

Abstract
The most commonly used dose–response models implicitly assume that accumulation of dose is a time-independent process where each pathogen has a fixed risk of initiating infection. Immune particle neutralization of pathogens, however, may create strong time dependence; i.e. temporally clustered pathogens have a better chance of overwhelming the immune particles than pathogen exposures that occur at lower levels for longer periods of time. In environmental transmission systems, we expect different routes of transmission to elicit different dose–timing patterns and thus potentially different realizations of risk. We present a dose–response model that captures time dependence in a manner that incorporates the dynamics of initial immune response. We then demonstrate the parameter estimation of our model in a dose–response survival analysis using empirical time-series data of inhalational anthrax in monkeys in which we find slight dose–timing effects. Future dose–response experiments should include varying the time pattern of exposure in addition to varying the total doses delivered. Ultimately, the dynamic dose–response paradigm presented here will improve modelling of environmental transmission systems where different systems have different time patterns of exposure.
Keywords: dose–response, anthrax, disease dynamics, survival analysis, microbial risk assessment
1. Introduction
Dose–response functions are central to microbial risk assessments. In transmission systems, dose–response modelling is important in evaluating risk given an environmental pathogen exposure. Exposure occurs when susceptible individuals contact a pathogen source, usually an environmental reservoir or an infected individual. These exposure events can be characterized by the frequency and magnitude of pathogens that reach a susceptible host. The route of transmission, exposure behaviours and physical aspects of the system will cause the dose–timing patterns of pathogen exposure to vary. For example, in influenza transmission, a direct pathogen exposure from a sneeze may be characterized as a large bolus exposure event while aerosolized exposure may be constant over a long period of time but exposure consists of a smaller number of pathogens at any given time.
Biologically, the immune system may handle varying exposure patterns with varying efficiency. Pathogen inoculations that occur very close together may carry a comparable risk of infection to an equivalent total dose occurring at a point in time if the immune response is slow compared with the period of exposure for the repeated doses. Longer periods between pathogen exposures, however, may allow the immune system to eliminate the pathogen and recover between each inoculation. The rates at which the immune system responds and clears the pathogen are clearly important in determining the accumulation of multiple inoculations. For these extreme instances, short versus long inoculation intervals, we can conclude that inoculations either accumulate as a sum or should be considered as separate events. However, it becomes unclear how accumulation of pathogen levels within the host may vary for patterns that occur on a time scale where the innate immune system has begun to respond but failed to clear all the pathogens.
Immune response is variable depending on many factors, such as pathogen type, location of pathogen and prior exposure. We specifically focus on the dynamics of the initial immune response. This includes the innate immune response, natural host barriers (e.g. mucosal clearance) and potentially standing elements of the acquired immune response. It is possible, however, that given long enough time frames of exposures that the adaptive immune response will also act as an initial response to future inoculations. Our hypothesis is that these initial host protections are not constant in nature, and that repeated inoculations affect the probability of any single pathogen initiating the infection process.
The classic dose–response models used for microbial risk assessment are the exponential and beta-Poisson distributional models [1]. These models calculate the risk of infection for a single-dose value. Parameters for these models are empirically informed using animal dosing experiments in which varying single-bolus pathogen doses are given to animals, and infection or disease is monitored [2,3]. In environmental infection transmission systems where the environment is not just a source but is a medium of pathogen transport between individuals, these models are justified for the extreme scenarios of very closely spaced or very distantly spaced exposures described above. In these scenarios, the probability of infection can be calculated independently for each dosing event. However, for other exposure patterns, total within host pathogen level at a given inoculation time is dependent on remaining pathogen levels from past inoculations. In these cases, the state of the system (i.e. the number of living pathogens and the number of immune elements available to fight them) after any defined interval is dependent on the clearance rate of pathogens and the destruction and recruitment rate of standing immune elements. Here, we define standing elements as those elements existing or that would have appeared on their own in the absence of new immune element generation owing to an acquired immune response.
The exponential and beta-Poisson models make implicit assumptions about how multiple pathogens interact to cause infection. Under the independent action hypothesis, any individual pathogen is capable of initiating infection with some independent probability [1]. The traditional dose–response models operate under this paradigm. This hypothesis, however, is generally considered only under single inoculation scenarios. We contend that, even though a single pathogen is capable of initiating infection, the infectivity of a pathogen may depend on the state of the immune system, which in turn is affected by prior inoculations. This is in contrast to the independent action hypothesis that pathogen risk probability is independent of other pathogens. It also deviates from a threshold model in that the risk of infection given any inoculation size is still never zero.
One aim for this model would be future integration into transmission models. Particularly, in transmission models, when we specifically consider pathogen exposure from the environment, we must translate an exposure event into a probability of infection. This could be done using the exponential or beta-Poisson models but these models potentially ignore exposure dynamics associated with different routes of exposures, as discussed in the exposure scenarios above. Although a more biologically motivated dose–response model was previously proposed [4], we present here a model that is computationally less intensive and therefore more suitable for integration into a transmission model. In the Pujol et al. [4] model, innate immune effector particles and pathogens are modelled in a stochastic competition model capturing both the growth of pathogens and diminishing immune response. In our model, we aim to capture these dynamics with a simple model that does not explicitly model the immune dynamics. This satisfies the goals of dose–response at the transmission or population level by allowing utilization of an exposure pattern (history of inoculations) into the calculation of the probability of infection. Our model provides a framework to realistically relax the assumption of dose independence in a biologically plausible yet computationally efficient manner that implicitly incorporates the dynamics of the immune system. Furthermore, we present a statistical method to analyse such time-series data where infection events are occurring amid inoculation events. Experimental data to inform a time-dependent dose–response model are extremely rare, but there are data from a 1966 study on inhalation anthrax in monkeys that incorporates varying exposure patterns and time to death data [5]. Even though this study was not specifically designed to study varying risk by exposure patterns, our analysis provides direction for more informative future dose–response experiments that will incorporate time-dependent dosing patterns.
2. Methods
2.1. Overview of dose–response model construction
The methods section will describe the construction of our time-dependent dose–response model followed by its application to time-series anthrax dose–response data. The first three sections describe the development of the model in a general framework focusing on the clearance of pathogens within a host and the probability of infection take-off during the clearance time frame. Section 2.1 mathematically describes the within host pathogen level at any given time after a point source inoculation and then §2.2 extrapolates this process to multiple inoculations. Section 2.3 describes the development of a hazard for infection at a given time post-inoculation. At this point, enough information is provided to use this time-dependent dose–response model to make risk calculations given a parametrization. Particularly, it could be used in a transmission model setting to translate. multiple exposure events into an infection risk calculation. The last three sections pertain to analysing data. Section 2.4 uses the hazard to develop a likelihood statistic suitable for analysing time-series dose–response data. To further analyse the data we have, we must make further assumptions concerning the data that are described in detail in §2.5. Section 2.6 describes our exploration of the results from §2.5 in more controlled experimental settings.
2.2. Time-dependent pathogen clearance
Our model aims to describe the clearance of pathogen until either the pathogen is eliminated or the pathogen establishes infection. To capture the dose–timing region between the extremes discussed above, a model should reflect decreasing ability of the immune system to inactivate pathogens as pathogens accumulate and immune elements are consumed. The differential equation in equation (2.1) illustrates such a model, where t represents time and P(t) is a function representing the total within host pathogen level at a given time. The parameter γ roughly approximates a net per-pathogen clearance rate. The pathogen clearance rate is also affected by a shaping parameter, α, which elicits different inoculation accumulation effects depending on its value.
| 2.1 |
To keep this model biologically plausible we consider the domain of γ to be in the interval (0, ∞) and the domain of α to be in the interval [0, 1]. When α = 1, γ becomes the per capita rate of decay of within host pathogen and the decay curve takes the exponential shape. In this case, the pathogen's die-out is a linear function of the total number of pathogens P, i.e. the immune system is equally effective in eliminating one pathogen regardless of the current number of pathogens in the system. The state of the immune system, therefore, is irrelevant since its efficacy to eliminate pathogens is constant. When α is less than one, this per capita change, γ, is attenuated by the factor, 1/P1−α. That is, the effectiveness of the immune system is dependent on the total number of within host pathogens. As α decreases and approaches zero, the shape of decay becomes more linear and slower for a fixed γ. Therefore, the parameter α can also be biologically described as the degree to which the immune system can be overwhelmed by pathogen level. For a single inoculation, the decay curve is illustrated in figure 1. Given a total dose of 100 pathogens, the curve represents the total within host pathogen level at any given time over the course of clearance for varying values of α over two fixed values of γ.
Figure 1.

The shape and rate of within host dose decay by levels of α and γ. While the shape of decay is dependent only on α, the rate of decay is dependent on both α and γ. (a) Dose over time (γ = 2); (b) dose over time (γ = 0.5). Dotted line, α = 1; grey line, α = 0.75; dashed line, α = 0.5; thin line, α = 0.25; thick line, α = 0.
By considering a negative differential equation, we are modelling under the assumption that the total within host pathogen level, or the infection hazard, is strictly decreasing. Biologically, our assumption is that after an inoculation, on average, the rate of pathogen reproduction is less than the rate of pathogen clearance. If this inequality reverses, that is pathogen reproduction becomes greater than pathogen clearance on average, this would correspond to the pathogen establishing infection. This assumption may not be suitable for all pathogens depending on biological traits, particularly the pathogen's ability to replicate within our time scale of interest.
2.3. Dose clearance and multiple dosing
We propose to use this function to calculate an effective dose for risk assessments when multiple inoculations occur within a biologically relevant time frame. To do this, we must first evaluate the solution to equation (2.1) with initial condition given at time 0, d = P(0), where d is a single inoculation given at time 0.
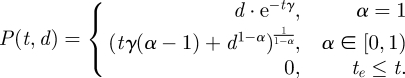 |
2.2 |
To ensure that P(t, d) > 0, we implement the last constraint in equation (2.2), where P(t, d) is absorbed at 0 after te, the time of extinction for a given inoculation. The closed-form solution for the time of extinction for a single inoculation is given by equation (2.3). Note that even though te is unbounded when α = 1, the dose function (an exponential decay function) takes on small values fairly quickly for a fixed γ as t increases, as illustrated in figure 1.
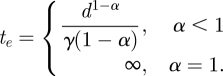 |
2.3 |
In multiple exposure scenarios, the input doses for this model are represented by a sequence of inoculations such as those illustrated in the top two graphs in figure 2. Each inoculation, di, is received instantaneously at a designated time, ti. Formally, we map a one to one correspondence between a sequence of n inoculations, 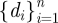 , and a sequence of n inoculation times,
, and a sequence of n inoculation times, 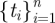 . In a study, we observe subjects in real time (or close to) and record a corresponding final observation time, T. This final observation time, T, can occur in any interval between inoculations, ti ≤ T ≤ ti+1 or after the final inoculation time, tn < T. Further, for a subject j, the total inoculations experienced before an infection event or censoring may be less than n, so we can denote the subject-specific sequence size to be nj with corresponding final observation time, Tj.
. In a study, we observe subjects in real time (or close to) and record a corresponding final observation time, T. This final observation time, T, can occur in any interval between inoculations, ti ≤ T ≤ ti+1 or after the final inoculation time, tn < T. Further, for a subject j, the total inoculations experienced before an infection event or censoring may be less than n, so we can denote the subject-specific sequence size to be nj with corresponding final observation time, Tj.
Figure 2.

(a,b) Example of two different inoculation patterns (c,d) and the time evolution of pathogen numbers within the host. The model's parameters are α = 0.5 and γ = 0.05. The patterns of inoculation correspond to two different multiple inoculation scenarios: (a) 20 inoculation events of two pathogens each, (b) and four inoculation events of 10 pathogens each. Although the total inoculated dose (40 pathogens) and the time of exposure (200 min) is the same, it is visually evident that pathogens from the four inoculation events persist longer in the immune system. (Online version in colour.)
Now that multiple dosing situations have been introduced, we can consider evaluation of equation (2.2) for multiple inoculations. Since past inoculations may still be present at the time of a new inoculation, the dose function must incorporate the sequence of all past inoculations up to time t. We can picture the multiple dose time function as a series of decay curves with discontinuity jumps occurring at each inoculation point, illustrated in the bottom two graphs of figure 2. Particularly, the total within host pathogen level at any inoculation time, ti, is the sum of the current inoculation, di, and the remaining within host pathogens pi. The remaining pathogen level is described in equation (2.4) recursively using equation (2.2). When α = 1, the remaining pathogen level can be defined as an independent accumulation of previous inoculations, this is derived in the online materials.
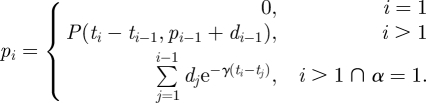 |
2.4 |
Since equation (2.4) describes the points of discontinuity at inoculation times, we can use it to reconstruct equation (2.2) to use multiple dose arguments. The following function is the multiple dose function.
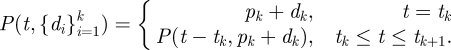 |
2.5 |
This function can be interpreted as the total within the host pathogen level at a given time, t, given all past inoculation up to that time. The function is jump-discontinuous in that it decreases and is absorbed at 0 (by construction in equation (2.2)) but has point increases at inoculation times, ti by an inoculation value, di. Note that, given a time sufficiently greater than the time of the last inoculation, this equation approaches 0 under the same conditions as equation (2.2). The time to extinction, te, after inoculation time ti, can still be calculated using equation (2.3) but with input dose, pi + di, instead of di. When α = 1, the function approaches zero quickly with decreasing error as t ≫ tk since the true convergence time is in the limit, t→∞, discussed in detail in the online electronic supplementary material.
2.4. Dose–response risk from multiple dose function
We consider an effective dose to be any value calculated from equation (2.5) that could contribute to an infection hazard at any given time. To evaluate the accumulated effective dose in a given host, we integrate equation (2.5) over a time period of interest. For a single inoculation, the closed-form solution for the effective dose from inoculation to extinction is.
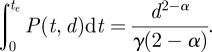 |
2.6 |
When there are multiple inoculations, the accumulated effective dose is a sum of integrals over the continuous sections of the multiple dose function. This is seen in equation (2.7), for any time, T, before the final within host pathogen extinction. If T = tk, or the upper bound of the integral is equivalent to an inoculation time, the last term of equation (2.7) is zero.
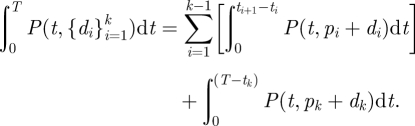 |
2.7 |
To evaluate the accumulated effective dose through final pathogen extinction, we simply replace the final term of equation (2.7) with equation (2.6) with pn + dn (the initial pathogen level at the final inoculation) substituted for d. For α = 1 and T ≫ tn, we can still substitute in equation (2.6) with small error, this is discussed in greater detail in the electronic supplementary material.
To consider how α determines the importance of dose–timing, consider an entire dose course over an exposure pattern, for example, the top two graphs in figure 2, where we have two distinct exposure patterns of 40 pathogens given over 200 min. The accumulated effective dose over this time can be calculated using the integral given in equation (2.7). When α = 1, the accumulation of inoculations is an independent process, as illustrated in equation (2.4) owing to the exponential memoryless property. Because of this property, the total effective dose over the exposure period is the sum of the inoculations divided by γ. This solution holds for all potential dosing patterns evaluated through effective extinction (T ≫ tn) when α = 1, discussed further in the online electronic supplementary material.
When α < 1, however, the pathogen clearance rate depends on the within host pathogen level. Under these conditions, the total effective dose is dependent on the timing of given inoculations. For α = 0.5, the accumulated effective dose (the area under the curve) differs across dosing patterns despite the sums of the inoculations both totalling 40 pathogens (see the bottom two graphs of figure 2). Although the total inoculated dose (40 pathogens) and the time of exposure (200 min) are the same, it is visually evident that pathogens from the four inoculation events persist longer.
The goal of our model is to analyse decaying hazard post inoculation, until either the pathogen is cleared or the pathogen takes hold and initiates infection prior to clearance. We now introduce the risk of infection owing to a single pathogen per unit time that is present in the immune system s. This formulation is a one-hit model of infection; i.e. a single pathogen unit is capable of initiating infection. This phenomenon has been shown empirically for pathogens such as influenza A [6] and intravenous Salmonella exposure [7]. However, unlike the exponential formulation of a one-hit model, each hit does not have identical and independent risk. Instead, risks are dependent upon prior hits and thus α and γ also contribute to the calculation of the risk.
For an instantaneous risk associated to a single pathogen, s, and the current number of pathogens within the host, P, we can calculate the force of infection, i.e. the probability of a susceptible individuals becoming infected, sP(t)dt. This is evaluated at each time step. For multiple inoculations, we insert our multiple dose function. We can interpret this as a hazard function, λ(T), given in equation (2.8).
| 2.8 |
By integrating and exponentiating the hazard over an interval time up to time, T, we can calculate the survival function, or the probability of not being infected by time, T, given as follows.
| 2.9 |
We now have a corresponding risk of 1 − S(T), which matches the familiar functional form of the exponential dose–response model. If we consider T ≫ tn, then this risk corresponds to the risk of an entire exposure pattern. When α = 1 and there is a single inoculation event and clearance, the single pathogen risk parameter, k, from the exponential model is equivalent to the ratio s/γ. Furthermore, continuing to assume complete pathogen clearance, this equivalence holds for all exposure patterns when α = 1. This relationship is lost when α < 1 as pathogen clearance, and thus risk, becomes dependent on the size and timing of inoculations. That is, when α < 1, the equivalence of the exponential function and our cumulative dose risk function is dependent on the inoculation size. Mathematical explorations of the relationship between the exponential model and our model when α = 1 are discussed in the online electronic supplementary material.
2.5. Likelihood statistic and parameter estimation from data
By multiplying the hazard and the survival function we can calculate the probability density for infection at a final observation time, T, standard in a survival analysis. This is given in equation (2.10).
| 2.10 |
To estimate the model parameters using time-dependent exposure data including time of infection, we propose a likelihood statistic derived from survival analysis framework. The likelihood is formulated depending on a subject j's infection status given by Δj. This is illustrated in equation (2.11) where we consider each subject to be independent. If Δj = 1 (infection occurs), then the likelihood is calculated from the probability of infection at time, Tj, given by the density function f(Tj). If Δj = 0 (no infection or censoring), then the likelihood is calculated from the probability of survival up to time, Tj. To estimate parameters, we propose a maximum-likelihood approach, that is, we find the best parameter values that maximize the likelihood values.
| 2.11 |
By taking the negative log of the likelihood and substituting in equation (2.10), we simplify equation (2.11) to equation (2.12).
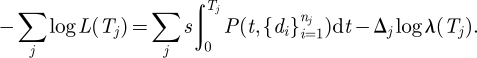 |
2.12 |
When evaluating time-dependent dose–response data, finding exact times of infection will generally be difficult if not impossible. We may know a time interval in which the process of infection began but not an exact time. For these scenarios we can adjust the likelihood formulation such that interval censoring can be incorporated. This adjustment is discussed in detail in the online electronic supplementary material.
2.6. Case study: inhalational anthrax data
Inhalation anthrax mortality data in monkeys were published by Brachman et al. [5] from an observational animal study conducted in a wool sorting mill in South Carolina in 1966 [5]. Cynomogus monkeys were placed in a laboratory trailer and air was ventilated into the trailer from the wool sorting mill. Air was periodically sampled to measure the anthrax concentration. A daily inhaled dose was estimated using these data and an estimated monkey respiration rate. We considered these inhaled spores as pathogens capable of initiating infection.
The experiment was conducted during five distinct time intervals and three of these were reported. During the third and fourth runs, the monkeys were continuously exposed to contaminated air during working hours. Air was intermittently sampled daily. These were the only two runs we were able to use to evaluate our model. As air was ventilated in from the wool sorting mill, exposure was based on the natural concentrations in the mill and therefore was not controlled by the researchers. For our purposes, dose levels were estimated from figures provided in the paper in which a single total daily exposure was recorded. We assumed that on days when exposures were recorded that single equivalent sized inoculation occurred at the beginning of each hour for the entire day. Monkeys' health was monitored over the exposure period and for a brief time after the terminal exposure. The experimenters checked in on the monkeys three times per day. Autopsies were conducted immediately after a death was discovered. Deaths owing to non-anthrax causes also occurred and were recorded. All monkeys were sacrificed shortly after the exposure period to determine the anthrax infection status.
During the third run, 32 monkeys were exposed for 47 days during which time 10 deaths owing to anthrax infection were recorded. On the 50th day, all the remaining live monkeys were sacrificed. At this time, two more monkeys were found to be infected bringing the infection total to 12 with observed risk to be 44 per cent. During the fourth run, 31 monkeys were exposed for 41 days during which time 10 deaths owing to anthrax infection were recorded. The remaining live monkeys were sacrificed on the 51st day. No additional monkeys were found to be infected and the observed risk was thus 23 per cent. Figure 3 is a graphical depiction of these data.
Figure 3.

Exposure (left axis and represented by lighter bars with width of 1 day) and Mortality (right axis and represented by black thin bars) results from the third and fourth runs of the anthrax dataset. Exposure is assumed to be given uniformly once every hour on days when exposures were recorded. In the third run, 12/32 monkeys died from anthrax, and in the fourth run, 10/31 monkeys died from anthrax. (a,b) Thin light blue lines, dose; thick black lines, deaths. (Online version in colour.)
By the end of the study, monkeys had become infected, died of other causes, or survived (did not become infected) over the duration of exposure. Survived subjects were sacrificed several days after the final inoculation, which technically elicits a right censoring. We assume that autopsies that were negative for anthrax are conclusive in that subjects would never have become infected. That is, we assume all non-infected sacrificed monkeys survived the entire dose course. We also assume that anthrax has a case-fatality rate of 100 per cent in monkeys (i.e. no monkey survived infection). A small set of subjects in these data died of causes not related to anthrax. We assume these censored subjects died from reasons that were completely random and unrelated to anthrax exposure and therefore evaluate their survival up to their final observed time, Tj. For subjects that were infected, only day of death was observed.
Since we were interested only in time to infection take-off, we considered a fixed population lag period, τ, which is the time between infection take-off (when the course of ongoing infection is assured to be progressive) and time of death. Given the observed time of death, Tj, the predicted time to infection is Tj − τ. We do not aim to dynamically model any processes during the lag time, τ, and thus treated it as a constant parameter. A previous study [8] showed that time between symptoms and death is on the scale of several hours for Cynomologus monkeys so we do not think symptom onset would aid in finding the total lag period.
This lag period, τ, was treated as a population parameter but it is probably individually probabilistic in nature. Based on the Vasconcelos study [8] that this lag is variable, a variety of τ values were initially implemented. However, the Vasconcelos experiments used significantly higher dosing in bolus (the ID50) and potentially a different strain of the Bacillus anthracis, and therefore lag times may not be comparable. Further, an experiment on several other pathogens showed relatively invariant latency periods for inoculations less than the ID50 [9].
To come up with a lag time estimates, we considered a previous study [10] that used lag time distributions with median lag times of 1–3 days. However, their lag period described the period from spore germination to symptoms. Anthrax infection occurs after germination in macrophages [11,12]. The biology and persistence of spores from time of exposure to infection is not well understood, but elimination of spores is mediated by epithelial cells and macrophages [13]. While epithelial cells are capable of eliminating anthrax spores alone [13], spores are also readily phagocytized by alveolar macrophages where they germinate. While, the macrophage is capable of eliminating the bacillus, B. anthracis toxins and defence mechanisms inhibit this ability as it attempts to use the macrophage to reach the regional lymph system where it can initiate infection as an extracellular pathogen [11]. This process implies that germination is an important step in the infection process but that it occurs before infection takes off. We therefore expect the lag period described in our model to be less than that described in Brookmeyer et al. [10] and used a period from 1 to 4 days. Treating τ as a nuisance parameter, we fixed it to a discrete uniform distribution; it was then integrated out of the likelihood using conditional expectation. Further discussion can be found in the online electronic supplementary material.
Model fitting was done by profiling over α while optimizing the parameters s and γ (in unit per hours) to minimize the negative log likelihood based on the Brachman experimental data for run 3 and run 4. By profiling over a fixed α, we are also analysing these data under different assumptions concerning accumulation of doses in terms of differing dose–timing. Inference was done by fitting a spline curve through the values of the negative log likelihood for each α value creating a smooth depiction of the log likelihood space. Using the minimum value of the spline (the overall maximum likelihood estimate (MLE) fit), we defined a critical cut-off using the likelihood ratio test [14]. That is, at the 95 per cent significance level, the confidence interval of α falls in the range of the minimum log likelihood ± ½ × χ2(0.95,d.f. = 1).
To evaluate consistency of our model with some past anthrax models, we calculated risks over a range of single dose values. This can be done for α = 1, reducing our model to an exponential model, in which the exponential risk parameter k is equivalent to the ratio of s over γ. For parameterizations involving α < 1 in which dose–timing patterns impact risk and the exponential model is no longer valid, our model is only comparable to past models under single inoculation scenarios. Risks for range of single dose values were calculated using equations (2.6) and (2.9).
Integrals were estimated using the adaptive Simpson quadrature method in Matlab. Optimization was done using minimax search algorithm in Matlab. Spline fitting was done using the default spline function in Matlab.
2.7. Dosing experiment design
To explore the model's ability to discern between different exposure patterns and to illustrate the results from the Brachman data optimization, exposure patterns were created representing two extremes; one large bolus and one evenly distributed set of smaller inoculations given once daily over 15 days. The sum of the inoculations is equivalent for both patterns at a value of 15 000 as it corresponds to the sum of the total dose in run 3. Using the parameter MLEs from the model, risks were calculated for each dosing pattern for a fixed α and corresponding MLE γ and s values. The varying values of α illustrate potential expected results from an animal experiment that incorporated dose–timing.
3. Results
Figure 4 shows the results of the optimization profile over α using data from the Brachman inhalational anthrax study runs 3 and 4. The likelihood space is a fairly smooth decreasing curve in the optimized log likelihood space over α as seen in figure 4a. A smoothed spline was fit through the points and the 95 per cent lower limit was connected to the upper boundary of α by a horizontal line. The optimal parameter fit occurred when α = 0.90 with respective MLE values for s and γ at 1.81 × 10−7 h−1 and 0.0097 h−1. For these overall MLE fits, the predicted risk for run 3 and run 4 is 50 and 16 per cent when compared with the observed attack rates of 44 and 23 per cent, respectively. The 95 per cent CI for α was (0.51, 1) where 1 is a bound owing to our imposed constraints on values of α. It is mathematically and statistically possible to extend α beyond our imposed upper limit constraint but the biological interpretation of our model becomes less intuitive. For α > 1, bolus exposures patterns have lower risks than evenly distributed exposure patterns when total dose is fixed. Results for α > 1 are discussed in the online electronic supplementary material.
Figure 4.

Results from the Brachman inhalational anthrax data analysis. (a) Results from optimization profile over α. A spline curve was fit to determine the minimum negative log likelihood (MLE α = 0.90) and to determine the 95% CI (0.51, 1) using the log likelihood ratio test. (b) Optimized s values for values of α within its 95% CI. (c) Optimized log(γ) values for values of α within its 95% CI. The minimum γ value in this range is 0.0046 h−1 and the maximum is 0.17 h−1. (d) Predicted risks for two exposure patterns using MLE values profiled over α. Two exposure scenarios were used, one bolus and one evenly distributed exposure pattern, both with the same total dose of 15 000. Parameter sets used for these calculations are (α = 0.50, s = 1.65 × 10−7 h−1, γ = 0.20 h−1); (α = 0.60, s = 1.72 × 10−7 h−1, γ = 0.093 h−1); (α = 0.70, s = 1.76 × 10−7 h−1, γ = 0.044 h−1); (α = 0.80, s = 1.78 × 10−7 h−1, γ = 0.021 h−1); (α = 0.90, s = 1.81 × 10−7 h−1, γ = 0.0097 h−1); and (α = 1.00, s = 1.84 × 10−7 h−1, γ = 0.0046 h−1). (d) Circles, exposure pattern 1 (bolus); crosses, exposure pattern 2 (evenly distributed).
Plotting of the MLE s and γ values over the significant region of α can be seen in figure 4b,c. The MLE values of γ are log linearly correlated with α. Recall that the shape and the rate of within host pathogen decay depends on both these parameters and, therefore, this relationship is not unexpected as the optimization is trying to find a relatively stable clearance curve and there are probably many sets of α and γ that could elicit such a curve. When given multiple exposure patterns of the same total dose, we would expect α parameter to determine the importance of dose–timing effects, i.e. whether the risk will differ between these exposure patterns. Therefore, without data containing more exposure patterns, it is difficult to independently estimate both α and γ. The MLE values of s are relatively insensitive for changing values of α. The data thus provide us with a consistent estimate of the instantaneous per pathogen infectivity parameter, s, for a given clearance pattern.
The attack rates for varying single doses were calculated using our MLE parameterization and equations (2.6) and (2.9). We compare our MLE results to another parameterization of our model and two previously analysed anthrax dose–response models by creating a risk curve over a large range of bolus dose values, all illustrated in figure 5. We consider results for α = 1, equating this parameterization of our model to the exponential model where k = s/γ = 3.95 × 10−5. The other two models were a previous analysis of the Brachman data assuming each day was an independent trial using an exponential dose–response model [15] and a separate analysis of anthrax infection in Rhesus monkeys, specifically developed to model anthrax clearance rates [10]. This model is similar to our model when α = 1, and produced a clearance rate and hazard rate of 0.0029 h−1 and 2.08 × 10−7 h−1.
Figure 5.

Comparison of our best-fit results with other anthrax models when modelling risk for a single bolus dose. To calculate this risk when α = 0.9, equations (2.6) and (2.9) were used in combination with MLE values, s = 1.81 × 10−7 h−1 and γ = 0.001 h−1. When α = 1, our model is equivalent to an exponential model with k = s/γ = 3.57 × 10−5. Previous exponential modelling of Brachman data assuming each day as an independent dosing event yielded k = 2.4 × 10−5 [15]. A model of anthrax outbreak in Rhesus monkeys which included clearance rate and hazard rate yielded an attack rate formula equivalent to an exponential model with k = 7.17 × 10−5 [10]. Thick line, Brachman results 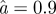 ; dashed line, Brachman results α = 1; dash-dotted line, Brachman results [15]; dotted line, Brookmeyer et al. [10].
; dashed line, Brachman results α = 1; dash-dotted line, Brachman results [15]; dotted line, Brookmeyer et al. [10].
Next, we calculated the corresponding risks for the exposure patterns given in §2.6. We chose the corresponding MLE parameter sets for several α values ranging from 0.5 to 1. Figure 4d depicts the predicted risks over this α range, comparing the bolus exposure to the evenly distributed exposure pattern. We can see that as α approaches 1, the gap between the predicted risks decreases. The largest gap presented occurs at α = 0.5 where the bolus exposure has a risk of 64 per cent and the distributed exposure has a risk of 59 per cent. When α = 0.9, the bolus risk is 47 per cent and the distributed exposure has a risk of 46 per cent indicating dose–timing effects that are small.
4. Discussion
4.1. Plausibility of our model as a dose–response model
Using a simple function that expresses cumulative dose dependence on the pathogen elimination rate, we are able to realistically relax the assumption that the risk of each pathogen dose is independent of the time of arrival of other pathogen doses. Further, through a survival analysis, we have presented a method for analysing dose–response time-series data of exposure and infection events. As a case study, we presented an analysis of inhalational anthrax infection in Cynomologus monkeys in an industrial setting [5]. Our optimization found the best-fitting parameters for these data as follows: α = 0.9, γ = 0.0097 h−1, and s = 1.81 × 10−7 h−1. This result indicates that there are very slight dose–timing effects, as indicated by the risk difference of 1 per cent from the simulated exposure experiments. This result also appears dependent on our lag time assumptions. The problem of non-identifiability between our clearance rate (γ) and lag period (τ) requires us to make some assumptions about the overall distribution of the lag period, a problem also found in a previous anthrax analysis [10], where they optimized a convolution of a time to germination likelihood and an exponential lag period. Our lag period describes a period beginning with infection take-off, which occurs sometime after germination within macrophages [11], and ending in death. The point at which infection has taken off is not provided in the Brachman experiment and thus we must rely on an assumed lag period. To test the sensitivity to lag period assumptions, we tried other distributions, such a larger range of lag period and a truncated exponential. If all times were weighted equally or if there were heavier weighting on longer lag periods (greater than 6 days), the MLE α was estimated at 1. However, a distribution that weights faster lag periods more (such as the exponential with mean of about 2 days) resulted in MLE α values between 0.9 and 1. While lag periods may be less variable and slower for doses under the ID50 [9], such as in this dataset, future anthrax modelling for larger doses may require careful distribution selection consistent with experiments done with higher dose levels [8]. The fitted values of s and γ decrease slightly as the lag period decreases. This result implies that our estimate of within host pathogen persistence and risk depends on our lag period selection and therefore we may be incorrectly classifying periods where dose–timing is important as the lag period.
Although our model is unique in its implementation of multiple doses, we can compare it to other dose–response models when considering the risk from a single dose. As illustrated in figure 5, our model produced consistent results with other anthrax models. Particularly, the ID50 of all these models are all within an order of magnitude. A review of several anthrax dose–response models [16] found that risk models that were similar to exponential distributions were the most successful at modelling anthrax risk. Our analysis is consistent with this result. To truly check consistency among other analyses, however, requires more data and models implementing multiple dosing.
Our 95 per cent CI of (0.51, 1) reflects imprecision in estimation of α values. Our confidence interval calculation is limited in this example as we implemented an artificial upper bound on α that is biological instead of statistical. If we allow α values greater than 1, we find this interval to extend as far as 1.48, as discussed in the online electronic supplementary material. If α > 1, we would have a paradigm where bolus exposures have lower risks than smaller, distributed exposures of the same total dose.
The strong functional relationship of α and γ implies that they should not be estimated independently. Particularly, they share a decreasing log linear relationship (figure 4c) showing that γ values decrease when α values increase. That is, when clearance decreases in speed, the shape of the clearance curve becomes more curvilinear. This illustrates a range of potential clearance curves that describe the pathogen decay. The s parameter then provides us with an instantaneous per pathogen infection risk over the clearance time. When estimating the parameters for our model, we suggest a profile optimization over α. Using this approach, inference can then be done to determine if α differs from 1, which would imply that risk estimation depends on dose–timing. When using the model to estimate exposure risks or infection timing, we assign the appropriately fitted γ value to a specified α value.
One limitation of our model owing to its simplicity is the use of abstract parameters. The α and s parameters are not readily biologically interpretable from the results and the parameter γ is only interpretable in the special case of exponential clearance. We can still present expected clearance curves and describe expected behaviour of the system with known parameters. For example, if we know we do not have exponential clearance (α < 1), then bolus exposures correspond to the highest risk of infection. However, given two different pathogens with this property (α < 1) but different α values, it is not clear what the differing values of α tell us specifically about each pathogen, especially if the other parameters vary also. Furthermore, extrapolating our results to human populations requires additional assumptions. For traditional dose–response experiments, we would need susceptibility of the host animal to be similar to humans. Additionally, for our model, we would also need the surrogate's immune response to occur at a similar rate with similar effectiveness to the human population. Cynomolgus monkeys and humans have similar anthrax infection pathology [8], which implies that we may expect the dynamics and risk assessments to be similar given these exposure patterns, however, it is not clear how the parameter sets might differ.
Another limitation of our model is that it does not take into account the reproduction of pathogens within host tissue, i.e. we only model pathogen clearance as a decreasing curve between inoculations. Ideally, a model of the infection process includes pathogen elimination by the immune system and the growth of the pathogen within the host. The persistence of pathogen would then be more realistically described as a stochastic process, with spikes both increasing and decreasing over clearance until either the pathogen level reaches zero or begins to reproduce to an unbounded level, as presented in a previous model [4]. This model, however, requires additional parameters and is computationally intensive that makes its implementation into complex models of environmental infection transmission systems difficult. By ignoring pathogen growth during innate clearance but before the infection takes off, the overall shape of pathogen decrease described by our model will be different than the more realistic model that takes into account both processes. However, by relaxing the assumption that infection risk must be time-independent, our model is a step forward in dose–response risk assessment.
4.2. Experiments to inform time-dependent dose–response models
Our proposed exposure patterns illustrate simple experimental structures that would elicit varying risk owing to dose–timing effects. It is important to note that our model is only one potential realization of time-dependence in dose–response models. Conducting the proposed experiments and observing a significant risk difference between exposure patterns might be enough to imply that risk depends on dose–timing. This discovery alone would illustrate how characterizing the risk of different routes of transmission is critical, and further, would have important ramifications on intervention policies.
To conduct such a time-dependent dose–response experiment, preliminary experiments would need to be conducted to find a viable dose; that is, doses that do not have risks near 0 or 100 per cent. Further, these preliminary experiments need to give insight into the time scale of interest. We used doses spaced by days since our results pointed to clearance rates on the scale of days. For a pathogen like the influenza virus, we may expect dose–timing effects to be on a shorter time scale. After preliminary experiments, dosing schemes similar to our proposed patterns should be implemented with fixed total doses. By using exposure patterns that differ so widely, we are better able to evaluate the impact of dose–timing on the effectiveness of the immune system. Simply observing varying risk by exposure pattern can provide enough insight to imply that risk depends on dose–timing. Simple analysis on differing risks would be sufficient to show dose–timing effects. One shortfall of these experiments is that they would probably require large subject sizes unless there are substantial dose–timing effects. If we look at the predicted risks of our exposure patterns from our analysis when α = 0.5, we would need 1550 subjects per pattern to find a statistically significant difference at the 0.05 level with 80 per cent power using Fisher's two-sided exact test. Naturally, if there are stronger dose–timing effects (risks differences are much higher), the necessary sample size drops. Other options would include designing experiments that are mechanistically specific to the pathogens of interest and monitoring the corresponding immune response. For anthrax, we may design an experiment monitoring macrophage loads over different dosing patterns and then analysing the data with an anthrax-specific mechanistic model. This scenario would reduce the emphasis on elucidating risk differences and therefore may not require such large sample sizes.
By using a sufficient amount of subjects, the time to infection distribution and overall risk would be stabilized for each dosing course and thus provide stable data to estimate our parameters. Further, using varying total doses is an additional method to introduce precision to parameter estimation in our model. There were many limitations in using the Brachman data to estimate our model parameters. The data had small sample size and only two exposure patterns. Furthermore, the exposure patterns are roughly similar for each run and the total doses differ. The estimation of α depends on the effect of dose–timing and thus would be best estimated by widely varying exposure patterns of the fixed total dose.
4.3. Time-dependence in the dose–response model paradigm
Through the use of our model, we have shown that exposure timing can be used in the calculation of risk and estimation of infection times. This is a distinct advantage over the risk calculation of classical dose–response models. In transmission systems, different routes of transmission lead to varying types of exposure patterns. We can now relax the assumption that the risk is invariant to different dose–timing and thus varying exposure patterns. Even if our model predicts invariant risks to exposure patterns (exponential clearance), it allows us to estimate when infections would occur over an exposure course.
Fundamentally, we are interested in whether dose–timing is an important factor in the calculation of risk. Our analysis shows that for anthrax, there may not be these effects on the time scale we examined, i.e. the accumulation of inoculations is an independent process with respect to immune clearance when time intervals are 1 day at the minimum. There might be such time dependence across shorter times.
Despite these findings, we still provide a reasonable estimate for the persistence of the pathogen in the host on the scale of days. We expect that the importance of dose–timing would be dependent on the infection process of a given pathogen. For other pathogens, such as non-respiratory bacteria or viruses, we may not expect these properties to remain constant, especially if the immune mechanisms of clearance differ biologically. For example, the B. anthracis bacillus uses macrophages as a transport to the lymph system in the course of the infection process [12]. This is a unique mode of infection that affects both immune effectiveness and clearance time scale that differs from infection processes of many other pathogens, e.g. the influenza virus.
Infectious disease transmission systems are time-dependent processes generally involving many different types of environmental exposure routes. In influenza transmission, the virus can be transmitted in the air, through direct contact, and through fomite surfaces [17]. In anthrax bioterrorism scenarios, we may wish to consider the risks of large release versus a small steady release of spores. For an enteric disease, like cholera, norovirus or pathogenic Escherichia coli, competing routes such as contaminated food or water contribute to varying exposure patterns. Each of these transmission routes could be characterized by distinct exposure patterns such as evenly distributed, small exposures (e.g. breathing dispersed pathogen in air) or a large bolus exposure (e.g. consuming a contaminated glass of water). Modelling these routes requires many assumptions, particularly when time-independent dose–response models are implemented to give a risk calculation for a given exposure pattern. If exposures occur in a time frame in which the immune system has begun to respond but has not cleared the pathogen, we may no longer have independent pathogen risk calculations. In this scenario, distinct exposure routes may elicit differing risk properties. To see these properties, dose–timing effects must be considered in dose–response experiments, such as we suggest, elucidating the time scale of clearance and the potential importance to risk calculations. We aim to develop a dose–response paradigm that readily includes time-dependence in both risk and infection time calculations.
Acknowledgements
This research was supported by US EPA Science to Achieve Results (STAR) Programme and by US Department of Homeland Security University Programmes (grant no. R83236201).
References
- 1.Haas C. N., Rose J. B., Gerba C. P. 1999. Quantitative microbial risk assessment. New York, NY: John Wiley [Google Scholar]
- 2.Haas C. N. 1983. Estimation of risk due to low doses of microorganisms: a comparison of alternative methodologies. Am. J. Epidemiol. 118, 1097–1100 [DOI] [PubMed] [Google Scholar]
- 3.Haas C. N. 1996. How to average microbial densities to characterize risk. Water Res. 30, 1036–1038 10.1016/0043-1354(95)00228-6 (doi:10.1016/0043-1354(95)00228-6) [DOI] [Google Scholar]
- 4.Pujol J. M., Eisenberg J. E., Haas C. N., Koopman J. S. 2009. The effect of ongoing exposure dynamics in dose response relationships. PLoS Comput. Biol. 5, e1000399. 10.1371/journal.pcbi.1000399 (doi:10.1371/journal.pcbi.1000399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brachman P. S., Kaufman A. F., Dalldorf F. G. 1966. Industrial inhalation Anthrax. Bacteriol. Rev. 30, 646–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lowen A. C., Mubareka S., Tumpey T. M., García-Sastre A., Palese P. 2006. The guinea pig as a transmission model for human influenza viruses. Proc. Natl Acad. Sci. USA 103, 9988–9992 10.1073/pnas.0604157103 (doi:10.1073/pnas.0604157103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srinivasan A., Foley J., Ravindran R., McSorley S. J. 2004. Low-dose Salmonella infection evades activation of flagellin-specific CD4 T cells. J. Immunol. 173, 4091–4099 [DOI] [PubMed] [Google Scholar]
- 8.Vasconcelos D., Barnewall R., Babin M., Hunt R., Estep J., Nielsen C., Carnes R., Carney J. 2003. Pathology of inhalation anthrax in Cynomolgus Monkeys (Macaca fascicularis). Lab. Invest. 83, 1201–1209 [DOI] [PubMed] [Google Scholar]
- 9.Meynell G. G., Meynell E. W. 1958. The growth of micro-organisms in vivo with particular reference to the relation between dose and latent period. J. Hyg. (London) 56, 323–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brookmeyer R., Johnson E., Barry S. 2005. Modeling the incubation period of anthrax. Stat. Med. 24, 531–542 10.1002/sim.2033 (doi:10.1002/sim.2033) [DOI] [PubMed] [Google Scholar]
- 11.Guidi-Rontani C. 2002. The alveolar macrophage: the Trojan horse of Bacillus anthracis. Trends Microbiol. 10, 405–409 10.1016/S0966-842X(02)02422-8 (doi:10.1016/S0966-842X(02)02422-8) [DOI] [PubMed] [Google Scholar]
- 12.Ruthel G., Ribot W. J., Bavari S., Hoover T. A. 2004. Time-lapse confocal imaging of development of Bacillus anthracis in macrophages. J. Infect. Dis. 189, 1313–1316 10.1086/382656 (doi:10.1086/382656) [DOI] [PubMed] [Google Scholar]
- 13.Radyuk S. N., Mericko P. A., Popova T. G., Grene E., Alibek K. 2003. In vitro-generated respiratory mucosa: a new tool to study inhalational anthrax. Biochem. Biophys. Res. Commun. 305, 624–632 10.1016/S0006-291X(03)00830-1 (doi:10.1016/S0006-291X(03)00830-1) [DOI] [PubMed] [Google Scholar]
- 14.He D., Ionides E. L., King A. A. 2010. Plug-and-play inference for disease dynamics: measles in large and small populations as a case study. J. R. Soc. Interface 7, 271–283 10.1098/rsif.2009.0151 (doi:10.1098/rsif.2009.0151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas C. N. 2002. On the risk of mortality to primates exposed to anthrax spores. Risk Anal. 22, 189–193 10.1111/0272-4332.00028 (doi:10.1111/0272-4332.00028) [DOI] [PubMed] [Google Scholar]
- 16.Wilkening D. A. 2006. Sverdlovsk revisited: modeling human inhalation anthrax. Proc. Natl Acad. Sci. USA 103, 7589–7594 10.1073/pnas.0509551103 (doi:10.1073/pnas.0509551103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber T. P., Stilianakis N. I. 2008. Inactivation of influenza A viruses in the environment and modes of transmission: a critical review. J. Infect. 57, 361–373 10.1016/j.jinf.2008.08.013 (doi:10.1016/j.jinf.2008.08.013) [DOI] [PMC free article] [PubMed] [Google Scholar]