

Abstract
Purpose
SR13668, a bis-indole with potent activity in vitro and in vivo against various cancers and promising cancer chemopreventive activity, was found to have very low oral bioavailability, <1%, in rats during pilot pharmacokinetic studies. The objective of these studies was to better understand the source of low oral exposure and to develop a formulation that could be used in preclinical development studies.
Methods
An automated screening system for determining solubility in lipid-based vehicles, singly and in combination, was used to identify formulations that might enhance absorption by improving solubility of SR13668, and these results were confirmed in vivo using Sprague–Dawley rats. Pharmacokinetics of SR13668 was then determined in male and female Sprague–Dawley rats administered 1 mg/kg iv, 1, 10, and 30 mg/kg po formulated in PEG400:Labrasol® (1:1 v/v). Blood was collected at time points through 24 h and the concentration of SR13668 determined using HPLC with UV and fluorescence detection.
Results
SR13668 was found to be resistant to plasma esterases in vitro and relatively stable to rat and human liver microsomal metabolism. SR13668 concentrates in tissues as indicated by significantly higher levels in lung compared to blood, blood concentrations ~2.5-fold higher than plasma levels, and apparent volume of distribution (V) of ~5 l/kg. A marked sex difference was observed in exposure to SR13668 with area under the curve (AUC) significantly higher and clearance (CL) lower for female compared to male rats, after both iv and oral administration. The oral bioavailability (F) of SR13668 was 25.4 ± 3.8 and 27.7 ± 3.9% (30 mg/kg), for males and females, respectively. A putative metabolite (M1), molecular weight of 445 in the negative ion mode (i.e., SR13668 + 16), was identified in blood samples from both the iv and po routes, as well as in vitro microsomal samples.
Conclusions
In summary, while SR13668 does undergo metabolism, probably by the liver, the oral bioavailability of SR13668 in rats was dramatically improved by the use of formulation that contained permeation enhancers and promoted better solubilization of the drug.
Keywords: SR13668, Formulation, Pharmacokinetics, Absorption enhancer, Bioavailability, Chemoprevention, Rat, Drug metabolism
Introduction
Indole-3-carbinol (I3C), a dietary component found in cruciferous vegetables such as cabbage, is known to induce apoptosis and suppress proliferation of many different cancer cell types and has been considered as a cancer chemopreventive agent in preliminary human clinical trials. I3C is unstable in stomach acid, where it is converted into more than 20 different condensation products, 4 of which have demonstrated anticancer activity in a variety of models. In an attempt to identify a more potent compound that would be stable in acid, a lead-based rational drug design program was undertaken that identified SR13668 (Fig. 1) as a drug candidate with both in vitro and in vivo activity against various cancers [1].
Fig. 1.
Structure of SR13668
In a pilot rat pharmacokinetic study, oral administration of SR13668, 50 mg/kg (suspension in 0.5% methyl cellulose), resulted in very low plasma drug concentrations; the highest concentrations were less than 10 ng/ml. Estimation of the absolute oral bioavailability of SR13668 under these conditions was much less than 1%. To continue the preclinical development of SR13668, eVorts to understand the source of low oral exposure were undertaken. The stability of SR13668 in plasma and to liver metabolic enzymes was investigated, as well as ways to increase the absorption of the drug. These investigations resulted in the development of a formulation that markedly enhances oral absorption of SR13668 in rats. The pharmacokinetics and disposition of a single dose of SR13668 in male and female rats were determined in the subsequent study.
Materials and methods
Chemicals
SR13668 (2,10-Dicarbethoxy-6-methoxy-5,7-dihydro-indolo [2,3-b]carbazole) was synthesized at SRI International [1]. The following vehicles were obtained from Sigma–Aldrich (St. Louis, MO): methylcellulose, PEG400, Tween 20, hydroxypropylmethylcellulose, glycerol, corn oil, cremophore EL, Span 85, soybean oil, sesame oil. Gelucire 44/14, Labrasol, Labrafil M1944, Labrafac, and Maisine 35-1 were obtained as gift samples from Gattefosse (Paramus, NJ). DMSO was obtained from Mallinckrodt Baker (Phillipsburg, NJ). PEG300 and propylene glycol were obtained from Spectrum (New Brunswick, NJ); 1-Methyl 2-pyrollidone was obtained from EMD (Gibbstown, NJ). Tween 80, Tween 20, and ethyl alcohol (ethanol) were obtained from Spectrum (Gardena, CA). Solutol HS-15 and Pluronic F127 were obtained from BASF (Mt. Olive, NJ). Miglyol 840 and 812 N were obtained from Creanova (Piscataway, NJ). Cavamax was obtained from EMD (Gibbstown, NJ). Acetonitrile was purchased from Mallinckrodt Baker (Phillipsburg, NJ). Sprague–Dawley rat plasma was purchased from Pelfreez (Rogers, AR). Chromatography C18 column (Luna, 5 μm, 50 × 2 mm) was purchased from Phenomenex (Torrance, CA).
High throughput solubility (HTS) screening
A TECAN FreedomEVO Liquid Handling Instrument (Tecan Systems, San Jose, CA) was used to prepare the excipient combinations. The TECAN dispenser has four tips that can travel in the x, y, and z directions. The FreedomEVOware (software) was programmed to aspirate solvents from the selected positions on the source deck and dispense them into the desired well locations in the flat bottom 96-well plates placed on the dispense deck.
Binary and ternary combinations of excipients were evaluated in n = 3 replicate wells. Baseline turbidity of the excipient combinations was determined using a Tecan GENios Microplate Reader for turbidimetric analysis (Tecan Systems, San Jose, CA) by reading absorbance at 500 nm. A stock solution of the drug was prepared in either absolute ethanol or DMSO and added to 96-well plates containing solvent combinations. The plates were again mixed by shaking at 175–200 rpm for 20–30 min after addition of the drug, and turbidity was measured. Solubility was assessed by evaluating the change in turbidity before and after addition of the drug to the solvent system. Solubility screening was performed at four levels, with the choice of excipients dependent on the behavior of SR13668 at the preceding level. The excipients and the concentrations used at each level of screening are presented in Table 1. Based on the data obtained from Level 4 screening, solubility was assessed at 5 mg/ml concentration of SR13668. To verify the results obtained by HTS screening at all four levels, confirmatory manual evaluation of 30 different cosolvent systems was performed, determining SR13668 concentration by UV spectroscopy at 310 nm.
Table 1.
List of solvents with concentrations used for the SR13668 solubility screening
| Level 1 |
Level 2 |
Level 3 |
Level 4 |
||||
|---|---|---|---|---|---|---|---|
| Vehicles | Concentration | Vehicles | Concentration | Vehicles | Concentration | Vehicles | Concentration |
| Ethanol | 50% v/v in water | Tween 20 | 25% v/v in water | PEG400 Tween 20 (8:2) | 100% | Span 85 | 50% v/v in Ethanol |
| Water | 100% | Labrafac-Maisin (4:1) | 50% v/v in ethanol | PEG400 Tween 80 (8:2) | 100% | Soy bean oil | 100% |
| PEG 400 | 100% | PEG400 | 100% | PG Tween 20 (8:2) | 100% | Sesame oil | 100% |
| Propylene Glycol | 60% v/v in water | Propylene Glycol | 100% | PG Tween 80 (8:2) | 100% | Caproyl PGMC | 100% |
| Tween 80 | 15% v/v in water | Tween 80 | 25% v/v in water | Labrasol PEG (1:1) | 100% | Labrafac | 100% |
| Glycerin | 50% v/v in water | Pluronic F127 | 12.5% w/v in water | Cremophore EL | 25% v/v in water | Labrafil | 100% |
| Labrafil M 1944 | 100% | Labrafac-Maisin (4:1) PEG400-N-Methyl-2- pyrrolidone (1:1) |
100% | Miglyol 840 | 100% | ||
| Labrasol | 100% | PEG400 | 100% | PEG400 | 100% | ||
| N-Methyl-2-pyrrolidone | 40% v/v in water | Propylene Glycol | 100% | Miglyol 812 | 100% | ||
| Solutol HS-15 | 20% w/v in water | Labrasol | 100% | Labrasol | 100% | ||
Plasma protein binding
SR13668 was incubated at concentrations of 2.5, 75, 250, and 1,750 ng/ml in rat plasma at 37°C for 2 h. Following incubation, 400 μl samples were transferred in triplicate at each concentration to upper reservoir of the washed Centri-free YM-30 ultrafiltration units (Millipore, Bedford, MA) and then centrifuged for 20 min at 2,000g. Aliquots of the ultrafiltrates and unfiltered incubation mixtures were analyzed for SR13668 by HPLC and the total protein concentration by the bicinchoninic acid assay for protein concentration (BCA assay; Pierce, Rockford, IL).
Plasma stability
Rat plasma was incubated at 37°C with 0.05 and 1 μg/ml SR13668 plus and minus sodium fluoride (50 μM) to inhibit plasma esterases for up to 60 min. The reaction was stopped with 4 volumes of acetonitrile, and aliquots were analyzed using HPLC with fluorescence detection, as described above.
Metabolism by human and rat liver microsomes
Human liver microsomes (HLM) were pooled from multiple donors (Gentest, Woburn, MA), and rat liver microsomes (RLM) were prepared at SRI by homogenization and centrifugation to collect the microsomal pellet. Liver microsomes were incubated at a final protein concentration of 0.5 mg/ml with 1 or 10 μM SR13668 (in DMSO, final concentration of DMSO in incubation was 0.1%). Incubations were in phosphate buffer, 100 mM, pH 7.4, and the reaction was started by the addition of 2.5 mM NADPH and 3.3 mM MgCl2. Heat-killed microsomes (100°C for 10 min) were also incubated under the same conditions as a control. Incubations were stopped at 0, 15, 30, and 60 min by the addition of an equal volume of acetonitrile. Samples were centrifuged to pellet the precipitated protein, and the clear supernatant was removed and stored at approximately −70°C until analysis.
Animals
Sprague–Dawley rats, male and female, were obtained from Charles River (Hollister, CA) and were 6–8 weeks old at study initiation. Rats were housed either 1 or 3 per cage, with a 12 h light/12 h dark cycle, 68°–72°C temperature, 39–59% humidity. Rats were fed Purina Rodent Chow #5002 and provided water (purified, reverse osmosis) ad libitum.
Protocols for all animal studies were approved by the Institutional Animal Care and Use Committee, and the studies were carried out in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health.
Pharmacokinetics studies in rats
To screen formulations, male Sprague–Dawley rats were administered a single dose of 50 mg/kg SR13668 in one of several formulations, n = 3 rats per treatment group (5 mg/ml). Blood was collected at a limited number of time points, 3 or 6, selected to span the time of maximum blood drug levels (~4 h) based on a pilot study and analyzed for concentration of SR13668. The lungs were also collected and analyzed at 4 h from rats in the first formulation screening study.
For the definitive oral bioavailability and pharmacokinetic study, male and female Sprague–Dawley rats, jugular vein catheterized, n = 3 per sex per treatment group, were administered a single dose of SR13668 by iv (1 mg/kg; 6mg/m2) via the tail vein or oral gavage (1, 10, or 30 mg/kg; 6, 60, 180 mg/m2) routes. The volumes administered were 1 ml/kg for iv and 10 mg/l po. The rats were fasted overnight prior to SR13668 administration. For iv treatment, SR13668 was solubilized in DMSO:PEG300 (15:85) at 1 mg/ml and for oral gavage, it was formulated in PEG400:Labrasol (1:1, v:1) at concentrations of 0.1, 1, and 3 mg/ml. Blood samples (~200 μl) were collected at multiple time points post-dose through 24 h from the jugular canula and transferred to tubes with EDTA to obtain whole anticoagulated blood. Plasma (~200 μl of blood collected at the 2-h time point) was prepared by centrifugation at 2–8°C and stored frozen at approximately −70°C until analysis to determine the concentration of SR13668.
Bioanalytical method for SR13668
Samples were processed by addition of 4 volumes of acetonitrile to 1 volume of plasma or whole blood and well mixed. Lung tissue was processed by adding 80% (v:v) acetonitrile in water to preweighed lung sample. The tissue was homogenized on ice using a polytron tissue grinder, followed by brief sonication. The suspensions (prepared from both blood, plasma or lung homogenates) were then clarified by centrifugation, and the resulting supernatants were transferred to fresh microfuge tubes, and evaporated under vacuum in a centrifugal evaporator without heat application. The dry residues were reconstituted in 200 μl of 80% acetonitrile in water, centrifuged to clarify the samples and the supernatants then transferred to HPLC vials fitted with glass inserts for chromatographic analysis. Study samples were quantitated using a set of SR13668 calibration standards prepared in blank matrix that were processed in parallel (plasma: 1, 5, 25, 50, 100, 500, 1,000, 1,500 and 2,000 ng/ml; whole blood: 0.5, 2.5, 5, 25, 100, 500, 1,000, 1,500 and 2,000 ng/ml). Quality control standards were also processed in parallel and analyzed concurrently with the experimental samples (plasma: 2.5, 75, 250 and 1,750 ng/ml; whole blood: 1, 50, 250 and 1,750 ng/ml). Lung samples were quantitated against a set of neat standard solutions.
Samples were analyzed by HPLC with fluorescence and UV detection using a Waters 2690 Separation Module (Waters, Milford, MA), autosampler at 4°C, Luna C18 [2] column (5 μm, 250 × 4.6 mm) at ambient temperature. The mobile phase consisted of 50 mM sodium acetate buffer, pH 5.0 (A) and acetonitrile (B). Flow rate was 1 ml/min and the elution mode used 65% A: 35% B from 0 to 6 min, 25% A: 75% B from 6.2 to 25 min, 65% A: 35% B from 25.2 to 30 min. Detection by UV absorbance was 307 nm, and by fluorescence, the excitation wavelength was 345 nm with emission wavelength of 480 nm. The retention time of SR13668 was ~16.9 min. Calibration standards and quality control, QC, standards were analyzed in triplicate with each day of study sample analysis and processed according to the extraction protocol. Lower limit of quantitation was 1.0 ng/ml plasma, whole blood or lung homogenate. Mean percent accuracies for whole blood and plasma were 107.87 and 107.45%, respectively. Relative standard deviations (RSD) for whole blood and plasma were 10.50 and 4.08%, respectively. QC results show that accuracy and precision values were well within ±15% of the corresponding nominal QC concentrations.
Metabolite characterization
Plasma was mixed with an equal volume of acetonitrile to precipitate plasma proteins, and then centrifuged (18,000g, 10 min) to pellet the precipitate. Twenty microliters of the resulting supernatants were injected directly onto the LC–MS/MS system described below. Chromatography (Waters 2795 separation module, Phenomenex Synergi Fusion 4 μ, 150 × 3 mm column, autosampler and column temperature 20°C, mobile phase water with 0.1% formic acid and acetonitrile with 0.1% formic acid) was monitored in tandem with a photodiode array detector and Micromass Ultima triple quadrupole mass spectrometer (Waters, Milford, MA) set to monitor in the full scan mode over a range of m/z values of 60–1,000. The mass spectrometer was configured as follows in the characterization of metabolites in the blood samples. The HPLC eluant was examined by full scan mass spectrometry in the negative ion mode set for unit mass resolution. The capillary voltage was 2.5 kV, the cone voltage was 40 v, the ion source temperature was 115°C, and the desolvation temperature was 350°C with desolvation gas at a sufficient flow rate to give an even spray at 0.4 ml/min HPLC flow. The chromatograms shown are the extracted ion chromatograms obtained from the total ion chromatograms at the expected m/z values for SR13668 (m/z 429) and a monohydroxylated metabolite of SR13668 (m/z 445).
Pharmacokinetics data evaluation
Pharmacokinetic analysis was performed on whole blood drug concentration data for individual animals using non-compartmental methods and WinNonlin® Professional (Version 5.2, Pharsight, Mountain View, CA). Pharmacokinetic parameters, calculated for each individual animal, included observed maximal plasma concentration (Cp), extrapolated plasma concentration at time 0 (C0; iv group only), area under the plasma concentration–time curve from time 0 to the last time point (AUClast) and extrapolated to infinity (AUCinf), terminal elimination half-life (t1/2), apparent volume of distribution (V), and total clearance (Cl). Mean residence time (MRT) was calculated by dividing area under the first moment curve (AUMC) by the AUC. Bioavailabilty (F) was estimated using the following formula: F = AUCinf po × Doseiv/AUCinf iv × Dosepo. In the oral treatment groups, V and CL are presented as V/F and CL/F. Terminal phase parameters (t1/2, AUCinf,V, Cl, and MRT) were not determined in animals with fewer than 3 non-zero points in the terminal phase.
Student’s t test was used to determine statistically significant differences in Cmax, AUC, CL for male and female rats.
Results
In vitro characterization of SR13668
The ester functionality on SR13668 suggested that the drug might be readily metabolized by plasma esterases; however, incubation of SR13668 with rat plasma showed that more than 90% of the parent drug was recovered unchanged at 60 min (Fig. 2a). The inclusion of NaF, a potent inhibitor of plasma esterase activity had no impact on SR13668 stability. An unknown peak that elutes earlier than SR13668 was detected in these plasma incubations, although it accounted for less than 1% of the SR13668. This putative metabolite was not present when plasma esterases were inhibited with NaF and it did not co-elute with an authentic standard of di-acid-SR13668 or a demethylated analog of SR13668, but it may be a mono-acid form of SR13668.
Fig. 2.

Metabolic stability of SR13668. a Rat plasma at 37°C for up to 60 min, + or −NaFl; b Rat and human liver microsomes + NADPH incubated at 37°C for 60 min
SR13668 was also relatively stable to liver microsomal metabolizing enzymes (Fig. 2b). After 60 min of incubation, 65 ± 9 and 89 ± 2% of the initial drug concentration, 1 or 10 μM, respectively, remained unchanged by RLM supplemented with NADPH. HLM metabolized less SR13668 than rats; 83 and 93 ± 4% remained unchanged at 60 min. A potential metabolite peak (M1) eluting earlier than the parent drug was detected in rat and human liver microsomal incubation samples. The concentration of this putative metabolite was higher in rat liver preparations than in human liver and was not present in incubations of SR13668 with heat-killed liver microsomes.
Evaluation of plasma protein binding by ultrafiltration indicated that SR13668 is essentially 100% plasma protein bound over a wide range of concentrations, from 2.5 to 1,750 ng/ml.
Oral formulation development
Vehicles known to improve permeability of other poorly absorbed drugs were screened for their impact on blood levels of SR13668 in rats. Sprague–Dawley male rats were administered, by oral gavage, 50 mg/kg SR13668 in five different formulations–0.5% methylcellulose (0.5% MC), 10% Gelucire (44/14) in water (10% Gelucire), 100% PEG400 (PEG400), 5% Cavamax: 40% 1-methyl-2-pyrrolidone in water (Cavamax:NMP), or propylene glycol:2.5% aqueous hydroxypropyl methyl cellulose (PG:2.5% HPMC). Marked differences were observed in blood levels of SR13668 with the different formulations (Table 2). The relative exposure of rats treated with SR13668 in 10% Gelucire or PEG400 was 23- and 28-fold higher compared to 0.5% MC as the vehicle. Tissue concentration of SR13668, as shown by measuring the drug in lung, was also significantly enhanced in the Gelucire and PEG400 formulation groups and was much higher than the blood concentration (Fig. 3).
Table 2.
Effect of formulation on blood levels of SR13668 in male rats
| Formulation | Experimenta | Cp (ng/ml) | AUC (h*n g/ml) | Relative exposureb |
|---|---|---|---|---|
| 0.5% MC | 1 | 2.8 | 15.4 | 1 |
| 10% Gelucire | 1 | 130 | 360 | 23 |
| PEG400c | 1 & 2 | 76 | 427 | 28 |
| PG: 2.5% HPMC | 1 | 4.8 | 23.8 | 2 |
| PEG400: Labrasol (3:1) | 2 | 577 | 3,007 | 195 |
| PEG400: Tween 20 (3:1) | 2 | 895 | 4,773 | 310 |
| PEG400: Labrasol (1:1) | 2 | 994 | 5,588 | 363 |
| PEG400/Labrasol/Tween 20 (2:1:1) | 2 | 1,174 | 6,572 | 427 |
Values calculated from SR13668 concentrations in blood collected at 2, 4, and 8 h after oral administration of 50 mg/kg to n = 3 rats per formulation, except where noted otherwise
Two experiments were conducted to screen formulations in rats. PEG400 was evaluated in both experiments
Relative exposure is calculated by dividing AUC for each formulation by AUC for 0.5% MC
Formulation repeated in two separate experiments and values represent the mean of these data
Fig. 3.
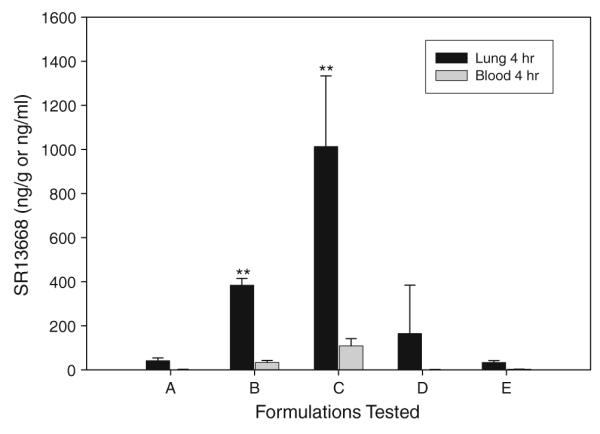
Concentration of SR13668 in lung tissue compared to blood 4 h after oral administration of the drug in four different formulations to male Sprague–Dawley rats. A =0.5% MC; B = 10% Gelucire; C = PEG400; D = 5% Cavamax; E = PG/2.5% HPMC. Double asterisks indicate values that are significantly different from 0.5% MC (P < 0.05%)
HTS screening of mixtures of excipients was then applied, focusing on PEG400 and Gelucire and related vehicles with the goal of achieving approximately 5 mg/ml in the vehicle. Multiple screening runs were performed, testing more than 125 binary and ternary combinations of excipients (Table 1). Level 1 screening evaluated combinations of ethanol, water, PEG400, propylene glycol (PG), Tween 80, and glycerin, but did not identify any solubilizing excipient combinations (data not shown). Level 2–3 identified several combinations of excipients that achieved 1.98 mg/ml solubility for SR13668. As shown in Fig. 4, the majority of the excipients that resulted in higher solubility were lipophilic. When tests were repeated at a drug concentration of 5.0 mg/ml, 16 solvent combinations achieved the desired solubility. Using manual solubility assessment of these 16 combinations by direct addition of the drug without the use of DMSO identified 2 combinations that resulted in close to 5 mg/ml—PEG400:Labrasol, at a 3:1 or 1:1 ratio, achieving concentrations of SR13668 of 4.89 ± 0.02 and 4.62 ±0.29 mg/ml, respectively. Six of the eight tested combinations solubilized greater than 3 mg/ml, and the solubility in the other two was less then 3 mg/ml (Table 3).
Fig. 4.

Solubility of SR13668 (1.98 mg/ml) Level 3 screening with binary combinations of ten solvents: PEG Tween 20 (8:2) (PEGT20), PEG Tween 80 (8:2) (PEGT80), PG Tween 20 (8:2) (PGT20), PG Tween 80 (8:2) (PGT80), Labrasol PEG (1:1) (LsolPEG), Cremophor EL (CRE; 25% v/v in water), Labrafac-Maisin (4:1) PEG (1:1) (NMP), polyethylene glycol (PEG), propylene glycol (PG), and Labrasol (Lsol)
Table 3.
Maximum concentrations of solubilized SR13668 as determined by UV-Spectrophotometric assay performed by manual solubility assessment
| Solvent combination | Solvent percent ratio |
Soluble concentration mg/mL |
|---|---|---|
| PEG400/labrasol | 3:1 | 4.89 ± 0.02 |
| PEG400/labrasol | 1:1 | 4.62 ± 0.29 |
| PEG400/labrasol/Tween 20 | 2:1:1 | 3.54 ± 0.03 |
| PEG400/Tween 20 | 3:1 | 3.18 ± 0.08 |
| Sesame oil/labrasol | 1:1 | 3.36 ± 0.14 |
| PEG400/labrafil | 1:1 | 2.89 ± 0.20 |
| PEG400/caproyl PGMC | 1:1 | 3.08 ± 0.19 |
| Caproyl PGMC/labrasol | 1:1 | 2.19 ± 0.01 |
From this list of vehicle combinations, four formulations in which SR13668 was soluble at 3 mg/ml or better were selected for comparison to PEG400—PEG400:Labrasol (3:1), PEG400:Tween 20 (3:1), PEG400/Labrasol (1:1), and PEG400/Labrasol/Tween 20 (2:1:1). Oral gavage of SR13668 (50 mg/kg) in these vehicles to male Sprague–Dawley rats was performed and all rats appeared normal during the study with no adverse clinical signs (e.g. diarrhea). The highest observed blood concentration of SR13668 was at 4 h for each formulation and all four formulations, designed to optimize solubility of SR13668, resulted in higher blood levels than PEG400 alone (Table 2). PEG400:Labrasol (1:1) and PEG400/Labrasol/Tween20 (2:1:1) gave the largest increase in relative exposure; the blood concentration of SR13668 was approximately 400-fold higher with these formulations compared to 0.5% MC and about 15-fold higher than PEG400 alone. Although all of the vehicles investigated have low toxicity and can be safely administered, the simpler formulation, PEG400/Labrasol (1:1 v/v) was selected for use in future safety and bioavailabilty studies of SR13668 because it has a lower concentration of PEG400 and no Tween 20.
Oral bioavailability using an optimized formulation
Administration of 1 mg/kg SR13668 iv resulted in blood drug levels at the first collected time point of approximately 1,000 ng/ml (~2,300 nM) in both male and female rats (Fig. 5). Drug concentrations in the iv group decreased to approximately 5 ng/ml by 24 h. After oral gavage administration of SR13668 in PEG400/Labrasol (1:1 v/v), drug concentrations in blood were markedly higher in female rats than males, and the concentrations increased in with dose for both genders, although the increase was not proportional to dose level. At 24 h, concentrations of SR13668 in the blood remained high, especially in the two higher po dose groups; for example at 30 mg/kg, the mean blood concentrations at 24 h were 118.6 ± 13.2 ng/ml (275.4 ± 30.7 nM) in males and 143.5 ± 14.6 ng/ml (333.3 ± 33.9 nM) for females.
Fig. 5.

Blood concentration of SR13668 in male and female Sprague–Dawley rats after iv and oral gavage administration. Plotted values represent mean ± SD of n = 3 rats
SR13668 was also measured in plasma collected at 2-h post-dose from each rat. The mean ratio of the concentration of SR13668 in blood/plasma varied from approximately 2.5–3.5 in the iv, low and mid-dose po groups, suggesting that SR13668 concentrates in blood cells. The mean ratio of blood/plasma concentrations was lower in the 30 mg/kg, at ~1.2. At doses of 30 mg/kg and above, binding sites in the cells may be saturated.
Pharmacokinetic parameters determined from iv blood concentrations are summarized in Table 4. The t1/2 values are 4.4 ± 0.4 and 5.6 ± 2.2 h, for male and female rats, respectively. The observed maximum at the first collected time point (5 min), Cp is similar for both male and female rats, ~930 ng/ml. Male rats have significantly lower AUCinf (31.8% lower) and higher Cl (46.9% higher) compared to female rats. V was 16% higher in male rats, but the difference was not statistically significant at 5.6 (males) and 4.8 l/kg (females).
Table 4.
Pharmacokinetic parameters for SR13668a
| iv, 1 mg/kg (6 mg/m2) |
po, 1 mg/kg (6 mg/m2) |
po, 10 mg/kg (60 mg/m2) |
po, 30 mg/kg (180 mg/m2) |
|||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Maleb | Female | Male | Female | |
| t1/2 (h) | 4.4 ± 0.4 | 5.6 ± 2.2 | 7.5 ± 1.3 | 7.1 ± 0.6 | 5.6 | 9.0 ± 4.4 | 9.6 ± 1.3 | 7.3 ± 1.1 |
| Tmax (h) | NA | NA | 4.0 ± 4.4 | 3.3 ± 1.2 | 5.7 ± 3.4 | 5.3 ± 1.2 | 6.3 ± 2.5 | 8.0 ± 1.7 |
| C0 (ng/ml) | 1,435.7 ± 233.2 | 1,557.8 ± 299.4 | NA | NA | NA | NA | NA | NA |
| Cp (ng/ml) | 925.8 ± 82.4 | 933.0 ± 113.4 | 17.1 ± 10.9 | 23.2 ± 18.4 | 130.1 ± 63.6c | 531.5 ± 216.8c | 455.5 ± 85.5c | 883.9 ± 82.4c |
| AUClast (ng h/ml) | 1,124.7 ± 99.2c | 1,601.0 ± 157.1c | 130.3 ± 26.2 | 236.6 ± 118.0 | 1,446.6 ± 277.6c | 6,560.6 ± 2,133.3c | 7,038.1 ± 1,347.3c | 12,365.0 ± 1,957.0c |
| AUCinf (ng h/ml) | 1,138.7 ± 100.5c | 1,668.8 ± 114.8 | 143.7 ± 22.6 | 262.37 ± 127.1 | 1,699.2 | 8,236 ± 2,797.3c | 8,676.9 ± 1,300.1c | 13,857.9 ± 1,961.5c |
| V or V/F (L/kg) | 5.6 ± 0.8 | 4.8 ± 2.0 | 77.7 ± 26.1 | 49.7 ± 32.4 | 49.4c | 16.9 ± 5.2c | 49.3 ± 12.4c | 23.2 ± 6.3c |
| Cl or Cl/F (ml/h/kg) |
883 ± 82c | 601 ± 41c | 7,085.3 ± 1,196.9 | 4,767.0 ± 3,002.3 | 5924c | 1,340.3 ± 559.7c | 3,507.0 ± 495.3c | 2,193.0 ± 295.3c |
| MRT (h) | 3.6 ± 0.3 | 5.8 ± 2.0 | 10.4 ± 2.2 | 10.6 ± 1.2 | 9.1 | 14.8 ± 7.0 | 14.9 ± 1.5 | 11.8 ± 0.8 |
| F (%) | 12.6 ± 2.0 | 15.7 ± 7.6 | 14.9 | 49.4 ± 16.8 | 25.4 ± 3.8 | 27.7 ± 3.9 | ||
Values represent the mean ± SD of n = 3 unless noted otherwise
Parameters requiring calculation of the terminal phase are for n = 2 rats, other parameters calculated with n = 3
Values of males and females signiWcantly diVerent, P < 0.05
A marked sex-related difference in exposure was apparent after oral gavage of SR13668, with female rats exhibiting higher Cmax and AUC values for all three doses than males; the difference was statistically significant for the two higher dose groups (P < 0.05). Figure 6 shows that the Cp and AUC for male rats increase linearly with dose. Cp and AUC also increase with dose level in female rats, but there is evidence of nonlinearity in these parameters. CL is significantly lower in females than males for all three oral dose groups.
Fig. 6.
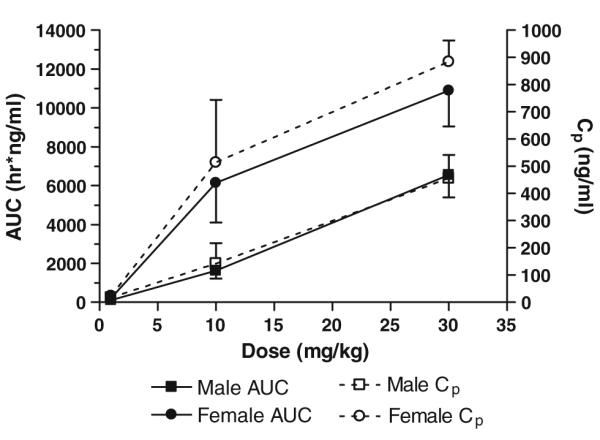
AUC and Cp versus oral dose of SR13668. Each data point represents the mean ± SD of n = 3 rats
The calculated MRT of SR13668 after oral gavage was longer, ~9–15 h, than the estimate obtained after iv treatment of ~5 h. This increase is related to the time required for the drug to be absorbed. Oral bioavailability of SR13668 was higher in the 10 and 30 mg/kg dose groups compared to the low dose. The highest mean oral bioavailability, 49.4 ± 16.8%, was observed in the female rats in the 10 mg/kg. Oral bioavailability values were similar for both males and females because the exposure (AUC) was higher in female rats for both the iv and oral routes.
A potential metabolite (M1) was detected in blood samples from treated rats. Figure 7 shows an example chromatogram from an extracted blood sample, male rat, 2 h after 30 mg/kg po dose. The quantity of M1 formed in vivo was estimated using HPLC with fluorescent detection and SR13668 calibration standards. The highest M1 blood concentration was observed at about 1 h after iv administration of SR13668; the ratio of AUC for SR13668 to M1 was ~4. After oral administration, the highest concentrations of M1 were present in blood collected from 2 to 9 h (Fig. 8). In general, female rats formed more M1 than males, and more M1 was present in samples containing higher concentrations of SR13668.
Fig. 7.

Chromatogram of extracted blood sample from male rat, 2 h after oral administration of 30 mg/kg SR13668. Fluorescent peak eluting before SR13668 at about 11 min is not present in blank blood samples while early eluting peaks (~2.5 and 5 min) were in blank (not shown)
Fig. 8.

Blood concentration of M1, putative metabolite of SR13668, in male and female Sprague–Dawley rats after iv and oral gavage administration
Metabolite characterization in vitro and in vivo
LC–MS characterization experiments indicated that the putative metabolite, M1, has a molecular weight of 445 in the negative ion mode (i.e., SR13668 + 16) and is formed in vitro, by liver microsomes and in vivo, based on its presence in blood and plasma samples from the rat pharmacokinetic study. Figure 9 shows extracted ion chromatograms obtained from the total ion chromatograms at the expected m/z values for SR13668 (m/z 429) and a monohydroxylated metabolite of SR13668 (m/z 445). The data obtained from these experiments did not allow assignment of the location of the additional oxygen. Authentic standards for potential metabolites of SR13668 with the addition of oxygen were not available.
Fig. 9.

a Extracted ion chromatograms for SR13668 parent ion (m/z 429–). b Extracted ion chromatograms for monohydroxy SR13668 ion (m/z 445–)
Discussion
SR13668 is the product of a computer-aided rational drug design program to identify a compound with the anti-cancer activity of I3H, and a better pharmacokinetic profile than I3H [1]. I3H is basically a prodrug that forms more than 20 acid condensation products in the stomach [2], including 3,3′-diindolylmethane (DIM). Under selected conditions, I3H can be detected in plasma and tissues following oral administration [3], but clinical studies found that DIM is the principal circulating product present after I3H treatment. Thus, the anti-cancer activity of I3H is generally attributed to DIM and other oligomeric condensation products of I3H [4]. The advantage of SR13668 is that the drug itself shows potent antitumor activity in xenograft models at doses as low as 10 mg/kg and is more active than I3H or DIM [1].
SR13668 is a highly planar, non-polar compound, and although the structure was designed to have better aqueous solubility than the first generation compounds that resulted from the program [1], the drug still is poorly water soluble (<2 μg/ml). Initial oral bioavailability estimates for SR13668 in mice and rats were less than 1%. Bioavailability of an orally administered drug is directly dependent on absorption through the gut wall and metabolism by intestinal tissue and liver. In vitro metabolism studies of SR13668 demonstrated that the drug is neither rapidly nor extensively metabolized, thus efforts to improve oral bioavailability of this drug focused on increasing absorption.
Lipid-based drug delivery systems have been proposed as a practical and effective approach for formulating poorly soluble drugs like SR13668 [5, 6]. Our evaluation of several formulations demonstrated that the relative oral bioavailability of SR13668 could be markedly enhanced, particularly by the use of lipid-based formulations containing PEG400 or Gelucire. Application of an automated approach to assess drug solubility made it possible to rapidly evaluate many different vehicles singly and in combination. These studies identified a formulation for SR13668 composed of PEG400:Labrasol (1:1) which substantially improved oral bioavailability of the drug candidate in rats. Additional formulation development studies in dogs and non-human primates have identified another lipid-based drug delivery approach containing Solutol to result in superior oral bioavailability in these species [7].
The pharmacokinetic behavior of SR13668 is consistent with that of a very lipophilic, dissolution-limited drug. Oral administration of SR13668, solubilized in PEG400:Labrasol, resulted in an extended plateau of high blood drug levels, suggesting that the lipid-based formulation increases the length of the absorption phase for SR13668, enhancing overall bioavailability. SR13668 also distributes readily to tissues and concentrates therein, as shown by higher concentrations in whole blood versus plasma, and lung concentrations that are several fold higher than the whole blood. The apparent V, ~5 l/kg, is also indicative of relatively high tissue binding.
A marked sex-related difference in exposure to SR13668 was demonstrated after both iv and oral administration. Male rats had significantly lower AUC and higher CL than female rats. Distribution of SR13668 in tissues and body fat, which may differ in quantity and location in female versus male rats, could be at least partially responsible for sex-dependent pharmacokinetics. The non-linearity of AUC and Cp in female but not male rats suggests clearance mechanisms or tissue binding sites become saturated in the female rats at higher dose levels The most common cause of sex-dependent differences in pharmacokinetics is related to drug metabolism and it is well documented that male rats have higher levels of some CYPs than females [8]. Recent research shows that a sex-dependent plasma growth hormone profile is responsible for sex-based differences in the hepatic expression of a large number of genes that regulate both drug metabolism and other physiological pathways [9]. Faster CL was observed in males compared to females, but whether this is due to a higher rate of metabolism by male rats, as has been reported for other drugs, cannot be determined based on the current study. One metabolite, whose molecular weight is consistent with the addition of an oxygen atom, i.e. hydroxylation, was observed to be present in the blood of both male and female rats. The level of this metabolite, based on its fluorescence peak area, suggested that in low dose groups, M1 and SR13668 may be present in blood at approximately equal concentrations. Although an authentic standard of M1 would be required to definitively determine the actual metabolite concentration, it is likely that SR13668 and SR13668 + 16 would have similar response factors for fluorescence detection. It is known that SR13668 is metabolized by other species; in dog pharmacokinetic studies, both oxidized and glucuronide conjugated metabolites have been observed (unpublished data). However, the pharmacokinetics of SR13668 in both beagle dogs and cynomolgus monkeys suggests that this drug is not subject to extensive presystemic metabolism [7].
In summary, SR13668 is a promising new cancer therapeutic and chemopreventive agent for which the identification of a formulation to enhance its oral bioavailability was critical for further development. Preclinical safety studies in both rats and dogs were performed on SR13668 using PEG400:Labrasol (1:1 v/v) as the vehicle and was found to be essentially free of significant toxicity in 28-day rat and 14-day dog subchronic toxicology studies [7, 10]. SR13668 was also negative in a genotoxicity battery [11].
Rapid first pass metabolism and solubility limitations were both considered as likely explanations for the low bioavailability observed in pilot studies. This research demonstrates that while SR13668 does undergo metabolism, probably by the liver, the oral bioavailability of SR13668 in rats was dramatically improved by use of a formulation that contained permeation enhancers and promoted better solubilization of the drug. Future work with SR13668 will focus on the tissue distribution, excretion, and metabolite profile of SR13668 in rats using radiolabeled compound. Such studies should help define more accurately the extent of absorption after oral treatment, as well as the impact of metabolism on drug clearance.
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Heath, Department of Health and Human Services, under Contract No. HHSN261200433005C.
Contributor Information
Carol E. Green, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA
Robert Swezey, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA.
James Bakke, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA.
Walter Shinn, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA.
Anna Furimsky, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA.
Naveen Bejugam, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA.
Gita N. Shankar, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA
Ling Jong, Biosciences Division, SRI International, 333 Ravenswood Avenue, Menlo Park, CA 94025, USA.
Izet M. Kapetanovic, National Cancer Institute, NIH CADRG, DCP, Bethesda, MD, USA
References
- 1.Chao W-R, Yean D, Amin K, Green C, Jong L. Computer-aided rational drug design: a novel agent (SR13668) designed to mimic the unique anticancer mechanisms of dietary indole-3-carbinol to block Akt signaling. J Med Chem. 2007;50(15):3412–3415. doi: 10.1021/jm070040e. [DOI] [PubMed] [Google Scholar]
- 2.Grose KR, Bjeldanes LF. Oligomerization of indole-3-carbinol in aqueous acid. Chem Res Toxicol. 1992;5(2):188–193. doi: 10.1021/tx00026a007. [DOI] [PubMed] [Google Scholar]
- 3.Anderton MJ, Manson MM. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin Cancer Res. 2004;10(15):5233–5241. doi: 10.1158/1078-0432.CCR-04-0163. [DOI] [PubMed] [Google Scholar]
- 4.Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, Mayo MS, Crowell JA, Hurwitz A. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3, 3′-diindolylmethane. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2477–2481. doi: 10.1158/1055-9965.EPI-06-0396. [DOI] [PubMed] [Google Scholar]
- 5.Porter CJH, Pouton CW, Cuine JF, Cahrman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deli Rev. 2008;60:673–691. doi: 10.1016/j.addr.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Gershkovich P, Wasan KM, Barta CA. A review of the application of lipid-based systems in systemic, dermal/transdermal, and ocular drug delivery. Crit Rev Ther Drug Carrier Syst. 2008;25(6):545–584. doi: 10.1615/critrevtherdrugcarriersyst.v25.i6.20. [DOI] [PubMed] [Google Scholar]
- 7.Kapetanovic IM, Muzzio M, Hu S-C, Crowell JA, Rajewski RA, Haslam JL, Jong L, McCormick DL. Bioavailability of candidate cancer preventative agent, SR13668 in dogs and monkeys. Cancer Chemother Pharmacol. 2010;65:1109–1116. doi: 10.1007/s00280-009-1116-4. [DOI] [PubMed] [Google Scholar]
- 8.Mugford CA, Kedderis GL. Sex-dependent metabolism of xenobiotics. Drug Metab Rev. 1998;30(3):441–498. doi: 10.3109/03602539808996322. [DOI] [PubMed] [Google Scholar]
- 9.Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol. 2009;76(2):215–228. doi: 10.1124/mol.109.056705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakke J, Furimsky A, Erickson R, Swezey R, Jong L, Kapetanovic IM, Crowell JA, Green CE. Pharmacokinetics and toxicity to rats of SR13668, a novel cancer chemopreventive agent. Soc Toxicol Meet Abstr. 2008;2008:392. [Google Scholar]
- 11.Doppalapudi RS, Riccio ES, Rausch LL, Shimon JA, Lee PS, Mortelmans KE, Kapetanovic IM, Crowell JA, Mirsalis JC. Evaluation of chemopreventive agents for genotoxic activity. Mutat Res. 2007;629:148–160. doi: 10.1016/j.mrgentox.2007.02.004. [DOI] [PubMed] [Google Scholar]