

Abstract
Protein–protein interaction (PPI) prediction method has provided an opportunity for elucidating potential biological processes and disease mechanisms. We integrated eight features involving proteomic, genomic, phenotype and functional annotation datasets by a mixed model consisting of full connected Bayesian (FCB) model and naive Bayesian model to predict human PPIs, resulting in 40 447 PPIs which contain 2740 common PPIs with the human protein reference database (HPRD) by a likelihood ratio cutoff of 512. Then we applied them to exploring underlying pathway crosstalk where pathways were derived from the pathway interaction database. Two pathway crosstalk networks (PCNs) were constructed based on PPI sets. The PPI sets were derived from two different sources. One source was strictly the HPRD database while the other source was a combination of HPRD and PPIs predicted by our mixed Bayesian method. We demonstrated that PCNs based on the mixed PPI set showed much more underlying pathway interactions than the HPRD PPI set. Furthermore, we mapped cancer-causing mutated somatic genes to PPIs between significant pathway crosstalk pairs. We extracted highly connected clusters from over-represented subnetworks of PCNs, which were enriched for mutated gene interactions that acted as crosstalk links. Most of the pathways in top ranking clusters were shown to play important roles in cancer. The clusters themselves showed coherent function categories pertaining to cancer development.
Keywords: Bayesian model, protein–protein interaction, pathway crosstalk network, cancer
1. Introduction
The widespread studies of large-scale protein–protein interactions (PPIs) has enabled opportunities for understanding complicated biological processes and molecular characterization of human diseases [1–6]. Many human protein interaction databases have been constructed based on curation of high-throughput data and literature, including the human protein reference database (HPRD) [7], DIP [8], MINT [9], IntAct [10], BioGrid [11], etc. However, these datasets cover only a considerable limited range of PPI data. Previous studies have made much effort in predicting protein interactions of different species, involving diverse types of data and methods.
The Bayesian probabilistic model has been widely used in predicting PPIs by integrating heterogeneous datasets. So far three main categories including sequence-based, high-throughput-based prediction methods and also a combination of them have been applied to PPI prediction. However, text-mining methods can provide a broad view of datasets and have not been thoroughly explored. Xia et al. [5] constructed a probabilistic model by integrating seven types of data, including physical interactions from model organisms based on orthology, genetic interactions and phenotype data of model organisms, coexpression, domain–domain interactions (DDIs), gene context information and biological function annotations. All the datasets were integrated by a simple naive Bayes model and resulted in 180 010 human PPIs, which were stored in IntNetDB. A recent study by Franke et al. [2] addressed an approach integrated by naive Bayesian method and FCB method, covering shared biological functions, coexpression, physical interactions derived from both human and model organisms. The Barton group predicted over 37 000 human PPIs and constructed the human protein–protein interaction prediction database (PIPs) by combing the features of gene expression, orthology of PPIs, subcellular localization, domain and post transcriptional modification (PTM) co-occurrence, disorder and topology of predicted PPI networks [12]. Besides human PPI prediction, a framework for predicting protein interactions of Arabidopsis thaliana has been proposed [1]. Similar to the approach proposed by Franke et al. [2] and McDowall et al. [12], a combination of naive Bayesian method and FCB method was used in our study. Gene expression, biological functional annotation, physical interactions from model organisms and human disease phenotype data are combined by naive Bayesian method, whereas some datasets derived from the same type of data were combined by the FCB method.
Though several research studies achieved the aim of PPI prediction, less attention has been paid to usage of those inferential PPI sets. A gene–gene interaction dataset has been applied to prioritizing disease candidate genes in susceptibility loci; the results showed good performance [2]. The most attractive character of the predicted PPI set is that these interactions can imply more potential information than insufficient interactions curated from many high-quality PPI databases when they are applied to the same analysis. Li et al. [3] proposed a method for identifying pathway interactions by combing pathway data and PPI data originated from HPRD, MINT, BIND and Reactome, which relied on the assumption that significantly more protein interactions would be found between two pathways than by chance. In order to explore underlying pathway interactions and test the effect of protein interaction set on this method, we use the combination of HPRD and part of the predicted PPI set with an accuracy of 80 per cent to identify pathway crosstalk between pathways extracted from the pathway interaction database (PID) [13], which involves signalling and regulatory pathways from NCI-Nature, Biocarta (http://www.biocarta.com/) and Reactome [14]. We further make the analysis of pathway crosstalk under disease conditions. Known mutated genes in cancers were mapped to protein interactions between significant pathway interaction pairs, the result indicated a key role of pathway crosstalk in the pathogenesis of cancer.
2. Material and methods
2.1. Bayesian network construction
2.1.1. Gold standard
We built the gold standard positive (GSP) dataset from human protein reference database (HPRD), which stored 34 998 interactions among 9303 proteins. The gold standard negative (GSN) dataset was constructed by randomly selected protein pairs from 56 059 human proteins in the Uniprot database; the ratio of negative and positive pairs is 100. As a result, we obtained a GSN set composed of 3 495 977 gene pairs by converting identifiers of 3 l499 800 protein pairs and removing redundant pairs or pairs overlapped with GSP. We used the likelihood ratio (LR) to evaluate the confidence level of gene pairs. The details are shown in §2.1.10.
2.1.2. Gene expression
We collected 32 published gene expression datasets comprising more than 5700 microarray profiles of diverse tissues and differentiation status from Gene Expression Ominibus (http://www.ncbi.nlm.nih.gov/geo/) and Oncomine [15] (see electronic supplementary material, table S1). Within each dataset, genes with more than 5 per cent missing values were filtered out and all expression values were log2 transformed. Each feature was mapped to the Entrez Gene identifier. For multiple features assigned to the same Entrez Gene identifier, we chose the one with the least missing values; for several features assigned to the same Entrez Gene identifier with the least number of missing values or without any missing values, the average value of these features was assigned to the gene. Missing values were imputed by the k-nearest neighbour imputation method.
Similar expression patterns of genes usually indicates the potential for protein interaction. Genes which are coexpressed always participate in the same biological process or constitute a transcription module. For each dataset, we calculated the Pearson correlation coefficient (PCC) between each pair of genes. We selected three high quality datasets which have a positive correlation with increasing coexpression and strongest LRs. The three datasets consisted of expression profiles of primary breast cancer (GSE12276) [16], acute myeloid leukaemia (GSE10358) [17] and various types of cancer samples (GSE2109) (http://www.intgen.org/). A meta-analysis method [18] was employed to independent datasets for different types of diseases. We measured the correlation between genes by effect size and transformed it back into the correlation coefficient. The average effect size is represented by μ, the observed effect size and sampling error for independent dataset k can be represented by zk and within-study variance 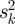 , where between-study variance τ2 represents the variability between datasets.
, where between-study variance τ2 represents the variability between datasets.
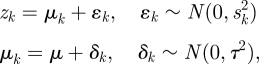 |
where 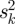 is given as
is given as 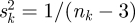 and τ2 is estimated by using the Cochran Q-statistic.
and τ2 is estimated by using the Cochran Q-statistic.
PCC of genes gx and gy is represented by rk, which is converted into zk by using Fisher's r to z transformation as follows:
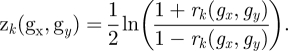 |
The average effect size zR and its variance wk are estimated as follows:
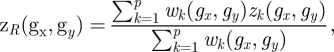 |
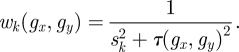 |
Finally, Fisher's z to r transformation is employed to reconvert the effect size back into the correlation coefficient as follows:
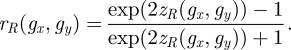 |
2.1.3. Physical protein–protein interactions
We collected high-throughput interaction data of three model organisms, including Sacchromyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster from DIP, MINT, IntAct and BioGRID. We mapped model organism protein interaction pairs to human orthologue protein pairs by using Inparanoid [19], which provided confidence scores (inparalogue value) for multiple orthologue proteins of diverse organisms in the same cluster. Moreover, many lower eukaryote genes have several co-orthologues in humans which were identified by Inparanoid. For each protein interaction pair presented in a model organism, the confidence scores of both proteins were summed up with the confidence scores of predicted orthologue human protein pairs, so the confidence score of a predicted human protein pair should range from 0 to 4. For protein interaction pairs obtained and predicted in every model organism, we classified the interaction pairs into the high confidence bin when they reached a perfect score of 4 so other pairs with a score less than 4 or missing in one of three organisms were treated as the low confidence bin. Then, we used the FCB method to divide all interaction pairs which were present in at least one organism dataset into four bins based on their frequencies of high confidence or low confidence score in three organisms.
2.1.4. Biological functional annotation
Based on the knowledge that proteins sharing more specific biological functional annotation are more likely to have interaction relationships, we used a traditional method for evaluating the similarity of gene function. The Gene Ontology Association file in the Gene Ontology Consortium [20] was downloaded in June 2009, which assigned 2698 molecular function terms and 4722 biological process terms to 15 298 and 14 256 genes, respectively. Then we found the smallest shared molecular function (SSMF) term and biological process (SSBP) term for each pair of proteins, and mapped the numbers of genes which are annotated to the SSMF or SSBP to the protein pair.
2.1.5. Human phenotype
Large-scale RNA interference screens have been used for predicting protein interactions for model organisms, e.g. D. melanogaster, C. elegans and S. cerevisiae. In the traditional method, phenotype data of model organisms were transferred to human genes by orthology mapping. In this research, we adopted a method without using phenotype data from other species but directly from humans. Using the method for humans phenotype similarity analysis based on text-mining [21], we obtained the phenotype similarity scores (range from 0 to 1) of all pair-wise combinations between 2055 disease phenotype records derived from the Online Mendelian Inheritance in Man database (http://www.ncbi.nlm.nih.gov/omim/). The causative genes or proteins of phenotype records were known. Then we calculated the phenotype similarity between a gene pair by taking the maximum value of the phenotype similarity value matrix, the columns and rows of which were composed of phenotype terms for each gene.
2.1.6. Domain–domain interaction
Domain–domain interactions (DDIs) can be inferred from protein interactions and they were also used to predict protein interactions in previous research [5]. The Interdom database provides potential domain interactions by combining data from multiple data sources, involving domain fusions, protein interactions, complexes and literature [22]. All the PPI pairs were transformed to DDI pairs with DDI values in InterDom. As many proteins contain more than one domain, a pair of proteins can be transformed into all possible combinations of domains derived from both proteins. Finally, we chose the maximum value to represent the domain interaction value of a PPI pair and grouped values into three bins.
2.1.7. Co-occurrence of post-translational modification pairs
PTM annotation was downloaded from dbPTM and HPRD. dbPTM compiles experimentally validated PTM sites from Swiss-Prot, PhosphoELM, O-GLYCBASE and Ubiprot. Each amino-acid categorizing PTM was mapped to the main types of PTM which were supplied by dbPTM and HPRD, resulting in 16 289 PTM annotations composed of 11 561 distinct genes and 61 PTM types. Similarly, the PTM enrichment values can be assigned to PPI pairs in the same way as described in §2.1.6. The LR was assessed by the PTM pair enrichment score which was introduced by Scott & Barton [23], all enrichment scores were grouped into three bins.
2.1.8. Genetic interaction
Synthetic genetic array (SGA) analysis has been performed on genome-wide scale mapping of yeast genetic interactions. In previous studies, genetic interactions have been found to exhibit a significant association with physical interactions [5,24]. We downloaded a SGA genetic interaction dataset with a lenient cutoff from DRYGIN [25], covering over 500 000 yeast genetic interactions with Array Genetic interaction scores. First, all the genetic interactions of yeast were mapped to human gene pairs. Second, we grouped genetic interaction pairs into three bins (lenient, intermediate and stringent) set by DRYGIN.
2.1.9. Regulation of common transcriptional factors
Based on the assumption that proteins which are close to each other in a PPI network are prone to be regulated by the same set of transcriptional factors (TFs) [26], the number of TFs shared by a pair of proteins was used to calculate the likelihoods. The experimentally proven binding sites or regions of regulated genes and TFs were downloaded from TRANSFAC. Three bins were defined for human protein pairs, the corresponding genes of which share common TFs in human, rat, mouse and fly, involving 442 TFs and 18 139 target genes in all.
2.1.10. Bayesian network model and performance evaluation
All the evidence was integrated by either a naive Bayesian model or an FCB model, for some evidence were dependent on each other and some were totally independent. In general, we integrated the evidence which derived from different data sources but belonged to the same data type by FCB method, including physical protein interactions from different organisms and biological functional annotation. Moreover, we integrated PTM co-occurrence and DDI by the FCB method [23]. Finally, we integrated the seven modules by the Naive Bayesian model.
As P(pos) is defined as the possibility of finding a interaction relationship between two proteins and P(neg) means the possibility of finding a pair of non-interaction proteins, the prior odds of finding an interaction pair is represented by Oprior; similarly, Oposterior means the odds of finding an interaction pair when evidence i to n is considered. The prior odds and posterior odds of finding a positive pair are calculated as follows:
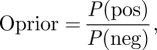 |
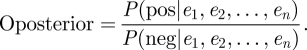 |
The likelihood ratio L is defined as below:
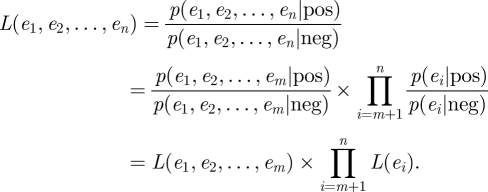 |
The relation between Oprior and Oposterior is defined as
We applied a 10-fold cross-validation to evaluating the performance of this PPI prediction method through randomly dividing the GSP set and GSN set into 10 sets separately. After each of the 10 sets was tested by training the other nine sets, we obtained the counts of true positive (TP), false positive (FP), true negative (TN) and false negative (FN). Apparently, sensitivity (TP/(TP + FN)), specificity (TN/(TN + FP)) and TP/FP ratios can be calculated.
2.2. Pathway crosstalk network construction
PID (http://pid.nci.nih.gov) is a growing collection of human signalling and regulatory pathways curated from peer-reviewed literatures and stored in a computable format [13]. PID aims to provide predefined pathways and allow novel networks composed computationally to be explored from the universe of interactions underlying the predefined pathways. The focus on signalling and regulatory pathways makes PID, HumanCyc [27] and KEGG [28] different. As our interest mainly pointed to the relationships between signalling pathways, the PID-specific XML format of the principle data source ‘NCI-Nature curated’ together with two other data sources, Biocarta and Reactome pathway collections, were downloaded and then processed.
Proteins, genes, RNAs and interactions among them in subnet pathways were integrated to their parent pathways according to the hierarchy structure of NCI-Nature curated pathways and Reactome pathways in PID. We obtained 99 non-subnet pathways and 65 subnet pathways from NCI-Nature curated, 254 non-subnet pathways from Biocarta, 70 non-subnet pathways and 827 subpathways from Reactome. All three sources of pathways cover a total of 5127 genes.
We combined 34 998 PPIs from HPRD with 40 447 predicted interactions (precision ≈ 80%, log2 (LR) > 9) and finally ended up with 72 705 interactions without self interactions or duplicate pairs.
The pathway crosstalk network was constructed by the method proposed by Li et al. [3]. The details are shown below.
First, we removed the pathways containing fewer than five genes, but no upper limit. This cutoff had the large pathways composed of several subnet pathways preserved; there was a slight difference from Li et al.'s method for they removed pathways containing more than 100 genes. Therefore, we preserved the interactions between large pathways, the subnet pathways of which might not be able to have interactions with other pathways. Evaluation of gene overlaps between the rest of the pathways was performed by Fisher's exact test and p-values were adjusted by false discovery rate (FDR) Benjamini–Hochberg (BH) procedure [29].
Second, the real interaction count n between a pair of pathways was calculated based on interactions between genes which were only contained in those two pathways separately; N represented the number of total interaction counts of all pathway pairs.
Third, the significance of interaction between every pair of pathways was estimated by randomly replacing all the genes that participated in at least one interaction with genes which have identical degrees; as both pathways were permutated 1000 times; the average count of interactions between two pathways was recorded as r. In addition, the average of total interaction counts of all pathway pairs was recorded as R to correspond to N. Then, we performed the one-sided Fisher's exact test on all pathway pairs by using the 2 × 2 contingency table, which consisted of n, N–n, r and R–r. The p-values were adjusted by performing the FDR BH procedure [29] and pathway pairs with significantly higher ratio of n to N compared to the ratio of r to R (p-value <0.05) were recognized as the final result. What is more, significant overlapped pairs should be excluded from this result for relatively high similarity of biological functions between two pathways if they had many genes in common.
In this section, we used two different interaction sets as randomization background. One set was the high quality 34 998 PPI set supplied by HPRD, the other one was the mixed interaction set which was collected from HPRD and our predicted human protein interaction set. The mixed set included nearly as much as twofold of interactions in HPRD set. We used different interaction datasets to figure out whether some underlying pathway crosstalk could be discovered and the possibility of finding false positive interactions between pathways owing to insufficient interactions.
Fourth, we processed the pathways which interacted with overlapped pathways or both subnet pathway and parent pathway. In order to remove the redundant relationships with similar pathways in different databases and preserve the crosstalk with large pathways, especially for parent pathways, we deleted the interaction between smaller pathways A and B when A and C shared the same partner B and more than 75 per cent of A genes belonged to C.
2.3. Identify pathway interactions enriched for disease gene interactions
For the purpose of detecting important pathway crosstalk in diseases, we found the significant pathway pairs which were linked by mutated genes in cancers and ranked by counts of mutated gene pairs or corrected p-values.The enrichment analysis was performed by the hypergeometric test, then p-values were adjusted by the FDR BH procedure [29].
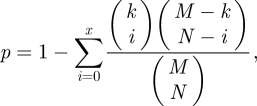 |
where x is the number of cancer protein pairs between a significant pathway pair, k the number of interactions between this pathway pair, M the number of interactions of PPI set used for pathway crosstalk analysis and N the total number of cancer gene pairs appearing in a PPI set.
The somatic mutated genes in cancers were obtained from the Sanger Institute Catalogue Of Somatic Mutations (http://www.sanger.ac.uk/cosmic), which is the most comprehensive public resource for information on somatic mutations in human cancer [30]. Part of the contents of COSMIC are derived from manual curation of the scientific literature for nominated genes from the Cancer Gene Census (CGC); the other genes are confirmed somatic mutations derived from the Cancer Genome Project (CGP), which focuses on tumour resequencing. We downloaded a full table of COSMIC genes which covered 3277 genes (including 481 CGC genes) by Biomart (http://www.sanger.ac.uk/genetics/CGP/cosmic/biomart) and extracted 25 mutated brain cancer genes in the CGC list.
3. Results
3.1. Bayesian network
We plotted the receiver operating characteristic (ROC) curve of the coexpression meta-analysis method; the area under the curve (AUC), which reached 60 per cent, measured it. A clear and strong correlation was observed between PCC and LR (see electronic supplementary material, table S2).
The performance of orthology mapping of physical protein interactions was shown by AUC of 62.4 per cent. We used the FCB method to divide all interaction pairs into four bins based on their frequencies of high confidence (score sum = 4) or low confidence score (score sum <4) in three organisms (see electronic supplementary material, table S3). When we classified interaction pairs in each organism into more complicated bins, such as a combination of a high confidence (score sum = 4), a medium-high confidence bin (3 = score sum <4) and a low confidence bin (score sum <3), the result did not perform better in 10-fold cross-validation. Therefore, we chose the simple classification method with lower calculation complexity in FCB method.
As we considered molecular function and biological process terms simultaneously, the FCB method was needed. The strong correlation which could not be shown by either SSBP or SSMF alone was found by considering both of them (see electronic supplementary material, table S4). The AUC was 75.4 per cent, compared with 65 per cent for the biological process only.
The correlation between human phenotype similarity and LR was very clear, so this method could be used for PPI prediction (see electronic supplementary material, table S5). Although the classifier did not work very well using genetic interactions of yeast (AUC = 57.3%) or phenotype similarity (62.3%) alone, they can be improved significantly by combining with orthologue mapping of the physical interaction of model organsisms (AUC = 72%) (figure 1a). What is more, there were only a small number of common pairs between physical protein interaction pairs and pairs with phenotype similarity scores, so the two prediction methods could be suitable for supplementating each other.
Figure 1.

Prediction performance. (a) ROC curve of different methods for PPI prediction. ‘Orth + Phen + Gene’ represents the combination of orthologue mapping of physical protein interactions from model organisms (Orth), phenotype similarity (Phen) and Genetics interaction (Gene); ‘TF + Coexp’ represents the combination of regulation of common transcriptional factors (TF) and meta-analysis of coexpression (Coexp); ‘Func + Dom + PTM’ represents the combination of shared biological functional annotation (Func), Interaction of domains (Dom), Co-occurrence of post translational modification pair (PTM); ‘All’ represents the integration of all methods. (b) log2 (LR) cutoffs versus precision of PPI prediction.
The LR of combination of DDIs and co-occurrence of PTMs method showed a clear correlation with DDI values and enrichment scores of co-occurrence of PTMs (AUC = 70.3%) (see electronic supplementary material, table S6). As this feature was combined with the functional annotation feature, the performance was improved by 7 per cent (figure 1a).
The number of common TFs shared by gene pairs exhibited a clear but still weak correlation with the LR; unsurprisingly, the prediction performance of this method is not very strong (AUC = 56.2%) (see electronic supplementary material, table S7). Therefore, we integrated the TF feature with the coexpression feature, resulting in a slight improvement in prediction performance (AUC = 62%; figure 1a).
Datasets which were used for coexpression meta-analysis covered almost 70 per cent of human genes, sharing over 15 000–16 000 genes with TF, functional annotation and DDI–PTM features. Obviously, it can be concluded that most protein pairs were supported by evidence of at least four interactions. The performance of all sources of data integrated by the naive Bayesian method showed a good performance (AUC = 80%) (figure 1a). The result showed that we could possibly obtain more accurate predictions if research covered more genes/proteins, such as human phenotype data.
The genetic interactions of yeast were assigned to three bins by scores obtained from DRYGIN which showed a clear but weak correlation with LR (see electronic supplementary material, table S8).
When we set the threshold of overall log2 (LR) as 9, a total of 40 447 predicted PPIs were obtained with a precision of 80 per cent. The correlation between precision and log2 transformed LR is shown (figure 1b).
As we set different LR cutoffs (LLR ranged from 6 to 12) for generating the predicted PPI sets with different confidence levels, the overlapped parts of our predicted PPIs and data derived from other databases are shown (figure 2a,b). Among four human protein interaction databases which supplied experimentally verified or literature-derived PPIs, HPRD had the most overlapped PPIs with our predicted PPI sets derived from all the LR cutoffs, followed by BioGrid. When the LR cutoff equals 512 (LR512 PPI set), a total of 2740 and 1875 PPIs were overlapped with HPRD and BioGRID, respectively (figure 2a). In addition, we also made a comparison of our results with other predicted PPI datasets, such as PIPs. After the conversion of identifiers, a total of 55 209, 27 133 and 18 827 PPIs with Entrez IDs were recovered from PIPLR100 (score = 0.25), PIPLR400 (score = 1) and PIPLR1000 (score = 2.5), which were downloaded from PIPs. As PIPLR100 includes the most PPIs in PIPs, it contains the most common PPIs with LR512 PPI set (12 297 overlapped PPIs; figure 2b). When we considered the comparative threshold for obtaining the predicted PPIs, PIPLR400 should be a good reference, which contains 6821 common PPIs with the LR512 PPI set (figure 2b). What is more, the numbers of PPIs obtained by setting the different cutoffs of LR were also shown (figure 2c).
Figure 2.

Overlap of PPIs in different databases and predicted PPIs. (a) Overlaps of predicted PPIs derived from different LR cutoffs and human PPI databases (HPRD, black bar; BioGRID, dark grey bar; MINT, white bar and IntAct, light grey bar). (b) Overlaps of Predicted PPIs derived from different LR cutoffs and Predicted PPIs with different posterior odds cutoffs in PIPs database; black bar, PIPLR100, dark grey bar, PIPLR400 and light grey bar, PIPLR1000 represent the PPIs datasets derived from the posterior odds cutoffs of 0.25, 1 and 2.5 separately. (c) The number of predicted PPIs under the condition of increasing LR cutoffs.
In a previous study, an interspecies comparison of PPI data from yeast, worm, fly and human was performed to identify conserved interactions; the result indicated that the overlap between those four species was relatively low [31]. In this paper, we used the PPI sets in §2.1.3 and treated the PPIs with summation of more than 3 (confidence score in Inparanoid) as high-confidence human orthologues, including 62 554 yeast, 11 021 fly and 3255 worm interactions. The orthologue PPIs from yeast, fly and worm were compared with the LR512 PPI set; 47 PPIs were common to the worm, fly and human while 20 PPIs were common to the all four species.
We collected 34 998 PPIs from HPRD and combined them with 40 447 predicted PPIs. Finally, there were 72 705 interactions without self-interactions and duplicated interactions. The mixed PPI set was used for constructing a pathway crosstalk network.
3.2. Pathway crosstalk network
Before the fourth procedure of pathway crosstalk network construction was performed, 13 148 pathway pairs (almost 2.5% of all possible pairs) overlapped significantly with p-values less than 0.05 after FDR adjustment. After removing all the significant overlapped pathway pairs and underlying redundant pathway pairs from the original result of significant pathway interactions, we obtained 10 081 interactions between 788 pathways based on the mixed PPI set (see the electronic supplementary material, table S9); similarly, 422 pathways comprised 1326 interactions based on the HPRD PPI set (see electronic supplementary material, table S10).
We obtained two pathway crosstalk networks based on mixed PPI set and HPRD PPI set, so we called them mixPCN and HPRDPCN for short. To make a comparison of these two networks, we analysed their topological characters, including average degree, average shortest path, average cluster coefficient and degree distribution fitting line parameters. As the PPI set increased from HPRD to mixed PPI, the average degree and cluster coefficient of pathways increased significantly, while the average shortest path decreased by nearly one. The degree of distribution of these two PCNs followed a power-law distribution (table 1).
Table 1.
Comparison of topological statistical of PCNs.
| PCN | mixLPCN | HPRDLPCN |
|---|---|---|
| average degreea | 25.586 | 6.280 |
| average SPb | 3.289 | 4.135 |
| average CCc | 0.188 | 0.083 |
| power-law γ (R2)d | −0.957(0.904) | −1.41(0.920) |
| edges (nodes)e | 10 081(788) | 1325(422) |
aThe average degree of pathways.
bThe average shortest path distance between pathway pairs.
cThe average clustering correlation of pathways.
dDegree distribution of pathways.
eThe counts of pathways and interactions they consist of.
3.3. Pathway pairs enriched for interactions between mutated genes of cancers
3.3.1. Top ranking and clusters of pathway pairs enriched for interactions between mutated genes in cancer
Most of the pathway pairs were enriched for mutated gene interactions which acted as crosstalk links between pathway pairs. In mixPCN and HPRDPCN, mutated gene pairs were significantly enriched in 6489 and 1096 pathway pairs, respectively (see electronic supplementary material, tables S11 and S12). We mainly paid attention to the pathway pairs which ranked top by decreasing counts of mutated gene pairs or by increasing p-values (tables 2 and 3). Then we applied MCODE algorithm [32] to get densely connected clusters in those over-represented subnetworks of mixPCN and HPRDPCN, resulting in 11 and eight clusters separately (see electronic supplementary material, tables S13 and S14). As MCODE was developed to detect molecular complexes based on vertex weighting by local neighbourhood density, we attempted to use this method to detect densely connected pathway clusters which might cooperate in special patterns under disease conditions.
Table 2.
Top 10 over-represented pathway interactions enriched for interactions between mutated genes in cancer in mixPCN (ranked by decreasing counts of mutated gene pairs).
| pathway A | pathway B | mutationa(allb) |
|---|---|---|
| IFN-γ pathway | class I PI3K signalling events | 238(555) |
| class I PI3K signalling events mediated by Akt | TGF-β receptor signalling | 225(624) |
| class I PI3K signalling events mediated by Akt | regulation of cytoplasmic and nuclear SMAD2/3 signalling | 225(624) |
| ErbB receptor signalling network | IL-1-mediated signalling events | 212(445) |
| proteogylcan syndecan-mediated signalling events | TRAIL signalling pathway | 202(557) |
| class I PI3K signalling events | coregulation of androgen receptor activity | 196(414) |
| proteogylcan syndecan-mediated signalling events | IFN-γ pathway | 187(573) |
| class I PI3K signalling events mediated by Akt | androgen-mediated signalling | 183(470) |
| BMP receptor signalling | integrins in angiogenesis | 177(354) |
| androgen-mediated signalling | IL-1-mediated signalling events | 173(442) |
aNumber of interactions between mutated genes in cancer existing between pathways A and B.
bNumber of all gene interactions existing between pathways A and B.
Table 3.
Top 10 over-represented pathway interactions enriched for interactions between mutated genes in cancer in mixPCN (ranked by increasing p-values of mutated gene pairs).
| pathway A | pathway B | mutationa(allb) |
|---|---|---|
| class I PI3K signalling events mediated by Akt | syndecan-1-mediated signalling events | 143(407) |
| sphingosine 1-phosphate (S1P) pathway | p75(NTR)-mediated signalling | 136(291) |
| sphingosine 1-phosphate (S1P) pathway | LPA receptor mediated events | 136(262) |
| p53 pathway | regulation of androgen receptor activity | 125(251) |
| ErbB receptor signalling network | BCR signalling pathway | 106(177) |
| angiotensin II mediated activation of JNK pathway via pyk2 dependent signalling | signalling events mediated by Stem cell factor receptor (c-Kit) | 103(153) |
| glypican pathway | IFN-γ signalling pathway | 103(153) |
| integrins in angiogenesis | keratinocyte differentiation | 97(166) |
| ephrinA-EPHA pathway | BCR signalling pathway | 89(139) |
| signalling events mediated by stem cell factor receptor (c-Kit) | growth hormone signalling pathway | 84(118) |
aNumber of interactions between mutated genes in cancer existing between pathways A and B.
bNumber of all gene interactions existing between pathways A and B.
The top cluster which was ranked by MCODE score was nearly fully connected (one included 29 nodes and 380 edges) in over-represented subnetworks of mixPCN. Unsurprisingly, the top ranking clusters in subnetworks were related to different cell signalling events in development of cancers. Cluster 1 was related to angiogenesis, inflammation and immune response; cluster 2 (27 nodes and 204 edges) was related to cell adhesion, migration and immune response. Clusters 3–5 might implicate the transcription regulation, differentiation, cell cycle and apoptosis process (see electronic supplementary material, table S13).
When our method was applied to HPRDPCN, more than half of all the pathways in the top two clusters (see electronic supplementary material, table S14) were conserved in cluster 1 of over-represented subnetworks of mixPCN (see electronic supplementary material, table S13). It showed that even the size of two networks differed a lot, the overlap between over-represented subnetworks of HPRDPCN and mixPCN which accounted for about 71 per cent (774/1096), implying a common network topology structure of crosstalk networks and conserved cooperation pattern between pathways. However, the result of ranking pathway crosstalk enriched for cancer-mutated gene pairs can be influenced a lot by using different PPI sets. For example, the crosstalk between ‘P53 pathway’ and ‘regulation of androgen receptor activity’ ranked fifth in the over-represented subnetwork of mixPCN while it only ranked 20th in the over-represented subnetwork of HPRDPCN by ranking the counts of mutated gene pairs. The difference might be owing to the different numbers of total protein interactions and mutated gene pairs (251 PPIs and 125 mutated gene pairs in mixPCN,167 PPIs and 76 mutated gene pairs in HPRDPCN) which were recognized between the two pathways based on mixed PPI set and HPRD set. In general, the size of the PPI set can play an important role in constructing PCN.
Class I PI3K signalling events, Class I PI3K signalling events mediated by Akt, IFN-γ pathway, proteogylcan syndecan-mediated signalling events, IL-1-mediated signalling events and androgen-mediated signalling pathways had crosstalk with equal or more than two pathways in the top 10 crosstalk through ranking counts of mutated gene pairs in over-represented subnetwork of mixPCN (table 2).
Most of the crosstalk in this list can be supported by publications. The PI3K signalling pathway is important in regulating the balance of decisions in cell growth, proliferation and survival. Recent study has shown that PI3K can regulate IFN signalling by controlling both transcription and translation of IFN-stimulated genes [33]; what's more, the process of PI3K regulating transcription of a subset of IFN-α-stimulated genes may be involved in the induction of apoptosis [34]. As the PI3K/Akt signalling pathway and the androgen receptor are both involved in regulation of prostate cancer cell proliferation and survival, recent research has suggested that these two pathways might cooperate to regulate prostate tumour development and progression [35]. The crosstalk between IL-1-mediated signalling events and androgen-mediated signalling has been implicated in some research. IL-1 can induce neuroendocrine differentiation, which is associated with androgen independence and survival in prostate cancer [36,37]. Syndecan-2, which functions in proteogylcan syndecan-mediated signalling pathways, has been reported to be highly expressed in the microvasculature of mouse glioma [38].
Besides the crosstalk enriched for most mutated gene pairs, the most significant crosstalk should be considered. For example, the p53 signalling pathway, which is one of the most famous cancer signalling pathways, can be ranked in the top 10 by p-value ranking method, but did not show up in top 10 of crosstalk enriched for most mutated gene pairs. In table 3, the Sphingosine 1-phosphate (S1P) pathway participated in two of the top three crosstalks, this might indicate its important role.
Previous research has proved that the uncontrolled increase in S1P, which has emerged as a growth promoting lipid, driven to a certain extent by lack of p53, may be a regulator of proliferation, apoptosis, migration and angiogenesis in tumour cells, such as glioblastoma cells and breast cancer cells in humans [39,40].
3.3.2. Top ranking pathway pairs enriched for interactions between mutated genes of brain cancer
To test the performance of a smaller set of disease genes, we selected 25 mutated genes in brain tumours, including neurofibroma, glioma, gliobastoma and astrocytoma from CGC. We mapped them to mixPCN, resulting in brain tumour-specific pathway interaction subnetworks which consisted of pathway pairs connected by at least one pair of those brain cancer-related mutated genes. The over-represented pathway pairs were ranked by number of mutated gene pairs and p-values first; if more than one pair had the same number of interactions, the pair with the larger fraction of mutated gene pairs was listed ahead of the smaller one (tables 4 and 5).
Table 4.
Top 10 over-represent pathway interactions enriched for interactions between mutated genes in brain cancer in mixPCN (ranked by decreasing counts of mutated gene pairs).
| pathway A | pathway B | mutationa(allb) |
|---|---|---|
| ctcf: first multivalent nuclear factor | cell cycle: G1/s check point | 4(33) |
| proteogylcan syndecan-mediated signalling events | role of brca1 brca2 and atr in cancer susceptibility | 4(49) |
| proteogylcan syndecan-mediated signalling events | BARD1 signalling events | 4(112) |
| proteogylcan syndecan-mediated signalling events | cell cycle: G2/m checkpoint | 4(125) |
| ErbB receptor signalling network | p75(NTR)-mediated signalling | 4(277) |
| ErbB receptor signalling network | IL-1-mediated signalling events | 4(445) |
| signalling by NGF | TGF-β receptor signalling | 4(482) |
| signalling by NGF | regulation of cytoplasmic and nuclear SMAD2/3 signalling | 4(482) |
| BARD1 signalling events | EGFR-dependent endothelin signalling events | 3(44) |
| proteogylcan syndecan-mediated signalling events | regulation of transcriptional activity by pml | 3(66) |
aNumber of interactions between mutated genes in brain cancer existing between pathways A and B.
bNumber of all gene interactions existing between pathways A and B.
Table 5.
Top 10 over-represented pathway interactions enriched for interactions between mutated genes in brain cancer in mixPCN (ranked by increasing p-values of mutated gene pairs).
| pathway A | pathway B | mutationa(allb) |
|---|---|---|
| the co-stimulatory signal during T-cell activation | regulation of cell cycle progression by plk3 | 1(1) |
| ctcf: first multivalent nuclear factor | cell cycle: G1/s check point | 4(33) |
| proteogylcan syndecan-mediated signalling events | role of brca1 brca2 and atr in cancer susceptibility | 4(49) |
| BARD1 signalling events | proteogylcan syndecan-mediated signalling events | 4(112) |
| proteogylcan syndecan-mediated signalling events | cell cycle: G2/m checkpoint | 4(125) |
| BARD1 signalling events | EGFR-dependent endothelin signalling events | 3(44) |
| proteogylcan syndecan-mediated signalling events | regulation of transcriptional activity by pml | 3(66) |
| regulation of telomerase | p53 signalling pathway | 3(68) |
| syndecan-1-mediated signalling events | regulation of cell cycle progression by plk3 | 3(76) |
| androgen-mediated signalling | role of brca1 brca2 and atr in cancer susceptibility | 3(77) |
aNumber of interactions between mutated genes in brain cancer existing between pathways A and B.
bNumber of all gene interactions existing between pathways A and B.
As CTCF, proteogylcan syndecan, ErbB receptor and NGF-mediated signalling pathways and cell cycle pathways which ranked in the top 10 of our results and crosstalk between them has been verified in many published studies (tables 4 and 5), we can deduce that this method can aid us in the explanation of mechanisms of complex diseases and supply a possibility of predicting disease-related pathways.
Several molecular studies have identified these critical signalling events in human brain tumours. TGF-β is an important mediator of the malignant phenotype of human gliomas [41]; meanwhile, TGF-β/SMAD signalling plays a role of upstream regulatory process of CTCF. Neurotrophins, which can activate survival signalling by binding to receptor tyrosine kinases (RTK), are important regulators for the survival, differentiation and maintenance of different peripheral and central neurons [42]. In contrast, proneurotrophins induce apoptotic signalling via p75NTR [43]. Moreover, ErbB2, as a member of the ErbB family of RTKs, has been treated as a critical growth factor receptor in development, and it can stimulate downstream signalling pathways in multiple cancers, e.g. malignant glioma [44]. So far as we know, the mechanism of coordination between RTK and p75NTR is unclear. However, the significant interaction between the ErbB receptor signalling network and p75 (NTR)-mediated signalling ranked in the top five. Nerve growth factor (NGF) withdrawal would evoke the p53-dependent apoptosis of primary neuronal cultures [44,45]. Actually, CTCF is central to signalling pathways in immature B cells elicited by cross-linking the BCR and stimulation with TGF-β, both of which can induce cell cycle arrest and apoptosis. Recent studies proved that CTCF is required for cohesin localization and enabled to insulate promoters from distant enhancers and controls transcription at the H19/IGF2 locus, while cohesin depletion has been demonstrated to have an important role in transcription during both G1 and G2 phases [46–48].
3.3.3. Clusters of over-represented pathway pairs enriched for interactions between mutated genes of brain cancer
After mapping 25 brain cancer-related mutated genes to mixPCN, 831 interactions which consisted of pathway pairs connected by at least one pair of those brain cancer-related genes were obtained as brain cancer-related subnetworks (see electronic supplementary material table S15). MCODE aided us in clustering this subnetwork into five clusters in the subnetwork of mixPCN. Three top-ranking clusters in subnetworks with highest scores are shown in figure 3.
Figure 3.

Top three clusters of over-represented subnetwork of mixPCN enriched for interactions between mutated genes in brain cancer. The light grey nodes represent pathways which ranked in top 50 crosstalk in over-represented subnetworks by either number of mutated gene pairs or p-value ranking methods; the triangle nodes indicate pathways which did not rank in top 50 crosstalk in over-represented subnetworks by two ranking methods; squares represent pathways which have been demonstrated to participate in brain cancers through interacting with other pathways in the same cluster in more than one publication; diamonds suggest that more than one interaction with this pathway in this cluster can be verified, but no direct evidence for their roles in brain cancer.
HDAC class I, proteogylcan syndecan, androgen- and p75 (NTR)-mediated signalling pathway were all ranked in the top 50 of over-represented subnetworks of mixPCN. Several reports have implicated protein family histone deacetylases (HDACs) in various neuronal processes, including the neuronal death programme. HDACs are also known to deacetylate several non-histone proteins such as p53 and E2F [49,50]. HDAC1 involvement in neuronal differentiation is further supported by studies indicating that the cell cycle modulating protein, retinoblastoma (Rb), mediates gene repression through recruitment of HDAC1 [51,52]. Although androgen (AR)-mediated signalling is always found to be involved in prostate cancer, the mechanisms contributed by AR can link to other pathways which existed in cluster 1 and have been verified by experiments. AR can play a role in telomere complex stability in prostate cancer cells; it suggests that cell death mediated by AR-antagonist may be induced by telomere complex disruption [53].
It was interesting to find out that several pathways which did not rank in the top 50 also have been verified to be involved in brain cancers, such as IL-12-mediated signalling pathway and HIF-1-α mediating pathway in cluster 1. HIF-1α is responsible for transcriptionally regulating adaptive responses to hypoxia in tumours. Expression of HIF-1α in neural cells is essential for normal development of the brain [54]. HIF-1α has been identified to have a potential role in effecting VEGF transcription and expression through BDNF in neuroblastoma cells [55]. In previous studies, IL-12 has been demonstrated to show effectiveness against brain tumours transplanted within the central nervous system [56].
4. Discussion
We applied a mixed Bayesian method which covers DNA, mRNA, protein and phenotype levels to predict human PPIs. At the DNA level, genetic interaction of yeast was used to imply underlying PPIs in humans. At the mRNA level, coexpression meta-analysis and TFs sharing the same method were integrated to improve performance. At the protein level, DDI and PTM features were integrated by the FCB method; moreover, orthologue mapping of PPIs from model organisms was considered. As a strong predictive feature, sharing of biological functional annotation was integrated in this work. Previous studies usually used the phenotype data of model organisms from gene knock-out or RNA interfere experiments, then gene pairs of model organisms with phenotype similarity score were mapped to human gene pairs, so it is hard to evaluate the effect of homology on phenotype similarity analysis. In this research, we evaluated the phenotype similarity of human genes by the text mining method, which eliminated the problem caused by homology and explored a new data source for phenotype similarity analysis in PPI prediction. In addition, we evaluated the overlap between our predicted PPIs and PPIs derived from other databases.
By integrating all pathway data sources (NCI-Nature, Biocarta and Reactome) from pathway interaction databases and PPI sets which were derived from the result of our mixed Bayesian method and HPRD, two different pathway crosstalk networks were obtained. The fact that the size of the background PPI set influenced the size and contents of PCN profoundly was shown in our research. To achieve the goal of mining disease-related pathways and relationships among them, we selected pathway pairs which realized crosstalk through disease gene pairs by mapping mutated genes in cancers to protein interactions between any pathway pair, then ranked pathway pairs by numbers of mutated gene pairs between pathways or by p-values which indicated significance of disease protein pair enrichment. This new method has been verified to be suitable for either small-scale or large-scale candidate gene sets, so it can perform better than traditional pathway enrichment analysis when only a small amount of candidate genes was supplied. In addition to detecting disease-related pathways, crosstalk relationships among them can also be obtained; therefore, the result has potential for elucidating the mechanisms of diseases. We assumed that if we replaced the PPI set with coexpression networks of different types or stages of disease, a disease-specific or a dynamic pathway crosstalk network can be constructed.
Besides the top ranking pathway pairs in over-represented subnetwork of PCN, we should pay more attention to pathways which were densely connected. Similar to the method for detecting protein complexes and predicting disease genes based on the knowledge of other known disease genes in the same complex, we applied MCODE to extracting highly connected pathway clusters. Pathways in the same cluster might cooperate with each other in disease conditions, so we can predict the candidate disease-related pathways if some known disease-related pathways are included in the same cluster. We used a small list of brain cancer genes to practice, resulting in some brain cancer-related pathway clusters in which most of the crosstalk was proved to be involved in brain cancers.
In conclusion, we demonstrated that integration of heterogeneous datasets for PPI prediction can indicate underlying pathway crosstalk and might play a role in mining cooperated disease-related pathways based on the knowledge of proven causative genes or candidate genes of diseases. Compared with single pathways, crosstalk of pathways which can be extended by increased PPI set will supply more information for uncovering the mechanisms of diseases.
Acknowledgements
This work is supported by Natural Science Foundation of China (Grant nos. 30871394), Post-doctorate Fund of Heilongjiang province (no. LBH-206 110), Scientific Research Fund of Heilongjiang Provincial Education Department (no.11531112) and Innovation Research Funds for Graduate Student of Harbin Medical University (HCXS2009010, HCXS2010009).
References
- 1.Cui J., et al. 2008. AtPID: Arabidopsis thaliana protein interactome database—an integrative platform for plant systems biology. Nucleic Acids Res. 36, D999–D1008 10.1093/nar/gkm844 (doi:10.1093/nar/gkm844) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franke L., van Bakel H., Fokkens L., de Jong E. D., Egmont-Petersen H., Wijmenga C. 2006. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 78, 1011–1025 10.1086/504300 (doi:10.1086/504300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y., Agarwal P., Rajagopalan D. 2008. A global pathway crosstalk network. Bioinformatics 24, 1442–1447 10.1093/bioinformatics/btn200 (doi:10.1093/bioinformatics/btn200) [DOI] [PubMed] [Google Scholar]
- 4.Rhodes D. R., Tomlins S. A., Varambally S., Mahavisno V., Barrette T., Kalyana-Sundaram S., Ghosh D., Pandey A., Chinnaiyan A. M. 2005. Probabilistic model of the human protein–protein interaction network. Nat. Biotechnol. 23, 951–959 10.1038/nbt1103 (doi:10.1038/nbt1103) [DOI] [PubMed] [Google Scholar]
- 5.Xia K., Dong D., Han J. D. 2006. IntNetDB v. 1.0: an integrated protein–protein interaction network database generated by a probabilistic model. BMC Bioinformatics 7, 508. 10.1186/1471-2105-7-508 (doi:10.1186/1471-2105-7-508) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong W., Sternberg P. W. 2006. Genome-wide prediction of C. elegans genetic interactions. Science 311, 1481–1484 10.1126/science.1123287 (doi:10.1126/science.1123287) [DOI] [PubMed] [Google Scholar]
- 7.Keshava Prasad T. S., et al. 2009. Human protein reference database–2009 update. Nucleic Acids Res. 37, D767–D772 10.1093/nar/gkn892 (doi:10.1093/nar/gkn892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salwinski L., Miller C. S., Smith A. J., Pettit F. K., Bowie J. U., Eisenberg D. 2004. The database of interacting proteins: 2004 update. Nucleic Acids Res. 32, D449–D451 10.1093/nar/gkh08632/suppl_1/D449 (doi:10.1093/nar/gkh08632/suppl_1/D449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ceol A., Aryamontri A. C., Licata L., Peluso D., Briganti L., Perfetto L., Castagnoli L., Cesareni G. 2010. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 38, D532–D539 10.1093/nar/gkp983 (doi:10.1093/nar/gkp983) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aranda B., et al. 2010. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 38, D525–D531 10.1093/nar/gkp878 (doi:10.1093/nar/gkp878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breitkreutz B. J., et al. 2008. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 36, D637–D640 10.1093/nar/gkm1001 (doi:10.1093/nar/gkm1001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDowall M. D., Scott M. S., Barton G. J. 2009. PIPs: human protein–protein interaction prediction database. Nucleic Acids Res. 37, D651–D656 10.1093/nar/gkn870 (doi:10.1093/nar/gkn870) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaefer C. F., Anthony K., krupa S., Buchoff J., Day M., Hannay T., Buetow K. H. 2009. PID: the pathway interaction database. Nucleic Acids Res. 37, D674–D679 10.1093/nar/gkn653 (doi:10.1093/nar/gkn653) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews L., et al. 2009. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 37, D619–D622 10.1093/nar/gkn863 (doi:10.1093/nar/gkn863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhodes D. R., et al. 2007. Oncomine 3.0: genes, pathways, and networks in a collection of 18 000 cancer gene expression profiles. Neoplasia 9, 166–180 10.1593/neo.07112 (doi:10.1593/neo.07112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bos P. D., et al. 2009. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 10.1038/nature08021 (doi:10.1038/nature08021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomasson M. H., et al. 2008. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 111, 4797–4808 10.1182/blood-2007-09-113027 (doi:10.1182/blood-2007-09-113027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi J. K., Yu U., Kim S., Yoo O. J. 2003. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 19(Suppl. 1), i84–i90 10.1093/bioinformatics/btg1010 (doi:10.1093/bioinformatics/btg1010) [DOI] [PubMed] [Google Scholar]
- 19.Berglund A. C., Sjölund E., Ostlund G., Sonnhammer E. L. 2008. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 36, D263–D266 10.1093/nar/gkm1020 (doi:10.1093/nar/gkm1020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashburner M., et al. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 10.1038/75556 (doi:10.1038/75556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Driel M. A., Bruggeman J., Vriend G., Brunner H. G., Leunissen J. A. 2006. A text-mining analysis of the human phenome. Eur. J. Hum. Genet. 14, 535–542 10.1038/sj.ejhg.5201585 (doi:10.1038/sj.ejhg.5201585) [DOI] [PubMed] [Google Scholar]
- 22.Ng S. K., Zhang Z., Tan S.-H., Lin K. 2003. InterDom: a database of putative interacting protein domains for validating predicted protein interactions and complexes. Nucleic Acids Res. 31, 251–254 10.1093/nar/gkg079 (doi:10.1093/nar/gkg079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott M. S., Barton G. J. 2007. Probabilistic prediction and ranking of human protein–protein interactions. BMC Bioinformatics 8, 239. 10.1186/1471-2105-8-239 (doi:10.1186/1471-2105-8-239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelley R., Ideker T. 2005. Systematic interpretation of genetic interactions using protein networks. Nat. Biotechnol. 23, 561–566 10.1038/nbt1096 (doi:10.1038/nbt1096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh J. L., Ding H., Costanzo M., Baryshnikova A., Toufighi K., Bader G. D., Myers C. L., Andrews B. J., Boone C. DRYGIN: a database of quantitative genetic interaction networks in yeast. Nucleic Acids Res. 38, D502–D507 10.1093/nar/gkp820 (doi:10.1093/nar/gkp820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagamine N., Kawada Y., Sakakibara Y. 2005. Identifying cooperative transcriptional regulations using protein–protein interactions. Nucleic Acids Res. 33, 4828–4837 10.1093/nar/gki793 (doi:10.1093/nar/gki793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romero P., Wagg J., Green M. L., Kaiser D., Krummenacker M., Karp P. D. 2005. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 6, R2. 10.1186/gb-2004-6-1-r2 (doi:10.1186/gb-2004-6-1-r2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M., et al. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 10.1093/nar/gkm882 (doi:10.1093/nar/gkm882) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y., Hociderg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B 57, 289–300 [Google Scholar]
- 30.Bamford S., et al. 2004. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 10.1038/sj.bjc.6601894 (doi:10.1038/sj.bjc.6601894) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandhi T. K., et al. 2006. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat. Genet. 38, 285–293 10.1038/ng1747 (doi:10.1038/ng1747) [DOI] [PubMed] [Google Scholar]
- 32.Bader G. D., Hogue C. W. 2003. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2. 10.1186/1471-2105-4-2 (doi:10.1186/1471-2105-4-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaur S., Sassano A., Joseph A. M., Majchrzak-Kita B., Eklund E. A., Verma A., Brachmann S. M., Fish E. N., Plantanias L. C. 2008. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J. Immunol. 181, 7316–7323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hjortsberg L., Lindvall C., Corcoran M., Arulampalam V., Chan D., Thyrell L., Nordenskjold M., Grandér D., Pokrovskaja K. 2007. Phosphoinositide 3-kinase regulates a subset of interferon-α-stimulated genes. Exp. Cell Res. 313, 404–414 10.1016/j.yexcr.2006.10.022 (doi:10.1016/j.yexcr.2006.10.022) [DOI] [PubMed] [Google Scholar]
- 35.Wang Y., Kreisberg J. I., Ghosh P. M. 2007. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 7, 591–604 10.2174/156800907781662248 (doi:10.2174/156800907781662248) [DOI] [PubMed] [Google Scholar]
- 36.Apte R. N., et al. 2006. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor–host interactions. Cancer Metastasis Rev. 25, 387–408 10.1007/s10555-006-9004-4 (doi:10.1007/s10555-006-9004-4) [DOI] [PubMed] [Google Scholar]
- 37.Hoosein N. M. 1998. Neuroendocrine and immune mediators in prostate cancer progression. Front. Biosci. 3, D1274–D1279 [DOI] [PubMed] [Google Scholar]
- 38.Fears C. Y., Gladson C. L., Woods A. 2006. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J. Biol. Chem. 281, 14 533–14 536 10.1074/jbc.C600075200 (doi:10.1074/jbc.C600075200) [DOI] [PubMed] [Google Scholar]
- 39.Ruckhaberle E., et al. 2008. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 112, 41–52 10.1007/s10549-007-9836-9 (doi:10.1007/s10549-007-9836-9) [DOI] [PubMed] [Google Scholar]
- 40.Taha T. A., Osta W., Kozhaya L., Bielawski J., Johnson K. R., Gillanders W. E., Dbaibo G. S., Hannun Y. A., Obeid L. M. 2004. Down-regulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53. J. Biol. Chem. 279, 20 546–20 554 10.1074/jbc.M401259200M401259200 (doi:10.1074/jbc.M401259200M401259200) [DOI] [PubMed] [Google Scholar]
- 41.Gramatzki D., Pantazis G., Schittenhelm J., Tabatabai G., Köhle C., Wick W., Schwarz M., Weller M., Tritschler I. 2009. Aryl hydrocarbon receptor inhibition downregulates the TGF-β/Smad pathway in human glioblastoma cells. Oncogene 28, 2593–2605 10.1038/onc.2009.104 (doi:10.1038/onc.2009.104) [DOI] [PubMed] [Google Scholar]
- 42.Gong Y., Cao P., Yu H.-J., Jiang T. 2008. Crystal structure of the neurotrophin-3 and p75NTR symmetrical complex. Nature 454, 789–793 10.1038/nature07089 (doi:10.1038/nature07089) [DOI] [PubMed] [Google Scholar]
- 43.Volosin M., Song W., Almeida R. D., Kaplan D. R., Hempstead B. L., Friedman W. J. 2006. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J. Neurosci. 26, 7756–7766 10.1523/JNEUROSCI.1560-06.2006 (doi:10.1523/JNEUROSCI.1560-06.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Contessa J. N., Bhojani M. S., Freeze H. H., Rehemtulla A., Lawrence T. S. 2008. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 68, 3803–3809 10.1158/0008-5472.CAN-07-6389 (doi:10.1158/0008-5472.CAN-07-6389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozniak C. D., Radinovic S., Yang A., McKeon F., Kaplan D. R., Miller F. D. 2000. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 289, 304–306 10.1126/science.289.5477.304 (doi:10.1126/science.289.5477.304) [DOI] [PubMed] [Google Scholar]
- 46.Parelho V., et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132, 422–433 10.1016/j.cell.2008.01.011 (doi:10.1016/j.cell.2008.01.011) [DOI] [PubMed] [Google Scholar]
- 47.Wendt K. S., Peters J. M. 2009. How cohesin and CTCF cooperate in regulating gene expression. Chromosome Res. 17, 201–214 10.1007/s10577-008-9017-7 (doi:10.1007/s10577-008-9017-7) [DOI] [PubMed] [Google Scholar]
- 48.Wendt K. S., et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801 10.1038/nature06634 (doi:10.1038/nature06634) [DOI] [PubMed] [Google Scholar]
- 49.Gu W., Roeder R. G. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 10.1016/S0092-8674(00)80521-8 (doi:10.1016/S0092-8674(00)80521-8) [DOI] [PubMed] [Google Scholar]
- 50.Marzio G., Wagener C., Gutierrez M. I., Cartwright P., Helin K., Giacca M. 2000. E2F family members are differentially regulated by reversible acetylation. J. Biol. Chem. 275, 10 887–10 892 10.1074/jbc.275.15.10887 (doi:10.1074/jbc.275.15.10887) [DOI] [PubMed] [Google Scholar]
- 51.Morrison B. E., Majdzadeh N., D'Mello S. R. 2007. Histone deacetylases: focus on the nervous system. Cell. Mol. Life Sci. 64, 2258–2269 10.1007/s00018-007-7035-9 (doi:10.1007/s00018-007-7035-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolas E., Morales V., Magnaghi-Jaulin L., Harel-Bellan A., Richard-Foy H., Trouche D. 2000. RbAp48 belongs to the histone deacetylase complex that associates with the retinoblastoma protein. J. Biol. Chem. 275, 9797–9804 10.1074/jbc.275.13.9797 (doi:10.1074/jbc.275.13.9797) [DOI] [PubMed] [Google Scholar]
- 53.Kim S. H., Richardson M., Chinnakannu K., Uma Bai V., Menon M., Barrack E. R., Reddy P.-V. 2010. Androgen receptor interacts with telomeric proteins in prostate cancer cells. J. Biol. Chem. (doi:10.1074/jbc.M109.098798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomita S., et al. 2003. Defective brain development in mice lacking the Hif-1α gene in neural cells. Mol. Cell Biol. 23, 6739–6749 10.1128/MCB.23.19.6739-6749.2003 (doi:10.1128/MCB.23.19.6739-6749.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakamura K., Martin K. C., Jackson J. K., Beppu K., Woo C.-W., Thiele C. J. 2006. Brain-derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1α in neuroblastoma cells. Cancer Res. 66, 4249–4255 10.1158/0008-5472.CAN-05-2789 (doi:10.1158/0008-5472.CAN-05-2789) [DOI] [PubMed] [Google Scholar]
- 56.Roy E. J., Gawlick U., Orr B. A., Rund L. A., Webb A. G., Kranz D. M. 2000. IL-12 treatment of endogenously arising murine brain tumors. J. Immunol. 165, 7293–7299 [DOI] [PubMed] [Google Scholar]