDGF-BB receptor (PDGF-BBr) and the genes for the three hyaluronan synthase isoforms (i.e. HAS1, HAS2, HAS3).
To define phenotypic changes, cell membrane receptors including integrins and PDGF were analysed alongside mechanisms of PDGF receptor internalization and HAS1, HAS2 and HAS3 gene expression. The study of the integrin and PDGF receptor distribution as well as the effect of blocking the PDGF stimulus highlighted the role of the various substrates in inducing phenotypic changes in the cells, while the assessment of the HAS1, HAS2 and HAS3 gene expression provided indication about the type of HA that the cells can synthesize when in contact with the substrates. Indeed, the different types of HA produced by these isoforms were considered indicative of the propensity to accumulate high- or low-molecular-weight HA within the neointimal tissue.
2. Material and methods
2.1. Substrate preparation
Substrates were prepared according to a method previously described [6]. Briefly, St discs (1.6 cm diameter, Goodfellow, Catalogue no. FI240300) were disinfected in ethanol 70 per cent (v/v) and washed three times in phosphate-buffered saline (PBS) pH 7.2 prior to use. Engelbreth–Holm–Swarm murine sarcoma (ECM) gel was purchased from Sigma, UK (Cat. no. E1270). ECM is a cell culture substrate extracted from animal source and containing primarily laminin, collagen type IV, heparin sulphate and entactin. The solution was obtained by dissolving ECM powder in ice-cold PBS following the manufacturer's instruction. The cold ECM solution (200 µl) was pipetted either into the wells of 24-well (1.6 cm diameter) tissue culture plates (TCPs) (Nunc, UK) or into 2-well Permanox chamber slides (Nunc). ECM coating of the well bottom surface was achieved by inducing the gelification of the ECM solution by equilibrating the liquid layer at room temperature. Alternatively, the wells were coated with 200 µl of polymerizing fibrin glue (Tissell, Baxter, UK) prepared according to manufacturer's instructions. Both ECM and Fib gels were prepared under sterile conditions, allowed to settle for at least 30 min and washed three times in PBS prior to use.
2.2. Cell study
Human aortic SMCs (ATCC no. CRL-1999, designation: T/G HA-VSMC, LCG Promochem, UK) were seeded at 3 × 103 cells ml−1 on the three substrates (15 mm diameter discs). The complete growth medium was an SmGM formulated medium (Cat. no. CC-3182, Lonza, USA) enriched with 0.05 mg ml−1 ascorbic acid, 0.01 mg ml−1 insulin, 0.01 mg ml−1 transferrin, 10 ng ml−1 sodium selenite, 0.03 mg ml−1 endothelial cell growth supplement, 10 mM HEPES buffer and 10 mM TES buffer. SMCs were cultured for 48 h in 10 per cent (v/v) foetal bovine serum (FBS)-enriched medium prior to a 24 h incubation in FBS-poor (5% by volume) medium. The adhering SMCs were cultured for a further 48 h in either the absence or presence of 2 ng ml−1 PDGF-BB [15]. Alternatively, prior to spiking, SMCs were treated with 0.2 M sucrose for 15 min to block the clathrin-mediated internalization of the PDGF-BB/PDGF-BBr complex upon PDGF spiking [15]. Finally, the cells were washed and incubated for a further 48 h with 10 per cent (v/v) FBS-supplemented medium as above. To relate cell morphology and receptor expression to cell counts, cell cytoskeleton staining and immunolabelling were performed at the same incubation time.
The morphology of the SMCs adhering on the three substrates was analysed by phalloidin-rhodamine staining. For this purpose, cells were washed three times in PBS, fixed with 3.7 per cent (v/v) formaldehyde in PBS for 30 min at room temperature and finally incubated with 1 : 100 diluted phalloidin-rhodamine staining for 15 min at room temperature. Cells were washed three times prior to microscopy.
SMC viability and proliferation were evaluated in a separate set of experiments on six samples for each substrate. In these experiments, cells were not fixed, but immediately stained with Hoescht propidium iodide staining. Within a few minutes of staining, viable and dead cells were counted by epi-fluorescent microscopy. Six fields per disc were counted and the data expressed as mean ± s.d. of alive and apoptotic cells and statistically analysed by the ANOVA (Dunnet's and Tukey's) test by Minitab 15 software (Minitab Ltd, UK). Samples were considered significantly different at p ≤ 0.01. All experiments were performed over a 72 h incubation to allow a significant cell proliferation. Previous optimization experiments established that the rate of proliferation of the SMC line was relatively slower during the first 24 h and that cell synchronization could not be performed at FBS concentrations lower than 5 per cent (v/v) (data not shown).
SMC expression of both PDGF-BBr and β1-integrin was also assessed by immunocytochemistry. The experiment was stopped by fixing the cells in 3.7 per cent (v/v) formaldehyde for 30 min at room temperature. Samples were washed three times in PBS and then washed in blocking buffer (1% (w/v) BSA (Sigma) in PBS) for 30 min at 37°C to minimize non-specific adsorption of the antibodies. Goat anti-human PDGF-BBr antibody (Calbiochem Cat. no. PC320L) was diluted 1 : 1000 in 10 µg ml−1 of blocking buffer and 200 µl was added to the samples and incubated for 1 h at room temperature. After incubation, primary antibody solution was removed and the samples washed three times with PBS. Rabbit anti-goat FITC-tagged IgG (Sigma, Cat. no. F4891), previously diluted to 1:2000 in blocking buffer to a final concentration of 5 µg ml−1, was added (200 µl) to samples and incubated at room temperature for 1 h. The secondary antibody solution was removed and the samples were washed three times in PBS. Similarly, SMCs adhering on the different substrates were immunolabelled to evaluate their β1-integrin expression. SMCs were fixed in formalin, washed in blocking buffer and incubated with mouse anti-human β1-integrin (Calbiochem Cat. no. CP26) diluted in blocking buffer at a final concentration of 5 µg ml−1. After washing in PBS, an anti-mouse FITC-tagged IgG (Calbiochem Cat. no. 401 214) was added at a concentration of 1 µg ml−1.
In all the experiments, TCP wells were used as control and the cells were analysed by epi-fluorescence microscopy. Sample slides were mounted in mounting medium (Vector Labs) and images captured at 40× magnification using a Nikon Eclipse TE2000-U inverted microscope fitted with a Nikon D1X digital camera.
In a separate set of experiments, after culturing on the different substrates and TCP in the presence and absence of PDGF, total RNA was extracted from SMCs using Trizol reagent according to the manufacturers' instructions (Invitrogen). RNA (1 µg) was reverse transcribed using a Quantitech cDNA kit (Qiagen, UK). Primers for human HAS isoform genes were the following:
— HAS1 forward: 5′-ggcttgtcagagctacttc-3′, HAS1 reverse: 5′-gccacgaagaaggggaa-3′
— HAS2 forward: 5′-atgcattgtgagaggtttct-3′, HAS2 reverse: 5′-ccatgacaactttaatcccag-3′
— HAS3 forward: 5′-gacgacagccctgcgtgt-3′, HAS3 reverse: 5′-ttgaggttcagggaaggagat-3′.
Amplification product sizes were 368, 354 and 335 bp, respectively. GAPDH was used as the housekeeping gene. Electrophoresis on 1 per cent (w/v) agarose gels was performed to resolve the PCR products that were visualized by ethidium bromide staining. All the experiments were repeated in duplicate on each substrate on different days (n = 6).
3. Results
Cell morphology studies clearly highlight the different effects of the three substrate materials on SMC phenotype. Figure 1b shows that SMCs adhering on ECM and on Fib (data not shown) substrates preserved a spindle-like morphology typical of the contractile phenotype. Conversely, SMCs adhering on TCP (figure 1a) and St (figure 1c) assumed a relatively more spread morphology. Figure 1b also highlights the integrity of the ECM coating at 72 h incubation; indeed, this coating is stable for up to 5 days whereby, at longer incubation times, its breakdown can be clearly identified at this magnification (data not shown). The change in cell morphology as highlighted in figure 1 reflected the different proliferation potential of the cells. Indeed, the proliferation of SMCs adhering on St was significantly higher than cells adhering on ECM and Fib (approx. 50% higher) and it was comparable to those cells that were cultured on TCP (figure 2). Remarkably, the spiking of the cells with PDGF-BB showed a pronounced effect only on St and TCP where a significant increase in cell proliferation was observed (approx. 20% higher than non-spiked cells). PDGF-BB spiking had a significant but limited effect on SMC proliferation on ECM and Fib; the proliferation rate of the cells under the PDGF-BB spiking was approximately 50 per cent lower than SMCs adhering on St. For all the three substrates, the number of apoptotic cells was significantly higher than the TCP control, but still represented a relatively low percentage (approx. 10% of the total cell number). For all the substrates, the inhibition of the PDGF-BB/PDGF-BBr complex internalization by the sucrose pre-treatment led to levels of cell proliferation comparable to the relative control. The inhibitory effect of the sucrose treatment showed to be specific for the PDGF-BB stimulus and did not alter the substrate surface effect.
Figure 1.

SMCs adhering on the different substrates. (a) TCP, (b) ECM, and (c) St.
Figure 2.

SMC proliferation on the different substrates with or without PDGF-BB spiking (2 ng ml−1). Suc indicates 0.2 M sucrose treatment. Asterisk indicates values significantly different from the relative control (<0.01).
Immunostaining of the SMCs with an antibody for the PDGF-BBr indicated an increase in receptor expression in SMCs adhering on the synthetic substrates; both TCP and St demonstrated greater immunostaining than ECM and Fib (figure 3a–c). Indeed, the images indicate that PDGF-BBr is localized into a few peri-nuclear areas in the case of SMCs adhering on the ECM (figure 3b, arrows) and Fib (data not shown) substrates, while the PDGF-BBr were more uniformly distributed throughout the spread morphology of SMCs adhering on St surface (figure 3a,c).
Figure 3.

PDGF-BBr immunolabelling of SMCs adhering on the different substrates. (a) TCP, (b) ECM, and (c) St. Arrows indicate areas of PDGF-BBr localization.
The distribution of β1-integrin receptors was also affected by the different substrate materials (figure 4a–c). In particular, SMCs adhering on the natural ECM substrate seemed to demonstrate receptors that were localized in proximity of focal adhesion points of the cell lamellipodia (figure 4b, arrows). By contrast, β1-integrin receptors appeared more localized in the nuclear region in the case of the SMCs adhering on TCP (figure 4a). The distribution of the β1-integrin receptors along the cell perimeter of the St-adhering cells may indicate an alteration in the cell membrane when in contact with this substrate (figure 4c, arrowheads). SMCs adhering on the Fib surface showed a pattern of the β1-integrin receptor with similarities to those observed on ECM (data not shown). All of the cellular studies were assessed over a 72 h time frame owing to the nature of the cells and their low proliferation rates observed in previous studies (data not shown).
Figure 4.

β1-Integrin immunolabelling of SMCs adhering on the different substrates. (a) TCP, (b) ECM, and (c) St. Arrows indicate areas of β1-integrin localization in SMCs adhering on ECM. Arrowheads show areas of β1-integrin localization in SMCs adhering on St.
In an attempt to provide further insights into the possible role played by SMCs in the formation of the HA-rich neointimal tissue associated with ISR, gene expression for HAS1, HAS2 and HAS3 was assessed. Figure 5 shows that the gene expression of the different HAS isoforms was differently affected by the various substrates and by PDGF-BB spiking. In particular, the gene for HAS1, the enzyme responsible for the synthesis of HA with a molecular weight up to 2 × 106, had similar levels of expression on all the substrates and it appeared to be increased by spiking the cells with PDGF-BB when the cells were adherent on Fib and St (figure 5, HAS1, lanes 6 and 8). A relatively inconsistent pattern of expression emerged when HAS2 gene expression was evaluated. TCP and Fib showed very faint bands both in the absence (figure 5, HAS2, lanes 1 and 2) and in the presence (figure 5, lanes 5 and 6) of PDGF-BB. In agreement, a consistent band was observed in the samples derived from SMCs adhering on St (figure 5, lanes 4 and 8). Unlike the HAS1 and HAS2, the gene for HAS3 showed no expression in cells that were grown on the synthetic surfaces, TCP (figure 3, HAS3, lanes 1 and 5) and St (figure 5, HAS3, lanes 4 and 8) with and without PDGF-BB spiking. Conversely, the two natural substrates Fib (figure 5, HAS3, lane 2) and ECM (figure 5, HAS3, lane 3) induced the expression of the HAS3 gene. This expression appeared to undergo a further stimulation with PDGF-BB spiking (figure 5, HAS3, lanes 6 and 7).
Figure 5.
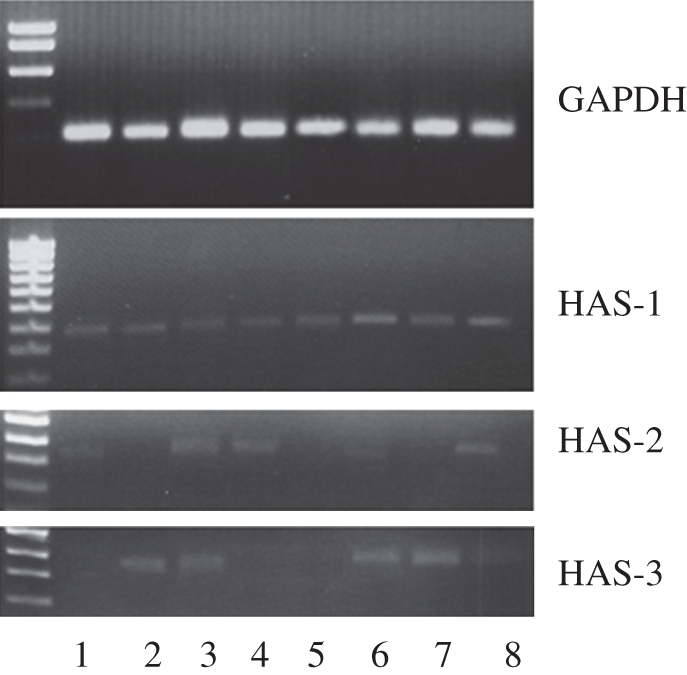
Gene expression of hyaluronan synthase isoforms (HAS1, HAS2, HAS3) in SMCs adhering on the different substrates with or without PDGF-BB (2 ng ml−1). Lane 1, TCP; lane 2, Fib; lane 3, ECM; lane 4, St; lane 5, TCP with PDGF-BB spiking; lane 6, Fib with PDGF-BB spiking; lane 7, ECM with PDGF-BB spiking; lane 8, St with PDGF-BB spiking. Ladder is shown on the left-hand side. GAPDH was used as the housekeeping gene.
4. Discussion
The expansion of both the balloon and the stent during angioplasty generates trauma, disrupting the integrity of the blood vessel endothelium [1,4,16]. While eliminating the occlusion produced by the atherosclerotic plaque, the device implantation triggers a repair process the consequence of which is the thickening of the media. Early blood clotting at the site of damage is followed by the activation of the inflammatory response [4,5]. Gradually, neutrophils and macrophages invade the traumatized tissue surrounding the stent struts and activate biochemical signalling pathways leading to a pathological pattern of SMC activation and de-differentiation [4]. This de-differentiation consists of a switch of the SMCs from a contractile to a secretory phenotype characterized by enhanced migration and proliferation activity. The final result is the thickening of the media and the formation of a clinically significant ISR [4].
Most investigations have focused on the role of biochemical signalling and shear stress in inducing this phenotypic switch in SMCs [8,13]. The effect of St on SMCs has been assessed as activation of thrombospondin-1-dependent TGF-β demonstrating the ability of the material ions to affect the activity of the SMCs [17]. In particular, it has been ascertained that PDGF is the most potent mitogen and chemoattractant for SMCs in cardiovascular pathogenesis [9,15]. PDGF is able to activate several signalling pathways that are known to transduce into the nucleus to stimulate cell proliferation [15]. All these signalling pathways are believed to be activated by the formation of a complex between the PDGF and its receptor and its internalization by the cell [15]. The binding of the ligand to its receptor initiates a series of events involving the synthesis of second messenger molecules, activation of small G proteins, protein phosphorylation and, finally, gene transcription. The downregulation of this mechanism is rather unclear. It has been postulated that it can be controlled in the short term by the ligand-induced internalization of the ligand/receptor complex and, later, by the ubiquitin-driven proteolysis of the internalized receptor and by the reduced expression of the PDGF-BBr mRNA [15]. Through this mechanism, SMCs seem to become less sensitive to PDGF-BB stimulus.
The effect of PDGF-BB stimulus on the ability of the SMCs to adhere to underlying substrates has also been investigated [11]. It has been found that spiking SMCs with PDGF-BB in vitro led to a decreased ability of the cell to adhere to laminin-5, a protein of the ECM. The adhesion of SMCs to the surrounding ECM is an important factor for the mutual regulation of ECM and cell functions. These functions are indeed controlled by specific adhesion receptors that serve as a transmembrane link between the ECM and the cytoskeleton [18]. As a consequence of this link, the ECM can control cell activity, while the cell can affect the remodelling of the ECM [18–24]. The most important adhesion receptors in this process are the integrins [18]. Although there is a large number of integrins expressed by the cells, in the case of SMCs, a key role seems to be played by the integrins α1β1, which are able to promote cell adhesion to collagen type I substrates [18]. In arterial SMCs, the β1 subunit of these integrins appears to be activated to enhance its affinity towards ECM ligands [21]. For these reasons, β1-integrin seems to play a fundamental role in the control of the SMC contractile phenotype and for the reorganization of the collagen matrix [18,21,23]. Conversely, in the case of injuries to the artery wall, this subunit may become deactivated, thus allowing the switch of the SMCs from a contractile to a migratory and proliferative phenotype [23]. Other studies such as integrin blocking have been considered but not performed in this study as they would have led to artefacts in cell morphology whilst offering no significant insight into cell behaviour.
In this paper, an in vitro model is presented that closely mimics the environment surrounding SMCs following stent implantation [5,16]. The model is based on the hypothesis that, following angioplasty-induced trauma, PDGF-BB stimuli could reach SMCs resident on the collagen-based ECM of the de-endothelialized artery wall as well as SMCs contacting the fibrin clot and the stent strut surface. The study of cell morphology supports the occurrence of a phenotypic switch in SMCs adhering on a typical stent material such as St. The experimental time point was set at 72 h to ensure adhesion and spreading of the cells on all the substrates as well as their proliferation following the synchronization of their cell cycle. Cytoskeleton staining of the SMCs adhering on St showed a loss of the spindle-like morphology by the cells, which is typical of their contractile phenotype; this morphology was preserved when the cells adhered on the natural ECM and Fib substrates. The de-differentiation of St-adhering SMCs into myofibroblast-like cells was further supported by their acquired proliferative potential. Although the cell numbers after 24 h of incubation were relatively low and not significantly different on the various substrates (data not shown), their density increased after 72 h. Indeed, SMCs adhering onto St had a density higher than that found on ECM and Fib. Moreover, SMCs adhering on St appeared more sensitive to the PDGF-BB stimulus. Therefore, it can be speculated that contact of SMCs with the stent surface under a PDGF stimulus may play a major role in the thickening of the neointima. This study indicates that neither the SMCs resident in the arterial wall nor those that accidentally contact thrombus material seem to be able to contribute significantly to the formation of the neointima.
The present work highlights the ability of St to induce morphological and molecular changes that, together with a higher proliferative capacity, infer a phenotypic change of the SMC. More specifically, an increase expression of PDGF-BB receptor and an altered distribution of the β1-integrin along the plasmalemma ridges were observed when cells were adhering on the metal substrate. The increased expression of PDGF-BBr would explain the significantly higher sensitivity of St-adhering SMCs to the PDGF-BB stimulus. The block in clathrin-mediated endocytosis of the PDGF-BB/PDGF-BBr complexes was achieved by the treatment of the cells in a hypertonic sucrose medium 15 min prior to PDGF-BB spiking [15]. This experiment showed that this blocking step was able to inhibit the effect of the PDGF-BB stimulus on all the tested surfaces. Furthermore, no downregulation of the cell proliferation was observed when the sucrose treatment was applied to the SMCs without PDGF-BB spiking. These results confirmed that the 0.2 M sucrose treatment specifically inhibits the effect of the PDGF-BB and not that exerted by the substrate. Previous studies have demonstrated that the ligand/receptor internalization specifically activates the mitogen-activated protein kinase (MAPK) pathways [15]. These studies have also elucidated that the phosphotyrosine dephosphorylation is the main inhibition pathway and that the receptor internalization plays a specific and positive role in MAPK transduction. The MAPK pathway is also known to be activated by the cell adhesion to the substrate through the integrins [25]. Therefore, it appears that the significantly higher rate of proliferation of the cells on both St and control TCP is a mechanism dependent on both the PDGF-BB/PDGF-BBr complex internalization and the integrin-mediated MAPK pathways. Data have been published linking the mutual effect of integrin and PDGF [19–21]. For example, it is well known that the PDGF-BBr undergoes inhibition in fibroblasts that have participated in the contraction of a collagen gel through integrin interactions [21]. Also, PDGF-stimulated fibroblasts show a rapid and transient change of the cytoskeleton organization and one of the main features of this change is the formation of membrane circular ruffles similar to those shown in figure 4b [26]. Furthermore, reports have suggested that integrin transducer signals converge with the responses stimulated by PDGF through its receptor [27]. In particular, it has been demonstrated that integrin-mediated adhesion processes induce an early and transient phosphorylation and internalization of the PDGF-BBr in fibroblasts [28].
HA has been shown to be one of the main components of the neointimal tissue formed in both atherosclerotic lesions and ISR [4,16]. HA is a polysaccharide composed of repeating d-glucuronic acid and β-1,3-N-acetylglucosamine-β1,4 units that is synthesized as polymers of different molecular weights by three isoforms of the enzyme hyaluronan synthase (HAS1, HAS2 and HAS3) [29–32]. In particular, HAS1 is constitutively expressed by the SMCs to produce high-molecular-weight HA (M.W. up to 2 × 106). HAS2 also synthesizes high-molecular-weight HA, but its expression in SMCs and myofibroblasts is found only when cells undergo specific external stimuli [29,30]. Conversely, HAS3 synthesizes a relatively lower molecular weight HA (M.W. from 2 × 105 to 3 × 105). It has been shown that HA of different molecular weights have different effects on tissue and immunocompetent cells [32]. High-molecular-weight HA has an anti-inflammatory and anti-proliferative action, whereas low-molecular-weight HA stimulates cell proliferation and it is involved in both angiogenesis and the inflammatory response [31,32]. Therefore, an alteration of the gene expression of the three HAS genes can play a fundamental role in cardiovascular pathology [33]. The data presented in this paper show that gene expression of the three HAS isoforms in SMCs respond differently both to the substrate stimuli and to the combined substrate/PDGF-BB stimuli [10,34]. HAS1 seemed to be equally expressed by SMCs irrespective of the substrate on which the cells were adhering and their expression seemed to be stimulated by the PDGF-BB. It is worth noting that HAS1 has been shown to synthesize and secrete only moderately low levels of HA [29]. Therefore, the contribution of this isoform to neointimal thickening might be limited. PCR experiments to evaluate HAS2 gene expression showed the presence of very low levels in SMCs on all substrates. Very faint or no band was visible in the case of SMCs adhering on TCP and Fib both with and without PDGF-BB spiking. In the case of cells adhering on ECM, spiking with PDGF-BB seemed to inhibit HAS2 gene expression. St-adhering SMCs were the only cells that showed a consistent expression of the HAS2 gene both with and without PDGF-BB stimuli. These results are not in agreement with those found by other research groups who indicate an induction of the HAS2 gene expression by PDGF-BB [34]. However, these experiments were performed at time intervals (0–24 h) relatively shorter than those evaluated in the present paper. Indeed, data by Evanko et al. [34], obtained from newborn human aorta SMC, seem to show a peak of HAS2 gene expression between 4 and 8 h in subconfluent cells grown on TCP. Later, a decline of HAS2 gene expression was observed. For these reasons, the expression of the HAS2 gene in proliferative SMCs adhering on St seems to emphasize even more the activation effect induced by the metal substrate. Finally, PCR analysis showed that the two artificial substrates, TCP and St, inhibited the expression of the HAS3 enzyme both in absence and in presence of PDGF-BB stimulus. The expression of the HAS3 gene was found in cells adhering on the two natural substrates, Fib and ECM, and stimulated by PDGF-BB stimulus. Overall, the PCR experiments seem to indicate that in the vascular environment altered by the angioplastic procedure, the contact of SMCs with the St stent struts can shift the balance towards the synthesis of high-molecular-weight HA. Previously published data showed that macrophages adhering on St acquire a post-inflammatory phenotype and undergo transdifferentiation into myofibroblast-like cells [5,6,31]. Therefore, it can be speculated that the synthesis of high-molecular-weight HA by St-adhering SMCs, in combination with a relatively low scavenger activity by the macrophages, may lead to an accumulation of this polysaccharide around the stent struts [35–38].
In conclusion, all the data collected in this paper seem to converge towards a substrate-dependent pathway of ISR development whereby SMC adhesion and phenotypic change by contact with St play an important role. It can be speculated that, following the trauma induced by both balloon expansion and the stent deployment, interactions between the stent material and the cells contacting its surface may lead to the stimulation of an HA-rich neointima. Indeed, the St clearly stimulated the SMCs to acquire a proliferative/secretory phenotype that is more likely to contribute to the deposition of excessive neointimal tissue. Conversely, the cells exposed to thrombus material do not seem to play a significant role in ISR.
References
- 1.Santin M., Colombo P., Bruschi G. 2005. Interfacial biology of in-stent restonosis. Exp. Rev. Med. Dev. 2, 429–443 10.1586/17434440.2.4.429 (doi:10.1586/17434440.2.4.429) [DOI] [PubMed] [Google Scholar]
- 2.Farb A., Sangiorgi G., Carter A. J., Walley V. M., Edwards W. D., Schwartz R. S., Virmani R. 1999. Pathology of acute and chronic coronary stenting in humans. Circulation 99, 44–52 [DOI] [PubMed] [Google Scholar]
- 3.McFadden E., et al. 2004. Late thrombosis in drug-eluting coronary stents after discontinuation of antiplatelet therapy. Lancet 364, 1519–1521 10.1016/S0140-6736(04)17275-9 (doi:10.1016/S0140-6736(04)17275-9) [DOI] [PubMed] [Google Scholar]
- 4.Burke A. P., Kolodgie F. D., Zieske A., Fowler D. R., Weber D. K., Varghese P. J., Farb A., Virmani R. 2004. Morphologic findings of coronary atherosclerotic plaques in diabetics: a post-mortem study. Arterioscler. Thromb. Vasc. Biol. 24, 1266–1271 10.1161/01.ATV.0000131783.74034.97 (doi:10.1161/01.ATV.0000131783.74034.97) [DOI] [PubMed] [Google Scholar]
- 5.Farb A., Weber D. K., Kolodgie F. D., Burke A. P., Virmani R. 2002. Morphological predictors of restenosis after coronary stenting in humans. Circulation 105, 2974–2980 10.1161/01.CIR.0000019071.72887.BD (doi:10.1161/01.CIR.0000019071.72887.BD) [DOI] [PubMed] [Google Scholar]
- 6.Stewart H. J. S., Guildford A. L., Lawrence-Watt D. J., Santin M. 2009. Substrate-induced phenotypical change of monocytes/macrophages into myofibroblast-like cells: a new insight into the mechanism of in-stent restenosis. J. Biomed. Mat. Res. 90A, 465–471 10.1002/jbm.a.32100 (doi:10.1002/jbm.a.32100) [DOI] [PubMed] [Google Scholar]
- 7.Harrison M., Siddiq A., Guildford A., Bone A., Santin M. 2007. Stent material surface and glucose activate mononuclear cells of control, type 1 and type 2 diabetes subjects. J. Biomed. Mat. Res. 83A, 52–57 10.1002/jbm.a.31204 (doi:10.1002/jbm.a.31204) [DOI] [PubMed] [Google Scholar]
- 8.Cospedal R., Abedi H., Zachary I. 1999. Platelet-derived growth factor-BB (PDGF-BB) regulation of migration and focal adhesion kinase phosphorylation in rabbit aortic vascular smooth muscle cells: role of phosphatidylinositol 3-kinase and mitogen-activated protein kinases. Cardiovasc. Res. 41, 708–712 10.1016/S0008-6363(98)00232-6 (doi:10.1016/S0008-6363(98)00232-6) [DOI] [PubMed] [Google Scholar]
- 9.Fu M., Zhang J., Zhu X., Myles D. E., Willson T. M., Liu X., Chen Y. E. 2001. Peroxisome proliferator-activated receptor γ inhibits transforming growth factor β-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with smad3. J. Biol. Chem. 276, 45 888–45 894 10.1074/jbc.M105490200 (doi:10.1074/jbc.M105490200) [DOI] [PubMed] [Google Scholar]
- 10.Stuhlmeier K. M., Pollaschek C. 2004. Differential effect of transforming growth factor β (TGF-β) on the genes encoding hyaluronan synthases and utilization of the p38 MAPK pathway in TGF-β-induced hyaluronan synthase 1 activation. J. Biol. Chem. 279, 8753–8760 10.1074/jbc.M303945200 (doi:10.1074/jbc.M303945200) [DOI] [PubMed] [Google Scholar]
- 11.Kingsley K., Rust W. L., Huff J. L., Smith R. C., Plopper G. E. 2002. PDGF-BB enhances expression of, and reduces adhesion to, laminin-5 in vascular smooth muscle cells. Biochem. Biophys. Res. Comm. 294, 1017–1022 10.1016/S0006-291X(02)00592-2 (doi:10.1016/S0006-291X(02)00592-2) [DOI] [PubMed] [Google Scholar]
- 12.Kosaki R., Watanabe K., Yamaguchi Y. 1999. Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 59, 1141–1145 [PubMed] [Google Scholar]
- 13.Li C., Xu Q. 2000. Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal. 12, 435–445 10.1016/S0898-6568(00)00096-6 (doi:10.1016/S0898-6568(00)00096-6) [DOI] [PubMed] [Google Scholar]
- 14.Werning F., Mayr M., Xu Q. 2003. Mechanical stretch-induced apoptosis in smooth muscle cells is mediated by β1-integrin signalling pathways. Hypertension 41, 903–911 10.1161/01.HYP.0000062882.42265.88 (doi:10.1161/01.HYP.0000062882.42265.88) [DOI] [PubMed] [Google Scholar]
- 15.Chiarugi P., et al. 2002. New perspective in PDGF receptor downregulation: the main role of phosphotyrosine phosphatises. J. Cell Sci. 115, 2219–2232 [DOI] [PubMed] [Google Scholar]
- 16.Guildford A. L., Colombo P., Bruschi G., Bonacina E., Klugmann S., Santin M. In press Direct comparison of the short-term clinical performance of Z Guidant and Taxus stents. Int. J. Cardiol. (doi:10.1016/j.ijcard.2008.12.161) [DOI] [PubMed] [Google Scholar]
- 17.Pallero M. A., Talbert Roden M., Chen Y. F., Anderson P. G., Lemons J., Brott B. C., Murphy-Ullrich J. E. 2010. Stainless steel ions stimulate increased thrombospondin-1-dependent TGF-beta activation by vascular smooth muscle cells: implications for in-stent restenosis. J. Vasc. Res. 47, 309–322 10.1159/000265565 (doi:10.1159/000265565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moiseeva E. P. 2001. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc. Res. 52, 372–386 10.1016/S0008-6363(01)00399-6 (doi:10.1016/S0008-6363(01)00399-6) [DOI] [PubMed] [Google Scholar]
- 19.Hollenbeck S. T., Itoh H., Louie O., Fariess P. L., Liu B., Kent K. C. 2004. Type I collagen synergistically enhances PDGF-induced smooth muscle cell proliferation through pp60src-dependent crosstalk between the α2β1 integrin and PDGFβ receptor. Biochem. Biophys. Res. Comm. 325, 328–337 10.1016/j.bbrc.2004.10.031 (doi:10.1016/j.bbrc.2004.10.031) [DOI] [PubMed] [Google Scholar]
- 20.Kappert K., Shmidt G., Doerr G., Wollert-Wulf B., Fleck E., Graf K. 2000. Angiotensin II and PDGF-BB stimulate β1-integrin-mediated adhesion and spreading in human VSMCs. Hypertension 35, 255–261 [DOI] [PubMed] [Google Scholar]
- 21.Lee R. T., Berditchevski F., Cheng G. C., Hemler M. E. 1995. Integrin-mediated collagen matrix reorganization by cultured human vascular smooth muscle cells. Circ. Res. 76, 209–214 [DOI] [PubMed] [Google Scholar]
- 22.Li S., Van den Diepstraten C., D'Souza S. J., Chan B. M. C., Pickering J. G. 2003. Vascular smooth muscle cells orchestrate the assembly of type I collagen via α2β1 integrin, RhoA, and fibronectin polymerization. Am. J. Pathol. 163, 1045–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen T. T. B., Ward J. P. T., Hirst S. 2005. β1 integrin mediate enhancement of airway smooth muscle proliferation by collagen and fibronectin. Am. J. Respir. Crit. Care Med. 171, 217–223 10.1164/rccm.200408-1046OC (doi:10.1164/rccm.200408-1046OC) [DOI] [PubMed] [Google Scholar]
- 24.Schwartz S. M. 1997. Smooth muscle cell migration in atherosclerosis and restenosis. J. Clin. Invest. 99, 2814–2817 10.1172/JCI119472 (doi:10.1172/JCI119472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howe A., Aplin A. E., Alahari S. K., Juliano R. L. 1998. Integrin signalling and cell growth control. Curr. Opin. Cell Biol. 10, 220231. 10.1016/S0955-0674(98)80144-0 (doi:10.1016/S0955-0674(98)80144-0) [DOI] [PubMed] [Google Scholar]
- 26.Xu J., Clark R. A. F. 1996. Extracellular matrix alters PDGF regulation of fibroblast integrins. J. Cell Biol. 132, 239–249 10.1083/jcb.132.1.239 (doi:10.1083/jcb.132.1.239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldschmidt M. E., McLeod K. J., Taylor W. R. 2001. Integrin-mediated mechanotransduction in vascular smooth muscle cells. Frequency and force response characteristics. Circ. Res. 88, 674–680 10.1161/hh0701.089749 (doi:10.1161/hh0701.089749) [DOI] [PubMed] [Google Scholar]
- 28.Sundberg C., Rubin C. 1996. Stimulation of β1 integrins on fibroblasts induces PDGF independent tyrosine phosphorylation of PDGFβ-receptors. J. Cell Biol. 132, 741–752 10.1083/jcb.132.4.741 (doi:10.1083/jcb.132.4.741) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allison D. D., Wight T. N., Ripp N. J., Braun K. R., Grande-Allen K. J. 2008. Endogenous overexpression of hyaluronan synthases within dynamically cultured collagen gels: implications for vascular and valvular disease. Biomaterials 29, 2969–2976 10.1016/j.biomaterials.2008.04.005 (doi:10.1016/j.biomaterials.2008.04.005) [DOI] [PubMed] [Google Scholar]
- 30.Jacobson A., Brinck J., Briskin M. J., Spicer A. P., Heldin P. 2000. Expression of human hyaluronan synthases in response to external stimuli. Biochem. J. 348, 29–35 10.1042/0264-6021:3480029 (doi:10.1042/0264-6021:3480029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang D., Liang J., Boble P. W. 2007. Hyaluronan in tissue injury and repair. Ann. Rev. Cell Dev. Biol. 23, 435–461 10.1146/annurev.cellbio.23.090506.123337 (doi:10.1146/annurev.cellbio.23.090506.123337) [DOI] [PubMed] [Google Scholar]
- 32.Volpi N., Schiller J., Stem R., Soltes L. 2009. Role, metabolism, chemical modifications and applications of hyaluronan. Curr. Med. Chem. 16, 1718–1745 10.2174/092986709788186138 (doi:10.2174/092986709788186138) [DOI] [PubMed] [Google Scholar]
- 33.Van den Boom M., et al. 2006. Differential regulation of hyaluronic acid synthase isoforms in human saphenous vein smooth muscle cells: possible implications for vein graft stenosis. Circ. Res. 98, 36–44 10.1161/01.RES.0000199263.67107.c0 (doi:10.1161/01.RES.0000199263.67107.c0) [DOI] [PubMed] [Google Scholar]
- 34.Evanko S. P., Johnson P. Y., Braun K. R., Underhill C. B., Dudhia J., Wight T. N. 2001. Platelet-derived growth factor stimulates the formation of versican–hyaluronan aggregates and pericellular matrix expansion in arterial smooth muscle cells. Arch. Biochem. Biophys. 394, 29–38 10.1006/abbi.2001.2507 (doi:10.1006/abbi.2001.2507) [DOI] [PubMed] [Google Scholar]
- 35.Deschrevel B., Tranchepain F., Vincent J. C. 2008. Chain-length dependence of the kinetics of the hyaluronan hydrolysis catalyzed by bovine testicular hyaluronidase. Matrix Biol. 27, 475–486 10.1016/j.matbio.2008.01.007 (doi:10.1016/j.matbio.2008.01.007) [DOI] [PubMed] [Google Scholar]
- 36.Evanko S. P., Angello J. C., Wight T. N. 1999. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 19, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 37.Sussmann M., Sarbia M., Meyer-Kirchrath J., Nusing R. M., Schror K., Fischer J. W. 2004. Induction of hyaluronic acid synthase 2 (HAS2) in human vascular smooth muscle cells by vasodilatory prostaglandins. Circ. Res. 94, 592–600 10.1161/01.RES.0000119169.87429.A0 (doi:10.1161/01.RES.0000119169.87429.A0) [DOI] [PubMed] [Google Scholar]
- 38.Wilkinson T. S., Bressler S. L., Evanko S. P., Braun K. R., Wight T. N. 2006. Overexpression of hyaluronan synthases alters vascular smooth muscle cell phenotype and promotes monocyte adhesion. J. Cell Physiol. 206, 378–385 10.1002/jcp.20468 (doi:10.1002/jcp.20468) [DOI] [PubMed] [Google Scholar]