

Abstract
Background
Mutations in the lamin A/C gene, LMNA, can cause dilated cardiomyopathy. We have shown abnormal activation of the extracellular signal-regulated kinase (ERK) and the c-jun N-terminal kinase (JNK) branches of the mitogen-activated protein kinase (MAPK) signaling cascade in hearts from LmnaH222P/H222P mice that develop dilated cardiomyopathy. We recently showed that partial inhibition of ERK and JNK signaling prior to the onset of cardiomyopathy in LmnaH222P/H222P mice prevented the development of left ventricle dilatation and decreased cardiac ejection fraction at a time when these occurred in untreated mice.
Methods and Results
To determine if pharmacological inhibitors of ERK and JNK signaling could be clinically useful to treat cardiomyopathy caused by LMNA mutation, we administered them to LmnaH222P/H222P mice after they developed left ventricular dilatation and decreased ejection fraction. LmnaH222P/H222P mice were treated with ERK and JNK signaling inhibitors from 16 to 20 or, in pilot experiments, 19 to 24 weeks of age. The inhibitors blocked increased expression of RNAs encoding natriuretic peptide precursors and proteins involved in sarcomere architecture that occurred in placebo-treated mice. Echocardiography and histological analysis demonstrated that treatment prevented left ventricular end systolic dilatation, increased ejection fraction and decreased myocardial fibrosis.
Conclusions
Inhibitors of ERK and JNK signaling could potentially be used to treat humans with cardiomyopathy caused by LMNA mutations.
Keywords: Cardiomyopathy, pharmacology, LMNA, mitogen-activated protein kinase
Dilated cardiomyopathy is characterized by ventricular dilatation and impaired systolic function with 20% to 48% of cases familial (1). Mutations in LMNA encoding A-type nuclear lamins have been shown to cause a several human diseases (2) with at least 3 having dilated cardiomyopathy as a predominant feature: autosomal Emery-Dreifuss muscular dystrophy (3), limb girdle muscular dystrophy type 1B (4) and dilated cardiomyopathy type 1A (5). Given the phenotypic overlap of these disorders, they can be described as LMNA dilated cardiomyopathy with variable skeletal muscle involvement (6). LMNA mutations appear to be responsible for approximately 8% of familial cardiomyopathies (7–10). The onset of symptoms in LMNA cardiomyopathy is variable, ranging from the first to sixth decade of life and occurring most frequently in the third decade (7–11). It has a natural history more aggressive than most other familial cardiomyopathies, with high rates of arrhythmias leading to sudden death and advanced heart failure necessitating cardiac transplantation (7,11,12).
To identify potential targets to treat cardiomyopathy caused by LMNA mutation, we have been examining cellular signaling pathways in hearts of Lmna H222P knock in mice, a model of the human disease. Male LmnaH222P/H222P mice develop left ventricular (LV) dilatation and depressed contractile function starting at approximately 8–10 weeks of age and invariably develop LV dilatation and decreased cardiac contractility at 16 weeks (13). We have shown abnormal activation of the extracellular signal-regulated kinase (ERK) and the c-Jun N-terminal kinase (JNK) branches of the mitogen-activated protein kinase (MAPK) signaling cascade in hearts of Lmna H222P knock in mice prior to the onset of clinically detectable cardiomyopathy (14). We have also shown that lamin A variants that cause cardiomyopathy activate ERK and JNK when expressed in cultured cells (14). Based on these results, we hypothesized that activation of ERK and JNK plays a primary pathogenic role in the development of cardiomyopathy. Our recent work has shown that small molecule inhibitors of ERK and JNK signaling administered to male LmnaH222P/H222P mice prior to the onset of detectable cardiomyopathy prevented LV dilatation and decreases in cardiac ejection fraction (EF) at an age when placebo-treated mice had significant abnormalities in these parameters (15,16).
A critical question relevant to potential treatment of human subjects with ERK and JNK inhibitors regards their effectiveness after the onset of cardiac dysfunction. It would be impractical to use such drugs as prophylactic treatment in asymptomatic humans with LMNA mutations, especially given the variable age of onset, usually in adulthood. To help answer this question, we initiated the present study to determine if inhibitors of ERK and JNK signaling would be beneficial in LmnaH222P/H222P mice after LV dilatation and decreased cardiac EF have already occurred.
Methods
An expanded Methods section is available in the Online Data Supplement.
LmnaH222P/H222P mice were generated and genotyped using polymerase chain reaction (PCR) primers as described (13). Drugs were dissolved in dimethyl sulfoxide (DMSO) are delivered into the peritoneal cavity by injection at 3 mg/kg/day for 5 days a week. Equal volumes of DMSO were administered as placebo. Cardiac structure and contractility were assessed by echocardiography. Representative stained cardiac sections were photographed using a Microphot SA (Nikon) light microscope attached to a Spot RT Slide camera (Diagnostic Instruments) with a 10x objective. Images were processed using Adobe Photoshop CS (Adobe Systems). RNA transcripts measured using real-time reverse transcription-polymerase chain reaction (RT-PCR) were quantified using iQ SYBR green super mix (Bio-Rad). Statistical comparisons were made using an unpaired Student’s t-test or a one-way analysis of variance with the Tukey post hoc test to evaluate the significance of differences between means.
Results
Rationale for Treatment of LmnaH222P/H222P Mice
Our hypothesis was that treatment with a MAPK/ERK kinase (MEK) 1/2 inhibitor, which inhibits activation of ERK, or a JNK inhibitor would improve cardiac structure and function in LmnaH222P/H222P mice when the compounds are administered after these parameters are significantly abnormal. Because the animal care facility at Columbia University Medical Center prohibits removal and re-entry of mice from its barrier facility, we could not obtain echocardiograms on individual subjects before and after treatment. To test our hypothesis, we therefore assigned male LmnaH222P/H222P mice 16 weeks of age to 3 different treatment arms (placebo DMSO, n=28; MEK1/2 inhibitor PD98059, n=22; JNK inhibitor SP600125, n=29) and examined parameters of cardiac structure and function at 20 weeks of age, after 4 weeks treatment. At 16 weeks, male LmnaH222P/H222P mice are known to have markedly increased LV end diastolic diameter (LVEDD) and LV end systolic diameter (LVESD) compared to Lmna+/+ mice (13,15,16). LmnaH222P/H222P mice also have depressed cardiac contractility, with fractional shortening (FS) decreased by 20%–40% compared to Lmna+/+ mice (13,15). Myocardial fibrosis occurs in LmnaH222P/H222P mice at 16 weeks of age (16). At 20 weeks, LVEDD and LVESD increase further in LmnaH222P/H222P mice and cardiac contractility also progressively deteriorates (16). During the 4-week treatment protocol, 6 mice in the DMSO group, 3 in the PD98059 group and 3 in the SP600125 group died prior reaching 20 weeks of age for evaluation.
Effect of PD98059 and SP600125 on ERK and JNK Signaling
Systemic administration of the MEK1/2 inhibitor, PD98059, and the JNK inhibitor, SP600125, to LmnaH222P/H222P mice from 16 to 20 weeks of age partially blocked the phosphorylation of ERK1/2 (Figure 1A) and JNK (Figure 1B), respectively, in hearts. At 3 mg/kg/day, PD98059 was highly selective for blocking ERK signaling, as phosphorylation of JNK was not significantly inhibited (Figure 1A). At 3 mg/kg/day, SP600125 was specific of the JNK signaling, as phosphorylation of ERK1/2 was not significantly inhibited (Figure 1B).
Figure 1.

(A) Representative immunoblots using antibodies against phophorylated ERK1/2 (p-ERK) and total ERK1/2 (ERK) and (B) against phophorylated JNK (p-JNK) and total JNK to probe proteins extracted from hearts from LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. Blots of proteins extracted from hearts of Lmna+/+ mice are shown for comparison. The graphs show quantification of (A) pERK/total ERK and (B) pJNK/total JNK. n=4 in each group. Comparison between DMSO-treated LmnaH222P/H222P mice and Lmna+/+ mice; *P<0.05. Comparison between PD98059-treated and SP600125-treated and DMSO-treated LmnaH222P/H222P mice; #P<0.05, ##P<0.005, n.s.: not significant.
Effect of the PD98059 and SP600125 on Cardiac Expression of Natriuretic Peptides and Myosin Light Chain
One of the features of dilated cardiomyopathy is the upregulation of cardiac hormones such as natriuretic peptides as a compensatory mechanism to maintain cardiac output (17,18). Upregulation of genes involved in sarcomere organization also occurs (19,20). We therefore assayed expression of Mlc-2a mRNA, encoding a cardiac isoform of myosin light chain, and NppA and NppB mRNAs, encoding natriuretic peptides precursors in hearts from Lmna+/+ mice, DMSO-treated LmnaH222P/H222P mice and inhibitor-treated LmnaH222P/H222P mice (Figure 2). In hearts from DMSO-treated LmnaH222P/H222P mice, expression of Mlc-2a mRNA was significantly increased approximately 30-fold compared to hearts of Lmna+/+ mice (Figure 2). Similarly, in hearts from LmnaH222P/H222P mice, NppA and NppB mRNA levels showed significant 36-fold and 17-fold increases in expression compared to hearts of Lmna+/+ mice (Figure 2). Treatment of LmnaH222P/H222P mice with PD98059 or SP600125 significantly decreased the expression of Mlc-2a, NppA and NppB mRNAs at 20 weeks of age (Figure 2). Hence, pharmacological inhibition of ERK or JNK signaling reversed molecular compensatory processes that occur in LmnaH222P/H222P mice with cardiomyopathy.
Figure 2.
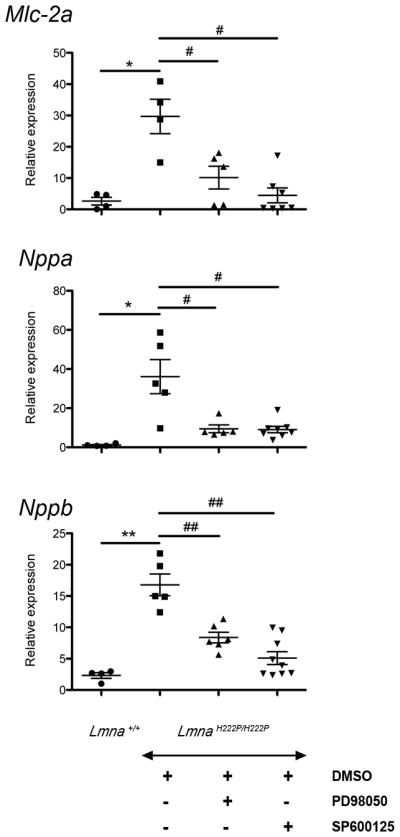
Effect of PD98059 and SP600125 on cardiac expression of natriuretic peptides and myosin light chain in LmnaH222P/H222P mice. Dot diagrams indicate the expression levels of Mlc-2a mRNA encoding the cardiac isoform of myosin light chain, Nppa mRNA encoding the atrial natriuretic factor and Nppb encoding the brain natriuretic peptide in hearts from Lmna+/+ mice and LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. n=4 in each group. Values were obtained using the ΔΔCT method using Gapdh as housekeeping gene (see Full Materials and Methods). *P<0.05, **P<0.005, #P<0.05, ##P<0.005.
Effect of PD98059 and SP600125 on LV Dilatation and Contractility in LmnaH222P/H222P Mice
After 4 weeks of treatment with DMSO, PD98059 or SP600125, LmnaH222P/H222P mice were anesthetized and cardiac dimensions and function measured by echocardiography. M-mode transthoracic echocardiography showed increased LVEDD and LVESD in LmnaH222P/H222P mice treated with DMSO compared to Lmna+/+ mice (Figure 3). LmnaH222P/H222P mice treated with PD98059 and SP600125 had significantly smaller LVESD compared to the DMSO-treated mice (Figure 3). FS and EF were reduced in LmnaH222P/H222P mice compared to Lmna+/+ mice but increased in the LmnaH222P/H222P mice treated with PD98059 or SP600125.
Figure 3.

Representative transthoracic M-mode echocardiographic tracings from LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. Tracings from Lmna+/+ mice are shown for comparison. LVESD and LVEDD are indicated.
Table 1 shows the composite echocardiographic data for the 3 treatment arms for LmnaH222P/H222P mice and Lmna+/+ mice for comparison. Compared to Lmna+/+ mice, LmnaH222P/H222P mice treated with DMSO had significantly increased LVEDD and LVESD. The EF of DMSO-treated male LmnaH222P/H222P mice at 20 weeks was 53.87% ± 2.58%, which was decreased by 28% compared to Lmna+/+ mice. LmnaH222P/H222P mice treated with PD98059 or SP600125 had a statistically significant reduction in the LVESD compared to mice treated with DMSO; however, LVEDD was not significantly different. LmnaH222P/H222P mice treated with PD98059 had an EF of 65.46% ± 2.64%, an increase of approximately 22% (P<0.005) compared to the DMSO-treated group. EF of LmnaH222P/H222P mice treated with SP600125 was 61.88% ± 1.66%, an increase of approximately 15% (P<0.005) compared to the DMSO-treated group. Overall, these results showed that PD98059 and SP600125 have positive effects on cardiac contractility when administered after cardiac dysfunction occurs in LmnaH222P/H222P mice.
Table 1.
Echocardiographic data at 20 weeks of age for Lmna+/+ mice and LmnaH222P/H222P mice treated with DMSO placebo or treated with SP600125 or PD98059
| Genotype (Treatment Group) | n | HR | LVEDD (mm) | LVESD (mm) | EF (%) | FS (%) |
|---|---|---|---|---|---|---|
| Lmna+/+ | 12 | 400 | 3.50 ± 0.06 | 2.07 ± 0.08 | 73.21 ± 1.17 | 41.71 ± 1.01 |
| LmnaH222P/H222P (DMSO) | 22 | 372 | 3.87 ± 0.11 * | 3.00 ± 0.13 *** | 53.87 ± 2.58 *** | 27.86 ± 1.54 *** |
| LmnaH222P/H222P (PD98059) | 19 | 350 | 3.55 ± 0.11 | 2.41 ± 0.11 ‡‡‡ | 65.46 ± 2.64 ‡‡ | 35.91 ± 1.88 ‡‡ |
| LmnaH222P/H222P (SP600125) | 26 | 363 | 3.73 ± 0.08 | 2.67 ± 0.10 ‡ | 61.88 ± 1.66 ‡‡ | 33.11 ± 1.16 ‡‡ |
Values are means ± SEM.
HR indicates heart rate in beats per minute.
Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice:
P<0.05,
P<0.0005.
Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P:
P<0.05,
P<0.005,
P<0.0005.
Effect of PD98059 and SP600125 on Myocardial Fibrosis in LmnaH222P/H222P Mice
Later-stage cardiomyopathy caused by LMNA mutations is characterized by myocardial fibrosis (21,22). Sirius Red and Gomori’s trichrome staining of hearts from LmnaH222P/H222P mice 20 weeks of age treated with DMSOhad a significant increase in fibrosis compared to hearts from Lmna+/+ mice (Figure 4A,B). In contrast, LmnaH222P/2PH222P mice treated with PD98059 or SP600125 had a lower degree of cardiac fibrosis than DMSO-treated mice (Figure 4A,B). We quantified the myocardial fibrotic area of each animal by determining the ratio of fibrotic tissue (blue stained with Gomori’s trichrome) to the total tissue area in each micrograph (Figure 4C). Hearts from DMSO-treated LmnaH222P/H222P mice had 15.01 ± 0.9% fibrotic tissue per total surface examined (Figure 4D). Systemic treatment with PD98059 or SP600125 significantly lowered the area of fibrotic tissue to 4.48% ± 1% (P<0.0005) and 5.86% ± 0.4% (P<0.0005), respectively (Figure 4D).
Figure 4.

(A) Sirius red and (B) Gomori’s trichrome staining of cross-sections of hearts from LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. A cross-section of heart from a Lmna+/+ mouse is shown for comparison. Scale bar: 50 μm. (C) Quantification of fibrotic area in hearts from mice. n=3 in each group. Y-axis corresponds to the area (pixels) and X-axis represents the color spectrum (red corresponds to the muscle tissue and blue corresponds to the connective tissue). (D) Bars indicate the percentage of fibrosis per surface area of myocardium examined in hearts from Lmna+/+ mice and LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. n=3 in each group. ***P<0.0005, ###P<0.0005.
Excessive extracellular matrix, predominantly collagen proteins, defines fibrotic tissue. We therefore determined expression of genes encoding a protein of the extracellular matrix (Fn1 encoding fibronectin) and genes encoding type I collagen (Col1a1 and Co1a2) using real-time RT-PCR. At 20 weeks of age, hearts from LmnaH222P/H222P mice treated with DMSO had a 5-fold increase of Col1a1, a 4-fold increase of Co1a2 and a 4-fold increase of Fn1 mRNAs compared to hearts from Lmna+/+ mice (Figure 5). Treatment with PD98059 and SP600125 significantly lowered the expression of Col1a1, Col1a2 and Fn1 (Figure 5). These results demonstrated that LmnaH222P/H222P mice treated with either MEK1/2 or JNK inhibitors had decreased progression of myocardial fibrosis.
Figure 5.
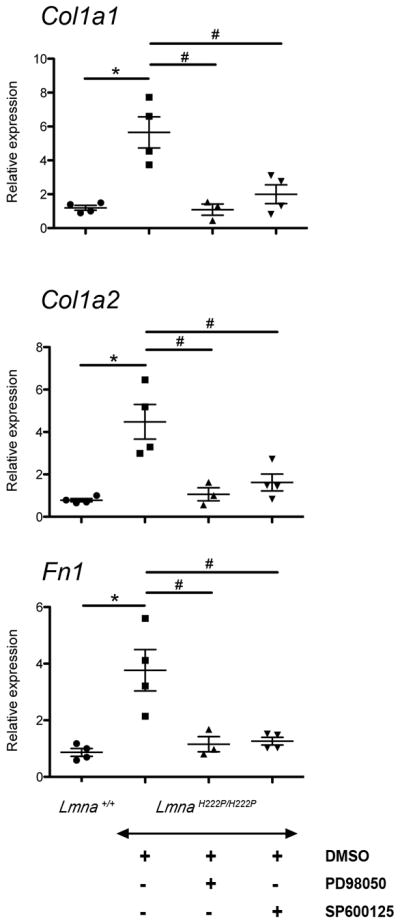
Effect of PD98059 and SP600125 on cardiac expression of genes encoding collagen and fibronectin in LmnaH222P/H222P mice. Dot diagrams indicate the expression of Col1a1, Col1a2 and Fn1 in heart from Lmna+/+ mice and LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. n=3 in each group. Values were obtained using the ΔΔCT method using Gapdh as housekeeping gene (see Full Materials and Methods). *P<0.05, #P<0.05.
Effect of PD98059 and SP600125 on Nuclear Shape in Cardiomyocytes in LmnaH222P/H222P Mice
We have reported abnormal elongation of nuclei in cardiomyocytes of LmnaH222P/H222P mice (15,16). Nuclei in cardiomyocytes in hearts from LmnaH222P/H222P mice treated with DMSO were elongated compared to those in Lmna+/+ mice (Figure 6A). Nuclei of cardiomyocytes in hearts of LmnaH222P/H222P mice treated with PD98059 or SP600125 LmnaH222P/H222P mice had an overall shape that was more “rounded” than those in hearts of mice treated with DMSO (Figure 6A). Mean length of cardiomyocyte nuclei in hearts of LmnaH222P/H222P mice treated with DMSO was significantly longer than in hearts from Lmna+/+ mice (P<0.0005) (Figure 6B). The mean lengths of nuclei in cardiomyocytes in hearts from LmnaH222P/H222P mice treated with PD98059 or SP600125 were significantly shorter than the in hearts of mice in the DMSO-treated group (P<0.0005) (Figure 6B). Similar nuclear elongation has also been reported in Lmna knockout mice, suggesting a role of lamins in determining nuclear shape in cardiomyocytes (23,24). While other abnormalities in nuclear morphology have been observed in hearts of LmnaH222P/H222P mice when cardiac tissue is examined by electron microscopy (13), we could not assess these ultrastructural alterations with the light microscopic methods we used.
Figure 6.

(A) Histological analysis of cross-sections of hearts from LmnaH222P/H222P mice treated with PD98059, SP600125 or DMSO. Heart from a Lmna+/+ mouse is shown for comparison. Sections are stained with hematoxylin and eosin. Yellow lines with arrowheads demonstrate the measurement of nuclear length. Scale bar: 25 μm. (B) Quantification of nuclear elongation in cardiomyocytes from mice. Cardiomyocyte nuclei were measured along the yellow lines with arrowheads. Bars indicate the length of cardiomyocyte nuclei in the indicated hearts. Values are means ± SEM for n=150, 290, 690 and 575 cardiomyocytes from Lmna+/+ mice, DMSO-treated LmnaH222P/H222P mice, PD98059-treated LmnaH222P/H222P mice, and SP600125-treated LmnaH222P/H222P mice, respectively. ***P<0.0005, ###P<0.0005.
Pilot Study of PD98059 and SP600125 to Treat More Advanced Heart Disease in LmnaH222P/H222P mice
In a pilot study, we assessed treatment of LmnaH222P/H222P mice with PD98059 and SP600125 at a more advanced stage of disease and for a longer time. We assigned male LmnaH222P/H222P mice at 19 weeks of age to 3 different treatment arms (placebo DMSO, n=4; MEK1/2 inhibitor PD98059, n=3; JNK inhibitor SP600125, n=3) and examined parameters of cardiac structure and function. Systemic administration of PD98059 and SP600125 to LmnaH222P/H222P mice partially blocked phosphorylation of ERK1/2 and JNK in hearts from 24 week-old mice (Figure IA in the online-only Data Supplement). At 24 weeks, LmnaH222P/H222P treated with PD98059 had decreased LV dilatation and increased FS compared to DMSO-treated mice (Figure IB in the online-only Data Supplement). There was also a trend toward decreased LV dilatation and increased FS in the LmnaH222P/H222P mice treated with SP600125 (Figure IB in the online-only Data Supplement). Cardiac expression of Mlc-2a, NppA, NppB, Col1a1 and Col1a2 mRNAs was also significantly reduced in the inhibitor-treated LmnaH222P/H222P mice at 24 weeks, except for NppB in those treated with SP600125 (Figure IC in the online-only Data Supplement).
Discussion
Our previous work has documented the effectiveness of inhibiting ERK and JNK signaling in preventing or delaying the onset of cardiomyopathy in LmnaH222P/H222P mice (15,16). In those studies, MEK and JNK inhibitors were administered prior the onset of any detectable structural or functional cardiac abnormalities. A critical remaining question was if MEK and JNK inhibitors would be effective in improving heart function in LmnaH222P/H222P mice when initiated after the onset of cardiac disease, which would be more analogous to potential treatment in human patients. In this study, we therefore tested the extent to which a treatment course starting after the onset of cardiac disease in LmnaH222P/H222P mice would be beneficial. Our results showed that pharmacological inhibitors of ERK and JNK signaling blocked increased expression of RNAs encoding natriuretic peptide precursors and proteins involved in sarcomere architecture, prevented LV end systolic dilatation, increased cardiac ejection fraction and decreased myocardial fibrosis. Two recent studies showed that either a calcium-sensitizing agent (25) or a β-blocker (24) also improved cardiac function in mouse models of Lmna-associated cardiomyopathy. Our work provides support for the possibility that MEK or JNK inhibitors could overcome the lack of definitive treatments for human patients suffering for cardiac disease caused by LMNA mutations.
Changes in myocardial structure and function in response to injury and proliferation of the non-myocyte cell populations of the heart, referred to as myocardial remodelling (26), alter cardiac performance over the long term. Part of such remodelling includes fibrosis, which results in exaggerated mechanical stiffness and causes systolic dysfunction (27). Established therapies for heart failure may also drive a significant part of their benefit from actions on cardiac fibroblasts. A beneficial effect on cardiac fibrosis has been reported for angiotensin converting enzyme inhibitors (28–30), angiotensin receptor blockers (31,32), diuretics (33) and aldosterone antagonists (34–36). Treatment of LmnaH222P/H222P mice with MEK or JNK inhibitors had a profound beneficial effect on myocardial fibrosis, a characteristic of later-staged cardiomyopathy caused by LMNA mutations (21,22). Activation of ERK and JNK signaling pathways by various stimuli have been correlated to several cellular processes such as cell proliferation and remodelling of extra-cellular matrix (37). Inhibition of ERK and JNK signaling pathways could therefore have a beneficial effect on cardiac function by also acting directly to decrease the proliferation of myocardial fibroblasts. Such a hypothesis needs to be tested. It also remains to be determined if simultaneous inhibition of both ERK and JNK signaling has additive effects in cardiomyopathy caused by Lmna mutation.
Our study in LmnaH222P/H222P mice was designed similar to a human clinical trial. It assessed primary endpoints (LV dilatation, EF) and “surrogate” secondary endpoints (expression of natriuretic peptide precursors) that are used in many human clinical heart failure trials. While mortality is a reasonable endpoint in phase III clinical trial for advanced heart failure, it is rarely if ever used in the initial drug assessment phase or in treatment of subjects with heart disease that is not end stage (38), as were both the case in our study. Furthermore, LmnaH222P/H222P mice have diaphragmatic muscle involvement (not reported in humans with LMNA mutations) and significant skeletal muscle pathology as they age, which may be non-cardiac causes of mortality (13). Nonetheless, the measurements of LV function we used correlate with prognosis in many human clinical trials and their behaviour parallels changes in mortality with treatment (38). For example, LV end-systolic volume, which is determined by measuring LVESD, is the major determinant of survival in human subjects after recovery from myocardial infarction and after coronary artery bypass grafting for impaired LV function (39,40). A study by Heywood et al. (41) also showed in human subjects with an EF less than 40% treated with angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers that an increase of more than 15% in EF resulted in mortality of only about 2% per year. In our study, PD98059 and SP600125 improved the EF of LmnaH222P/H222P mice approximately 22% and 15%, respectively, compared to placebo. Taking into account that EF improvement is an important predictor for survival in human subjects with systolic dysfunction, we speculate that small molecules inhibitors of the ERK and JNK signaling pathways could have a positive effect on survival of patients with LMNA mutations. While not an endpoint or our study, during the 4-week treatment protocol starting 16 weeks of age, 6 mice in the DMSO group, 3 in the PD98059 group and 3 in the SP600125 group died prior to reaching 20 weeks of age, suggesting that treatment with MEK1/2 or JNK inhibitors trended towards improved survival. Furthermore, our pilot study treating LmnaH222P/H222P mice up to 24 weeks of age, when they have a mortality rate of approximately 25% (13), showed improvements in echocardiographic and cardiac biochemical parameters.
The choice of therapeutic agents in clinical trials is predicated, at least in part, on the efficacy of drugs studied in murine models of disease (42–44). Both PD98059 and SP600125, which we used in this study to respectively inhibit ERK and JNK signaling, are tool compounds and are not suitable for use in humans secondary to problems with bioavailability and toxicity (45). Therefore, any future clinical trial of MEK or JNK inhibitor in human subjects with cardiomyopathy caused by LMNA mutations would require the use of superior drugs, including possibly those that are already entered the pipeline of pharmaceutical companies for other indications. For example, a second-generation oral MEK inhibitor, PD0325901 (Pfizer), has markedly improved properties, including better potency against MEK, better bioavailability, increased metabolic stability and a longer time of MEK suppression (46). PD0325901 has been administered to humans and has entered a phase II clinical trial to treat advanced non-small cell lung cancer (47,48). Similarly, AZD6244/ARRY-142886 (AstraZeneca/Array Biopharma) is in phase II clinical trials for patients with cancers (49). Superior JNK inhibitors are also in preclinical development for use in humans (50). Hence, our results in LmnaH222P/H222P mice with cardiac dysfunction could lay the foundation for clinical trials of MEK and JNK inhibitors, currently being developed for cancer and inflammatory conditions in human subjects with cardiomyopathy caused by LMNA mutations.
CLINICAL PERSPECTIVE.
Heart failure is responsible for considerable morbidity and mortality and dilated cardiomyopathy (DCM) is a major cause. Molecular genetic studies have revealed mutations in various genes in patients with familial DCM but the precise mechanisms of how they lead to heart muscle damage remain largely unknown. Mutations in LMNA encoding A-type nuclear lamins appear to be responsible for approximately 8% of cases of familial DCM and patients withLMNA mutations have a poorer prognosis than those with DCM caused by mutations in most other genes. We have previously shown an abnormal activation of the extracellular signal-regulated kinase (ERK) and the c-jun N-terminal kinase (JNK) branches of the mitogen-activated protein kinase signaling cascade in hearts of mice with DCM caused by a mutation in Lmna. We now establish that treating these mice with chemical inhibitors of ERK and JNK after the onset of left ventricular dilatation and decreased cardiac ejection fraction, a time when human patients would be considered for therapy, improves cardiac function and significantly decreases myocardial fibrosis. These results provide proof of concept that pharmacological inhibitors of ERK and JNK signaling, some of which are currently in clinical development for other indications, could be studied in human clinical trials of patients with DCM caused by LMNA mutations.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by Grants from the National Institutes of Health [AR048997] and Muscular Dystrophy Association [MDA4287].
List of Non-standard Abbreviations
- LV
left ventricular
- ERK
extracellular signal-regulated kinase
- JNK
c-jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- DMSO
dimethyl sulfoxide
- MEK
MAPK/ERK kinase
- SEM
standard error of mean
- PCR
polymerase chain reaction
- RT-PCR
reverse transcription-polymerase chain reaction
- LVEDD
left ventricular end diastolic diameter
- LVESD
left ventricular end systolic diameter
Footnotes
Disclosures
H.J.W. and A.M. are inventors on a pending PCT patent application on methods for treating and/or preventing cardiomyopathies by ERK and JNK inhibition filed by the Trustees of Columbia University in the City of New York.
References
- 1.Taylor MRG, Carniel E, Mestroni L. Cardiomyopathy, familial dilated. Orphanet J Rare Dis. 2006;13(1):27. doi: 10.1186/1750-1172-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119:1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 4.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 5.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Müehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 6.Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101:473–476. doi: 10.1161/01.cir.101.5.473. [DOI] [PubMed] [Google Scholar]
- 7.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L Familial Dilated Cardiomyopathy Registry Research Group. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–780. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 8.Vytopil M, Benedetti S, Ricci E, Galluzzi G, Dello Russo A, Merlini L, Boriani G, Gallina M, Morandi L, Politano L, Moggio M, Chiveri L, Hausmanova-Petrusewicz I, Ricotti R, Vohanka S, Toman J, Toniolo D. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet. 2003;40:e132. doi: 10.1136/jmg.40.12.e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, Wilde AA, van Langen IM, Nannenberg EA, van der Kooi AJ, Kraak M, van Gelder IC, van Veldhuisen DJ, Vos Y, van den Berg MP. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007;154:1130–1139. doi: 10.1016/j.ahj.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 10.Cowan J, Li D, Gonzalez-Quintana J, Morales A, Hershberger RE. Morphological analysis of 13 LMNA variants identified in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:6–14. doi: 10.1161/CIRCGENETICS.109.905422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005;83:79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- 12.Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, Agozzino M, Campana C, Gavazzi A, Febo O, Marini M, Landolina M, Mortara A, Piccolo G, Viganò M, Tavazzi L, Arbustini E. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–1260. doi: 10.1016/j.jacc.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 13.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Malisses M, Schwartz K, Bonne G. Mouse model carrying H222P-lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–169. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 14.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathway links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–1293. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulate kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–247. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W, Shan J, Bonne G, Worman HJ, Muchir A. Pharmacological inhibition of c-Jun N-terminal kinase prevents cardiomyopathy caused by mutation in LMNA gene. Biochim Biophys Acta. 2010;1802:632–638. doi: 10.1016/j.bbadis.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshimine K, Horiuchi M, Suzuki S, Kobayashi K, Abdul JM, Masuda M, Tomomura M, Ogawa Y, Itoh H, Nakao K, Osame M, Saheki T. Altered expression of atrial natriuretic peptide and contractile protein genes in hypertrophied ventricles of JVS mice with systemic carnitine deficiency. J Mol Cell Cardiol. 1997;29:571–578. doi: 10.1006/jmcc.1996.0300. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi T, Allen PD, Izumo S. Expression of A-, B-, and C-type natriuretic peptide genes in failing and developing human ventricles. Correlation with expression of the Ca(2+)-ATPase gene. Circ Res. 1992;71:9–17. doi: 10.1161/01.res.71.1.9. [DOI] [PubMed] [Google Scholar]
- 19.Hwang JJ, Allen PD, Tseng GC, Lam CW, Fananapazir L, Dzau VJ, Liew CC. Microarray gene expression profiles in dilated and hypertrophic cardiomyopathic end-stage heart failure. Physiol Genomics. 2002;10:31–44. doi: 10.1152/physiolgenomics.00122.2001. [DOI] [PubMed] [Google Scholar]
- 20.Yung CK, Halperin VL, Tomaselli GF, Winslow RL. Gene expression profiles in end-stage human idiopathic dilated cardiomyopathy: altered expression of apoptotic and cytoskeletal genes. Genomics. 2004;83:281–297. doi: 10.1016/j.ygeno.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Van Tintelen PJ, Tio RA, Kerstjens-Frederikse WS, van Berlo JH, Boven LG, Suurmeijer AJH, White SJ, den Dunnen JT, te Meerman GJ, Vos YJ, van der Hout AH, Osinga J, van den Berg MP, van Verhuisen DJ, Buys CHCM, Hofstra RMW, Pinto YM. Severe myocardial fibrosis caused by a deletion of the 5′ end of the lamin A/C gene. J Am Coll Cardiol. 2007;49:2430–2439. doi: 10.1016/j.jacc.2007.02.063. [DOI] [PubMed] [Google Scholar]
- 22.Raman SV, Sparks EA, Baker PM, McCarthy B, Wooley CF. Mid-myocardial fibrosis by cardiac magnetic resonance in patients with lamin A/C cardiomyopathy: possible substrate for diastolic dysfunction. J Cardiovac Magn Res. 2007;9:907–913. doi: 10.1080/10976640701693733. [DOI] [PubMed] [Google Scholar]
- 23.Nikolova V, Leimena C, McMahon AC, Tan JC, CHandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandar S, Yeo LZ, Leimena C, Tan JC, Xiao XH, Nikolova-Krstevski V, Yasuoka Y, Gardiner-Garden M, Wu J, Kesteven S, Karlsdotter L, Natarajan S, Carlton A, Rainer S, Feneley MP, Fatkin D. Effects of mechanical stress and cardvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ Res. 2010;106:573–582. doi: 10.1161/CIRCRESAHA.109.204388. [DOI] [PubMed] [Google Scholar]
- 25.Arimura T, Sato R, Machida N, Bando H, Zhan DY, Morimoto S, Tanaka R, Yamane Y, Bonne G, Kimura A. Improvement of left ventricular dysfunction and of survival prognosis of dilated cardiomyopathy by administration of calcium sensitizer SCH00013 in a mouse model. J Am Coll Cardiol. 2010;55:1502–1508. doi: 10.1016/j.jacc.2009.10.065. [DOI] [PubMed] [Google Scholar]
- 26.Swynghedauw B. Molecular mechanisms of myocardial remodelling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 27.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblasts: therapeutic target in myocardial remodelling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 28.Sleight P. Angiotensin II and trials of cardiovascular outcomes. Am J Cardiol. 2002;89:11–16. doi: 10.1016/s0002-9149(01)02322-0. [DOI] [PubMed] [Google Scholar]
- 29.Fox KM. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPE study) Lancet. 2003;362:782–788. doi: 10.1016/s0140-6736(03)14286-9. [DOI] [PubMed] [Google Scholar]
- 30.Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–1393. doi: 10.1161/01.cir.102.12.1388. [DOI] [PubMed] [Google Scholar]
- 31.Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergen J, Yusuf S, Pocock S. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–766. doi: 10.1016/s0140-6736(03)14282-1. [DOI] [PubMed] [Google Scholar]
- 32.Diez J, Querejeta R, Lopez B, Gonzalez A, Larman M, Martinez Ubago JL. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–2517. doi: 10.1161/01.cir.0000017264.66561.3d. [DOI] [PubMed] [Google Scholar]
- 33.Lopez B, Gonzalez A, Beaumont J, Querejeta R, Larman M, Diez J. Identification of a potential cardiac antifibrotic mechanism of torasemide in patients with chronic heart failure. J Am Coll Cardiol. 2007;50:859–867. doi: 10.1016/j.jacc.2007.04.080. [DOI] [PubMed] [Google Scholar]
- 34.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 35.Pitt B, Remme W, Zammad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 36.Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES) Circulation. 2000;102:2700–2706. doi: 10.1161/01.cir.102.22.2700. [DOI] [PubMed] [Google Scholar]
- 37.Yoon S, Seger R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 38.Zanolla L, Zardini P. Selection of endpoints for heart failure clinical trials. Eur J Heart Fail. 2003;5:717–723. doi: 10.1016/s1388-9842(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 39.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 40.Hamer AW, Takayama M, Abraham KA, Roche AH, Kerr AR, Williams BF, Ramage MC, White HD. End-systolic volume and long-term survival after coronary artery bypass graft surgery in patients with impaired left ventricular function. Circulation. 1994;90:2899–2904. doi: 10.1161/01.cir.90.6.2899. [DOI] [PubMed] [Google Scholar]
- 41.Heywood JT, Elatre W, Pai RG, Fabbri S, Huiskes B. Simple Clinical Criteria to determine the prognosis of heart failure. J Cardiovasc Pharmacol Therapeut. 2005;10:173–180. doi: 10.1177/107424840501000305. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda Y, Ross J. Models of dilated cardiomyopathy in the mouse and the hamster. Curr Opin Cardiol. 2000;15:197–201. doi: 10.1097/00001573-200005000-00013. [DOI] [PubMed] [Google Scholar]
- 43.Khurana TS, Davies KE. Pharmacological strategies for muscular dystrophy. Nat Rev Drug Discov. 2003;2:379–390. doi: 10.1038/nrd1085. [DOI] [PubMed] [Google Scholar]
- 44.Bhatnagar S, Kumar A. Therapeutic targeting of signalling pathways in muscular dystrophy. J Mol Med. 2010;88:155–166. doi: 10.1007/s00109-009-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen LF, Sebolt-Leopold J, Meyer MB. Cl-1040 (PD184352), a targeted signal transduction inhibitor f MEK (MAPKK) Semin Oncol. 2003;30:105–116. doi: 10.1053/j.seminoncol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 46.Brown AP, Carlson TCG, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–679. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- 47.Lorusso P, Krishnamurthi S, Rinehart JR, Nabell L, Croghan G, Varterasian M, Sadis SS, Menon S, Leopold J, Meyer MB. A phase 1–2 clinical study of a second generation oral MEK inhibitor, PD0325901 in patients with advanced cancer. J Clin Oncol (Abstract) 2005;23:3011. [Google Scholar]
- 48.Menon SS, Whitfield LR, Sadis S, Meyer MB, Leopold J, Lorusso PM, Krishnamurthi S, Rinehart JR, Nabell L, Croghan G. Pharmacokinetics (PK) and pharmacodynamics (PD) of PD0325901, a second generation MEK inhibitor after multiple oral doses of PD0325901 to advanced cancer patients. J Clin Oncol (Abstract) 2005;23:3066. [Google Scholar]
- 49.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, Maloney L, Gordon G, Simmons H, Marlow A, Litwiler K, Brown S, Poch G, Kane K, Haney J, Eckhardt SG. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;3:463–475. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.