

Abstract
Acidic and basic fibroblast growth factors (FGF-1/FGF-2) promote angiogenesis in cancer. Angiogenesis is integral to cardiac repair following myocardial infarction (MI). The potential regulation of FGF-1/FGF-2 in cardiac angiogenesis postMI remains unexplored. Herein, we examined the temporal and spatial expression of FGF-1/FGF-2 and FGF receptors (FGFR) in the infarcted rat heart at day 1, 3, 7, and 14 postMI. FGF-1/-2 gene and protein expression, cells expressing FGF-1/-2 and FGFR expression were examined by quantitative in situ hybridization, RT-PCR; western blot, immunohistochemistry and quantitative in vitro autoradiography. Compared to the normal heart, we found that in the border zone and infarcted myocardium 1) FGF-1 gene expression was increased in the first week postMI and returned to control levels at week 2; FGF-1 protein levels were, however, largely reduced at day 1, then elevated at day 3 peaked at day 7 and declined at day 14; and cells expressing FGF-1 were primarily inflammatory cells; 2) FGF-2 gene expression was significantly elevated from day 1 to day 14; the increase in FGF-2 protein level was most evident at day 7 and cells expressing FGF-2 were primarily endothelial cells; 3) FGFR expression started to increase at day 3 and remained elevated thereafter; and 4) FGF-1/FGF-2 and FGFR expression remained unchanged in the noninfarcted myocardium. Thus, FGF-1/FGF-2 and FGFR expression is enhanced in the infarcted myocardium in the early stage after MI, which is spatially and temporally coincident with angiogenesis, suggesting that FGF-1/FGF-2 are involved in regulating cardiac angiogenesis and repair.
Keywords: Myocardial infarction, Angiogenesis, FGF-1, FGF-2, FGFR
INTRODUCTION
Angiogenesis is the formation of new capillary blood vessels from existent microvessels. It plays a critical role in various biological processes such as wound healing, embryological development, the menstrual cycle, and inflammation and the pathogenesis of various diseases such as cancer, diabetic retinopathy, and rheumatoid arthritis. Promoting angiogenesis can aid in accelerating various physiological processes requiring increased vascularization such as the healing of wounds, fractures, and burns, and the treatment of inflammatory diseases, ischemia, peripheral vascular disease, and myocardial infarction (MI). Conversely, inhibition of angiogenesis can aid in the treatment of diseases such as cancer, diabetic retinopathy, and rheumatoid arthritis, where increased vascularization contributes to their progression.
The sequence of events that lead to the sprouting of new vessels is fairly well documented. Initially, a degradation of basement membrane associated with an increase in vessel permeability and proteolysis of the surrounding extracellular matrix leads, upon action of angiogenic factors, to initiate endothelial cell migration and proliferation. As endothelial cells proliferate and assemble, they receive instructions to mature the newly formed sprouts and form a lumen.
The family of Fibroblast growth factors (FGFs) is multifunctional proteins with a wide variety of effects. FGFs are key players in the processes of proliferation and differentiation of wide variety of cells and tissues. FGFs are critical during normal development of both vertebrates and invertebrates and any irregularities in their function leads to a range of developmental defects. FGFs have been reported to promote angiogenesis in pathological conditions, such as cancer. In the FGF family, acidic FGF (FGF-1) and basic FGF (FGF-2) are considered most important in stimulating endothelial cell proliferation and the physical organization of endothelial cells into tube-like structures, thus promoting angiogenesis in cancer. FGF exerts its biologic effect through interaction with cell surface FGF receptors (FGFR). The mammalian FGFR family has four members, FGFR-1, FGFR-2, FGFR-3, and FGFR-4. FGF-1 and FGF-2 are ligands of all different FGFR.
Myocardial infarction (MI) has emerged as a major health problem during the past two decades. Following MI, cardiac repair occurring at the site of myocyte loss preserves structural integrity and is essential to the heart’s recovery. Angiogenesis is central to cardiac repair and numerous studies have demonstrated that stimulation of angiogenesis is beneficial to both the ischemic and the infarcted heart. In a previous study, we have shown that following MI, angiogenesis begins at the border zone and then extends into the infarcted myocardium). Microvascular density in the infarcted myocardium peaks at day 7 postMI and subsequently declines, indicating that angiogenesis is most active in the early stages of cardiac repair and then gradually becomes quiescent when cardiac repair is completed. Local factors regulating the angiogenic process in the infarcted heart are not well understood, and the involvement of FGFs in cardiac angiogenesis is unknown. The current study is undertaken to investigate the involvement of FGF-1/FGF-2 in cardiac angiogenesis following MI. Using molecular and cellular approaches and imaging techniques, we explored the temporal and spatial expression of cardiac FGF-1/FGF-2 and FGFR following experimental MI in rats.
METHODS
Animal Model
The study was approved by University of Tennessee Health Science Center Animal Care and Use Committee. Left ventricular anterior transmural MI was created in 8-week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN) via permanent ligation of left coronary artery. Animals were anesthetized with 1.5% isoflurane, intubated and ventilated with a rodent respirator. The heart was exposed via a left thoractomy and the left anterior descending artery was ligated with a 6-0 silk suture. The chest was then closed and lungs reinflated using positive end-expiratory pressure. Only hearts with large free wall infarction (40–45% of left ventricle) were used in the study. Unoperated age and sex matched rats served as controls (n = 8). Sham-operated rats were not used as controls, since sham-operation causes pericardial injury/angiogenesis, thus affects data interpretation. Rats with MI were sacrificed at postoperative days 1, 3, 7 and 14 (n = 8 survived rats for each time point). Hearts were removed and kept frozen at −80 °C until use.
Quantitative In Situ Hybridization
The localization and density of cardiac FGF-1 mRNA was analyzed by quantitative in situ hybridization. Briefly, coronal cryostat sections (16μm) were fixed in 4% formaldehyde for 10 min (min) and incubated in 0.25% acetic anhydride in 0.1M TE-HCL for 10 min. Sections were then hybridized (overnight at 56°C) with a random primed 35S-UTP-labeled FGF-1 RNA probe. Sections were washed, dried, and subsequently exposed to Kodak Biomax X-ray film. Exposure time was based on signal intensity. Quantification of mRNA optical density was performed using a computer image analyzing system (NIH Image, 1.60).
RT-PCR
Total RNA was extracted from cardiac tissue using Trizol Reagent (Invitrogen, Carlsbad, CA). The RNA was treated with DNase by using TURBO DNA-free kit (Ambion, Austin, TX), and purified with RNeasy Mini Kit (Qiagen Inc. USA, Valencia, CA). The purification, concentration and integrity of the RNA were examined with NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE), and Angilent Bioanalyzer (Angilent Technologies, Foster City, CA), respectively. cDNA was prepared from total RNA using a High Capacity cDNA reverse transcriptase kit (Applied Biosystems Foster City, CA). The gene-specific probes and primer sets for FGF-2 were deduced using Universal ProbeLibrary Assay Design software (https://www.roche-applied-science.com). FGF-2 mRNA levels were detected and analyzed on an LightCycler 480 System (Roche, Indianapolis, IN) under the following cycling conditions: 1 cycle at 95°C for 5 min and then 45 cycles at 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 10 seconds. The PCR mix contained 0.2μl of 10μM primers, 0.1μl of 10μM Universal library probe, 5μl of LC 480 master mix (2X), 2μl of template cDNA and RNase-free water to 10μl. TATA box-binding protein has been selected as endogenous quantity control. Fold change were used to compare the different between groups.
Western Blotting
FGF-1 and FGF-2 protein levels were measured by western blot. Briefly, the normal, noninfarcted left myocardium (septum), border zone and infarcted myocardium were dissected and homogenized. The supernatant was collected and separated by 10% SDS-PAGE. After electrophoresis, samples were transferred to PVDF membranes and incubated with antibody against FGF-1 or FGF-2 (Santa Cruz Biotechnology, Santa Cruz, CA). Blots were subsequently incubated with peroxidase-conjugated secondary antibody (Sigma, St. Louis, MO). After washing, the blots were developed with the enhanced chemiluminescence method. The amount of protein detected was assessed by means of quantitative densitometry analysis with a computer image analyzing system.
Immunohistochemistry
Cells expressing FGF-1 and FGF-2 were detected by immunohistochemical labeling. Cryostat coronal sections (6μm) were air-dried, fixed in 10% buffered formalin for 10 min, and washed in phosphate-buffered saline (PBS). Endogenous peroxidase activity was blocked by immersion of the sections in 0.3% H2O2 for 10 min at room temperature. Sections were blocked with 5% goat serum in PBS. Tissues were incubated with primary antibodies against FGF-1 or FGF-2 for 1 hr at room temperature. Sections were then incubated with peroxidase conjugated secondary antibody for 1 hr at room temperature, washed in PBS for 10 min, and incubated with 0.5 mg/ml diaminobenzidine tetrahydrochloride 2-hydrate + H2O2 for 1 min. Negative control sections were incubated with secondary antibody alone. Sections were then dehydrated, mounted, and viewed by light microscopy.
Quantitative In Vitro Autoradiography
The localization and density of FGFR were detected by quantitative in vitro autoradiography. Cryostat 16μm coronal sections of heart were preincubated for 15 min in PBS buffer containing 0.2% bovine serum albumin, then incubated for 1 hr in a fresh volume of the same buffer containing 0.2μCi/mL 125I-FGF-2 (PerkinElmer, Waltham, MA). Nonspecific binding was measured in the presence of 1μM unlabeled FGF-2. After incubation, sections were washed, dried and exposed to Kodak NMB-6 film. Quantification of FGFR binding density was performed using a computer image analysis system. FGFR binding is expressed as optical density.
Statistical Analysis
Statistical analysis of data obtained from in situ hybridization, RT-PCR, western blot and in vitro autoradiography was performed using analysis of variance. Values are expressed as mean±SEM with P<0.05 considered significant. Multiple group comparisons among controls and each group were made by Scheffe’s F-test.
RESULTS
Cardiac FGF-1 Expression
Localization and Density of Cardiac FGF-1 Gene Expression
Using quantitative in situ hybridization, which provides the localization and density of targeted mRNA (Figure 1), we found that FGF-1 mRNA was present in the normal heart with a relatively low density (panel A). Following MI, FGF-1 mRNA in the border zone was significantly increased at day 1 (panel B) compared to controls, remained elevated till day 7 (panel C), and then returned to controls levels at day 14 (panel D). FGF-1 mRNA levels in the infarcted myocardium were significantly reduced at day 1 (panel B), increased at days 3 and 7 (panel C) and then declined to control levels at day 14. FGF-1 mRNA in the noninfarcted myocardium remained not significantly unchanged. Quantitative FGF-1 mRNA density in the border zone and infarcted myocardium is shown in panel E and F, respectively.
Figure 1.
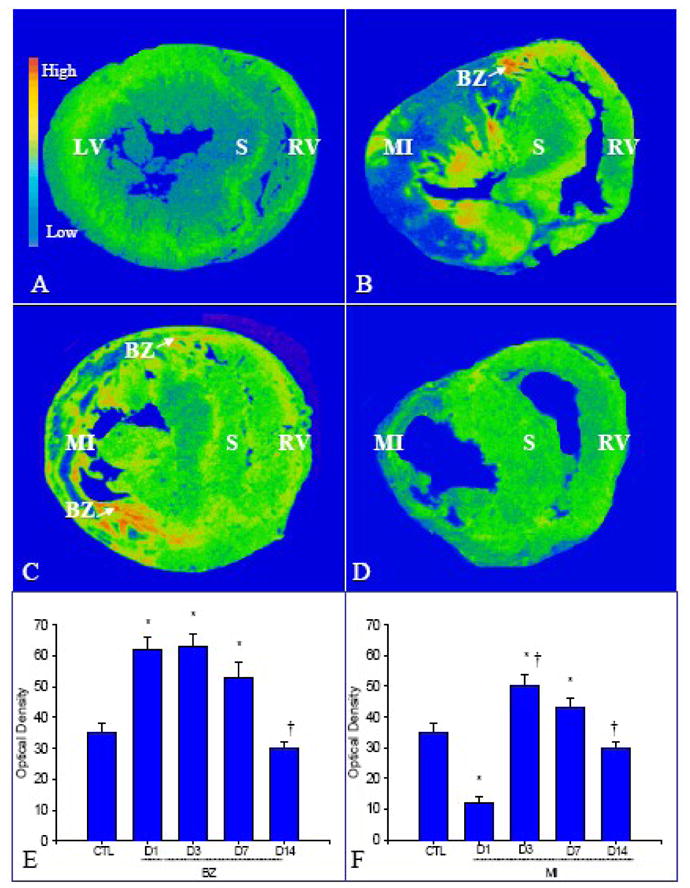
Temporal and spatial FGF-1 gene expression in the infarcted heart: FGF-1 mRNA was normally present in both left and right ventricles (LV, RV) (panel A). Following MI, increased FGF-1 mRNA levels were seen at the border zone at day 1 (panel B), followed by appearance in the infarcted myocardium at day 3 (panel C) and 7, and declined to control levels at day 14 (panel D). FGF-1 mRNA levels remained unchanged in the noninfarcted septum (S). Panels E and F show quantitative FGF-1 mRNA levels in the border zone (BZ) and infarcted myocardium at different time points postMI. *p<0.05 vs controls; †p<0.05 vs previous time point.
Cardiac FGF-1 Protein Levels
Western blot analysis was performed to quantitate the FGF-1 protein levels in the normal myocardium and various regions of the infarcted heart including border zone, infarcted and noninfarcted myocardium at the different time points postMI (Figure 2). FGF-1 was normally present in the rat heart. Following MI, FGF-1 protein levels at the border zone (panel A) and infarcted myocardium (panel B) were significantly reduced at day 1, elevated at day 3, peaked at day 7, then declined, but still remained significantly higher than in controls at day 14. FGF-1 protein levels remained unchanged in the noninfarcted myocardium at all time points (not shown).
Figure 2.
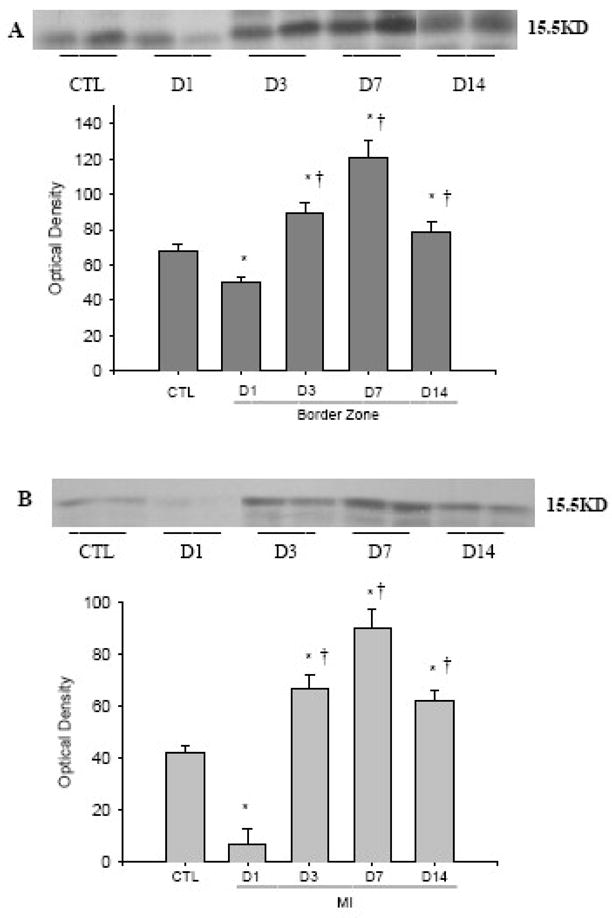
Temporal expression of cardiac FGF-1 postMI: Following MI, FGF-1 protein levels at the border zone (panel A) and infarcted myocardium (panel B) were significantly reduced at day 1, elevated at day 3, peaked at day 7, then declined, but still remained significantly higher than in controls at day 14.
Cells Expressing Cardiac FGF-1
By immunohistochemistry, we observed positive FGF-1 staining in the normal heart, where cells expressing FGF-1 were primarily interstitial cells (Figure 3, panel A, brown spots), while in the infarcted heart, strong staining was observed at the border zone (Figure 3, panel B) and infarcted myocardium (not shown) at day 7, where cells expressing FGF-1 were primarily inflammatory cells and fibroblast-like cells.
Figure 3.
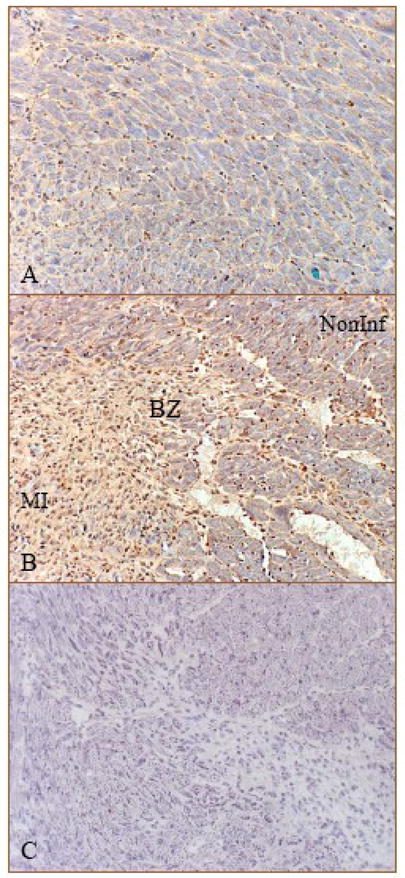
Cells expressing FGF-1 in the normal and infarcted heart: Immunohistochemistry shows positive staining in the interstitial cells of the normal heart (panel A). Following MI, strong staining was seen in the border zone at day 7, where cells expressing FGF-1 were primarily inflammatory cells and fibroblast-like cells (panel B, brown). Panel C: negative control; Noninf: noninfarcted myocardium; X200
Cardiac FGF-2 Expression
Cardiac FGF-2 Gene Expression
As detected by RT-PCR (Figure 4), compared to controls, FGF-2 mRNA was significantly increased in both border zone (panel A) and infarcted myocardium (panel B) from day 1 to day 14 postMI, but the increase was greatest at day 1.
Figure 4.

Quantitative FGF-2 gene expression in the border zone (panel A) and infarcted myocardium (panel B).
Cardiac FGF-2 Protein Levels
FGF-2 contains two isoforms with high and low molecular weight (22kD and 18kD). As detected by western blot (Figure 5) and compared to controls, high molecular weight FGF-2 at the border zone (panel A) was slightly, but not significantly reduced at day 1, significantly increased at day 3, peaked at day 7 and declined to normal levels at day 14. However, in the infarcted myocardium, it was decreased at days 1 and 3 and increased only at day 7 (panel B). The expression of low molecular weight FGF-2 showed similar pattern in the border zone and infarcted myocardium (not shown). However, FGF-2 protein levels remained unchanged in the non infarcted myocardium (not shown).
Figure 5.

Temporal expression of cardiac FGF-2 postMI. Compared to controls, FGF-2 at the border zone (panel A) was slightly reduced at day 1, significantly increased at day 3, peaked at day 7 and declined to normal levels at day 14, while in the infarcted myocardium, it was decreased at days 1 and 3 and increased only at day 7 (panel B).
Cells Expressing Cardiac FGF-2
Immunohistochemical staining revealed that FGF-2 was homogeneously distributed in the normal rat heart (Figure 6, panel A, brown), while in the infarcted heart, FGF-2 expression was increased at day 7 postMI and cells expressing FGF-2 were primarily en-dothelial cells in the newly formed vessels at the border zone (Figure 6, panel B) and infarcted myocardium (not shown).
Figure 6.

Cells expressing FGF-2 in the normal and infarcted heart: Positive FGF-2 staining was primarily distributed in myocytes of the normal heart (panel A). Following MI, strong staining was seen at the border zone at day 7, where cells expressing FGF-2 were primarily newly-formed microvessels (panel B, brown and arrows). Panel C: negative control. X400
The Localization and Density of Cardiac FGFR
The temporal and spatial changes of FGFR in the infarcted heart were detected by quantitative in vitro autoradiography (Figure 7). In the normal heart, we observed low density of FGFR in the left and right ventricles (panel A), and the blood vessels expressed higher density of FGFR (panel A, arrows). Following MI, FGFR binding density was unchanged in the infarcted myocardium at day 1 (panel B), significantly increased at the site of MI at day 3 (panel C) and remained elevated at day 7 (Panel D) and 14 (panel E). FGFR binding density was unchanged in the noninfarcted myocardium compared to controls.
Figure 7.
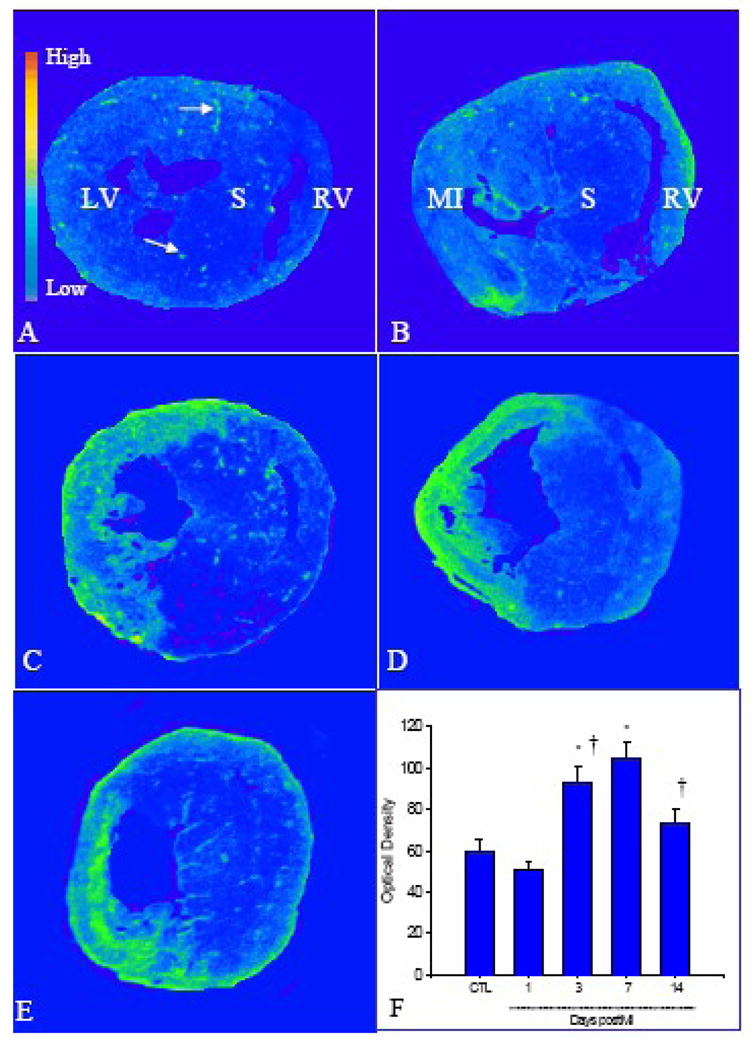
Temporal and spatial expression of cardiac FGFR postMI: As detected by quantitative in vitro autoradiography, low levels of FGFR were present in the normal myocardium (panel A), while blood vessels contained higher levels of FGFR (arrows). Following MI, FGFR levels remained unchanged in the infarcted myocardium at day 1 (panel B), increased at day 3 (panel C), reached a peak at day 7 (panel D), and declined, but remained higher than controls at day 14 (panel E). FGFR level remained unchanged in the noninfarcted septum. Panel F shows quantitative FGFR levels in the infarcted myocardium at different time points postMI.
DISSCUSSION
To determine whether FGF-1/FGF-2 are involved in cardiac angiogenesis after infarction, we investigated the spatial and temporal response of cardiac FGF-1/FGF-2 and FGFR postMI in the current study.
Firstly, we examined the FGF-1 expression in the infarcted heart and its relation to angiogenesis. Angiogenesis is initiated at the border zone between infarcted and noninfarcted myocardium and then extends into the infarcted myocardium. It is most evident in the first week postMI. We observed rapid increase in FGF-1 gene expression at the border zone, starting as early as day 1 postMI. The early rise of FGF-1 expression suggests that FGF-1 is involved in initiating angiogenesis in response to cardiac injury. FGF-1 gene and protein expression remained elevated in the border zone for a week before declining to the normal level. FGF-1 expression in the infarcted myocardium, however, was significantly reduced at day 1. This is likely due to the degradation of preexisting FGF-1 in the necrotic myocardium. Elevated FGF-1 gene and protein expression in the infarcted myocardium peaked at day 7 and then declined. The pattern of FGF-1 expression is coincident with angiogenesis in the infarcted myocardium, suggesting the involvement of FGF-1 in cardiac new vessel formation. It has been reported that when delivered to the myocardium in dogs with compromised coronary, FGF-1 causes vascular smooth muscle cell hyperplasia in areas of ischemic injury, also supporting the regulatory role of FGF-1 in cardiac angiogenesis.
Our immunohistochemical study has further revealed that inflammatory and fibroblast-like cells are the primary cells responsible for the elevated FGF-1 expression in the infarcted heart. In addition to FGF-1, these cells release other proangiogenic growth factors, such as vascular endothelial growth factor and transforming growth factor-beta in the repairing tissue, playing key roles in angiogenesis during tissue repair.
The involvement of FGF-1 in angiogenesis has been explored in other pathological conditions. Sustained angiogenesis is considered to be one of central hallmarks of cancer and FGF-1 serves as the predominant regulator of the pathological angiogenic process. It has been shown that FGF-1 is most closely linked to cancer progression in various organs. The anti-FGF signaling system has been considered an attractive therapeutic target for cancers.
Secondly, we quantitated the cardiac temporal and spatial FGF-2 expression in response to MI. Following MI, FGF-2 mRNA started to increase at the border zone and in the infarcted myocardium at day 1 and remained elevated over the course of 14 days, while the increased in FGF-2 protein was most evident at day 7. Immunohistochemistry revealed that vascular endothelial cells were the major cells expressing FGF-2. Thus, like FGF-1, elevated FGF-2 expression is coincident with the angiogenic response in the infarcted myocardium, suggesting that FGF-2 also plays a role in cardiac angiogenesis following MI. Many studies have shown that intracardiac or systemic delivery of FGF-2 to animals with MI increases vascular density in the infarcted myocardium. It has been also reported that deletion of FGF-2 in mice decreased endothelial proliferation in the infarcted heart. These data further demonstrated the importance of FGF-2 in cardiac angiogenesis postMI.
Thirdly, we mapped the localization and density of FGFR in the normal heart and the temporal changes of cardiac FGFR following MI. FGF-1/-2 stimulates cellular responses by binding to FGFR on the cell surface, causing them to become activated through transphosphorylation. Using 125I-FGF-2 as a ligand, we observed a low FGFR binding density in the normal rat heart, and blood vessels expressed the highest density of FGFR. A high density of FGFR in the blood vessels suggests that FGFs regulate vascular endothelial proliferation in the normal heart. Following MI, FGFR binding density was greatly increased in the infarcted myocardium in the early stage of MI and colocalized with elevated local FGF-1/-2. These observations imply that FGF-1/-2 play a role in cardiac angiogenesis/repair in an autocrine/paracrine manner.
Several factors have been reported to induce the expression of FGFs in the repairing tissue. Hypoxia is found to stimulate the expression of FGF-1 and FGF-2 as well as FGFR in the brain after injury. In addition, nitric oxide is shown to contribute to angiogenesis through activation of FGF-2 production. In the infarcted heart, inducible nitric oxide synthase, a major source of nitric oxide in the repairing tissue, is largely increased in the early stage of MI, which is colocalized with the enhanced FGF expression. Furthermore, FGF-2 expression can be mediated by increases in superoxide levels via NADPH oxidase activation. Our previous study has shown increased NADPH oxidase in the infarcted myocardium, particular in the early stage of MI, which is coincident with cardiac FGF expression.
In summary, this is the first exploring the spatial and temporal expression of FGF-1/-2 (mRNA and protein) and FGFR in the infarcted rat heart. FGF-1/-2 and FGFR expression were significantly increased at the border zone and infarcted myocardium in the early stages of MI, which is coincident with angiogenesis. These data suggest that FGF-1/-2 plays a role in cardiac angiogenic response following acute MI in an autocrine and paracrine manner.
Acknowledgments
The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology.
This work was supported by NIH Heart, Blood, and Lung Institute (RO1-HL77668, Yao Sun).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Tieqiang Zhao, Division of Cardiovascular Diseases, Department of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee.
Wenyuan Zhao, Division of Cardiovascular Diseases, Department of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee.
Yuanjian Chen, Division of Cardiovascular Diseases, Department of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee.
Robert A Ahokas, Department of Obstetrics and Gynaecology, University of Tennessee Health Science Center, Memphis, Tennessee
Yao Sun, Division of Cardiovascular Diseases, Department of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee
References
- 1.Timar J, Dome B, Fazekas K, Janovics A, Paku S. Angiogenesis-dependent diseases and angiogenesis therapy. Pathol Oncol Res. 2001;7:85–94. doi: 10.1007/BF03032573. [DOI] [PubMed] [Google Scholar]
- 2.Ren G, Dewald O, Frangogiannis NG. Inflammatory mechanisms in myocardial infarction. Curr Drug Targets Inflamm Allergy. 2003;2:242–56. doi: 10.2174/1568010033484098. [DOI] [PubMed] [Google Scholar]
- 3.Kang DH, Kanellis J, Hugo C, Truong L, Anderson S, Kerjaschki D, et al. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 2002;13:806–16. doi: 10.1681/ASN.V133806. [DOI] [PubMed] [Google Scholar]
- 4.Wheatley-Price P, Shepherd FA. Targeting angiogenesis in the treatment of lung cancer. J Thorac Oncol. 2008;3:1173–84. doi: 10.1097/JTO.0b013e318187220f. [DOI] [PubMed] [Google Scholar]
- 5.Roccaro AM, Russo F, Cirulli T, Di Pietro G, Vacca A, Dammacco F. Antiangiogenesis for rheumatoid arthritis. Curr Drug Targets Inflamm Allergy. 2005;4:27–30. doi: 10.2174/1568010053622911. [DOI] [PubMed] [Google Scholar]
- 6.Engelmann GL, Dionne CA, Jaye MC. Acidic fibroblast growth factor and heart development. Role in myocyte proliferation and capillary angiogenesis. Circ Res. 1993;72:7–19. doi: 10.1161/01.res.72.1.7. [DOI] [PubMed] [Google Scholar]
- 7.Itoh N. The Fgf families in humans, mice, and zebrafish: their evolutional processes and roles in development, metabolism, and disease. Biol Pharm Bull. 2007;30:1819–25. doi: 10.1248/bpb.30.1819. [DOI] [PubMed] [Google Scholar]
- 8.Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9:639–51. doi: 10.2174/156800909789057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen GJ, Forough R. Fibroblast growth factors, fibroblast growth factor receptors, diseases, and drugs. Recent Pat Cardiovasc Drug Discov. 2006;1:211–24. doi: 10.2174/157489006777442478. [DOI] [PubMed] [Google Scholar]
- 10.Cenni E, Perut F, Granchi D, Avnet S, Amato I, Brandi ML, et al. Inhibition of angiogenesis via FGF-2 blockage in primitive and bone metastatic renal cell carcinoma. Anticancer Res. 2007;27:315–9. [PubMed] [Google Scholar]
- 11.Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–24. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- 12.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–97. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 13.Ahn A, Frishman WH, Gutwein A, Passeri J, Nelson M. Therapeutic angiogenesis: a new treatment approach for ischemic heart disease--Part II. Cardiol Rev. 2008;16:219–29. doi: 10.1097/CRD.0b013e3181620e50. [DOI] [PubMed] [Google Scholar]
- 14.Pandya NM, Dhalla NS, Santani DD. Angiogenesis--a new target for future therapy. Vascul Pharmacol. 2006;44:265–74. doi: 10.1016/j.vph.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 15.Boodhwani M, Sodha NR, Laham RJ, Sellke FW. The future of therapeutic myocardial angiogenesis. Shock. 2006;26:332–41. doi: 10.1097/01.shk.0000225318.08681.a7. [DOI] [PubMed] [Google Scholar]
- 16.Nelson MA, Passeri J, Frishman WH. Therapeutic angiogenesis: a new treatment modality for ischemic heart disease. Heart Dis. 2000;2:314–25. [PubMed] [Google Scholar]
- 17.Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Reactive oxygen species promote angiogenesis in the infarcted rat heart. Int J Exp Pathol. 2009;90:621–9. doi: 10.1111/j.1365-2613.2009.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Weber KT. Angiotensin II receptor binding following myocardial infarction in the rat. Cardiovasc Res. 1994;28:1623–8. doi: 10.1093/cvr/28.11.1623. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–69. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- 20.Wolanska M, Bankowska-Guszczyn E, Jaworski S. Fibroblast growth factor gene expression in uterine leiomyomas. Ginekol Pol. 2008;79:555–9. [PubMed] [Google Scholar]
- 21.Kamalov G, Varma BR, Lu L, Sun Y, Weber KT, Guntaka RV. Expression of the multifunctional Y-box protein, YB-1, in myofibroblasts of the infarcted rat heart. Biochem Biophys Res Commun. 2005;334:239–44. doi: 10.1016/j.bbrc.2005.06.082. [DOI] [PubMed] [Google Scholar]
- 22.Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Reactive oxygen species promote angiogenesis in the infarcted rat heart. Int J Exp Pathol. 2009 doi: 10.1111/j.1365-2613.2009.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao T, Zhao W, Chen Y, Ahokas RA, Sun Y. Vascular endothelial growth factor (VEGF)-A: Role on cardiac angiogenesis following myocardial infarction. Microvasc Res. doi: 10.1016/j.mvr.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banai S, Jaklitsch MT, Casscells W, Shou M, Shrivastav S, Correa R, et al. Effects of acidic fibroblast growth factor on normal and ischemic myocardium. Circ Res. 1991;69:76–85. doi: 10.1161/01.res.69.1.76. [DOI] [PubMed] [Google Scholar]
- 25.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997;57:963–9. [PubMed] [Google Scholar]
- 27.Jouanneau J, Moens G, Montesano R, Thiery JP. FGF-1 but not FGF-4 secreted by carcinoma cells promotes in vitro and in vivo angiogenesis and rapid tumor proliferation. Growth Factors. 1995;12:37–47. doi: 10.3109/08977199509003212. [DOI] [PubMed] [Google Scholar]
- 28.Song S, Wientjes MG, Gan Y, Au JL. Fibroblast growth factors: an epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proc Natl Acad Sci U S A. 2000;97:8658–63. doi: 10.1073/pnas.140210697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takehara N, Tsutsumi Y, Tateishi K, Ogata T, Tanaka H, Ueyama T, et al. Controlled delivery of basic fibroblast growth factor promotes human cardiosphere-derived cell engraftment to enhance cardiac repair for chronic myocardial infarction. J Am Coll Cardiol. 2008;52:1858–65. doi: 10.1016/j.jacc.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 30.Bougioukas I, Didilis V, Ypsilantis P, Giatromanolaki A, Sivridis E, Lialiaris T, et al. Intramyocardial injection of low-dose basic fibroblast growth factor or vascular endothelial growth factor induces angiogenesis in the infarcted rabbit myocardium. Cardiovasc Pathol. 2007;16:63–8. doi: 10.1016/j.carpath.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Virag JA, Rolle ML, Reece J, Hardouin S, Feigl EO, Murry CE. Fibroblast growth factor-2 regulates myocardial infarct repair: effects on cell proliferation, scar contraction, and ventricular function. Am J Pathol. 2007;171:1431–40. doi: 10.2353/ajpath.2007.070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM. Chronic hypoxia up-regulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience. 2002;112:977–91. doi: 10.1016/s0306-4522(02)00060-x. [DOI] [PubMed] [Google Scholar]
- 33.Ziche M, Morbidelli L. Nitric oxide and angiogenesis. J Neurooncol. 2000;50:139–48. doi: 10.1023/a:1006431309841. [DOI] [PubMed] [Google Scholar]
- 34.Wildhirt SM, Dudek RR, Suzuki H, Bing RJ. Involvement of inducible nitric oxide synthase in the inflammatory process of myocardial infarction. Int J Cardiol. 1995;50:253–61. doi: 10.1016/0167-5273(95)02385-a. [DOI] [PubMed] [Google Scholar]
- 35.Black SM, DeVol JM, Wedgwood S. Regulation of fibroblast growth factor-2 expression in pulmonary arterial smooth muscle cells involves increased reactive oxygen species generation. Am J Physiol Cell Physiol. 2008;294:C345–54. doi: 10.1152/ajpcell.00216.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu L, Quinn MT, Sun Y. Oxidative stress in the infarcted heart: role of de novo angiotensin II production. Biochem Biophys Res Commun. 2004;325:943–51. doi: 10.1016/j.bbrc.2004.10.106. [DOI] [PubMed] [Google Scholar]
- 37.Coats AJ. Ethical authorship and publishing. Int J Cardiol. 2009;131:149–50. doi: 10.1016/j.ijcard.2008.11.048. [DOI] [PubMed] [Google Scholar]