

Abstract
Vascular inflammation has traditionally been thought to be initiated at the luminal surface and progress through the media toward the adventitial layer. In recent years, however, evidence has emerged suggesting that the vascular adventitia is activated early in a variety of cardiovascular diseases and that it plays an important role in the initiation and progression of vascular inflammation. Adventitial fibroblasts have been shown to produce substantial amounts of NAD(P)H oxidase-derived reactive oxygen species (ROS) in response to vascular injury. Additionally, inflammatory cytokines, lipids and various hormones, implicated in fibroblast proliferation and migration, lead to recruitment of inflammatory cells to the adventitial layer and impairment of endothelium-dependent relaxation. Early in the development of vascular disease, there is clear evidence for progression toward a denser vasa vasorum which delivers oxygen and nutrients to an increasingly hypoxic and nutrient-deficient media. This expanded vascularization appears to provide enhanced delivery of inflammatory cells to the adventitia and outer media. A combined adventitial fibroblast and inflammatory cell-derived ROS therefore is expected to synergize their local effect on adventitial parenchymal cells leading to further cytokine release and a feed-forward propagation of adventitial ROS production. In fact, data from our laboratory and others suggest a broader paracrine positive feedback role for adventitia-derived ROS in medial smooth muscle cell hypertrophy and neointimal hyperplasia. A likely candidate responsible for the adventitia-derived paracrine signaling across the vessel wall is the superoxide anion metabolite hydrogen peroxide, which is highly stable, cell-permeant and capable of activating downstream signaling mechanisms in smooth muscle cells. This leads to phenotypic modulation of smooth muscle cells. This review addresses the role of adventitial NAD(P)H oxidase-derived ROS from a non-traditional, perivascular vantage of promoting vascular inflammation and will discuss how ROS derived from adventitial NAD(P)H oxidases may be a catalyst for vascular remodeling and dysfunction.
1. Introduction
Hypercholesterolemia, smoking, hypertension, obesity, hyperglycemia, and aging are major risk factors for cardiovascular diseases (CVD) including myocardial infarction, stroke, congestive heart failure and renal vascular disease [1-4]. The precise mechanisms responsible for the initiation and progression of CVD are not entirely known, however there is compelling evidence that vascular wall inflammation and increased production of reactive oxygen species (ROS) play a key role in this process. Traditionally vascular wall inflammation has been considered an “inside-out” response during which cardiovascular risk factors promote accumulation of leukocytes in the sub-endothelial space and increase the endothelial production of ROS [5]. In this widely accepted paradigm, the inflammatory reaction, which involves complex interactions between vascular cells and different classes of leukocytes (T lymphocytes, neutrophils and monocytes/macrophages) as well as increased production of ROS, is initiated in the intima and progresses outward toward the adventitia [5, 6].
Over the past decade, however, evidence has accumulated to support a new paradigm of “outside-in” hypothesis, in which the vascular wall inflammation is initiated in the adventitial layer and progresses through the media inward toward the intima [7]. Hallmarks of the “outside-in” hypothesis include a relatively high adventitial infiltration of leukocytes, increased numbers of fibroblasts and myofibroblasts, increased vasa vasorum neovascularization and enhanced production of adventitia-derived ROS [8-11]. All of these processes appear to converge to exacerbate the attraction and delivery of inflammatory cells to the adventitia. Furthermore, findings of increased adventitial hyperplasia and thickness precede medial hypertrophy and neointimal hyperplasia in various manifestations of CVD. These findings appear to suggest that the adventitia is the “first-responder” and initiator of vascular remodeling in large and small vessels [12-14].
All vascular cell types are capable of producing ROS and possess several enzymatic sources of superoxide – including xanthine oxidase, cytochrome P-450, uncoupled nitric oxide synthase (NOS) and NAD(P)H oxidase - which are implicated in vascular pathophysiology. The adventitial NAD(P)H oxidases appear to be a major contributor to vascular ROS in CVD [15-18]. These findings combined with the growing awareness that adventitial remodeling is an early harbinger of progressive vascular disease provide the basis for this review. Both superoxide and its metabolite hydrogen peroxide (H2O2), generated from the former by dismutation and superoxide dismutase (SOD), have emerged as important players in vascular pathophysiology due to their signaling roles in a range of phenotypes ranging from cell proliferation, migration and hypertrophy to vascular tone dysfunction. ROS participate in vascular inflammation from early imperceptible stages involved in immune defense to severe ultimate complications including thrombosis, tissue hypoxia and necrosis. Some subtle and ostensibly innocuous changes observed in the vascular wall in CVD, such as hypertrophic remodeling, are also expected consequences. Therefore, the objective of this review is to explore a range of ROS effects in CVD-associated vascular inflammation from a non-traditional viewpoint of the role of adventitial NAD(P)H oxidase-derived ROS.
2. Phagocyte NADPH oxidase
Because the phagocyte oxidase is the prototype for the vascular oxidases, we will briefly describe the main features and regulation of this well-characterized enzyme. Phagocyte NADPH oxidase-derived ROS have long been accepted as important antimicrobial agents as well as potential culprits of inflammation in a defense system gone awry [19]. The multi-component phagocyte NADPH oxidase is unassembled and inactive in resting cells, but becomes rapidly activated by several stimuli, including bacterial lipopolysaccharides (LPS) and cytokines [20-22]. Upon activation, the assembled phagocyte NADPH oxidase catalyzes the transfer of one electron from NADPH to molecular oxygen, resulting in the formation of the forerunner for all biological ROS superoxide [23]. The phagocyte oxidase enzyme complex is comprised of flavocytochrome b558, an integral membrane heterodimer comprised of gp91phox and p22phox, four cytoplasmic protein subunits, p47phox, p67phox, p40phox and the regulatory low molecular weight GTPases, Rac1 or Rac2 [24-26]. During activation of the phagocyte NADPH oxidase, the cytoplasmic components become phosphorylated and translocate to the membrane-bound flavocytochrome b558, in a highly controlled manner. The catalytic core of the phagocyte NADPH oxidase, gp91phox, contains two hemes in the N-terminal transmembrane region, and NADPH-binding and FAD-binding domains in the C-terminal cytoplasmic region. Upon activation, NADPH binds to a site on the assembled oxidase on the cytoplasmic side of the membrane, releasing two electrons, which are transported by FAD and two hemes, and accepted by two molecules of oxygen on the opposite side of the membrane to form two molecules of superoxide anion.
3. Vascular NAD(P)H oxidases
Vascular NAD(P)H oxidases as a family of enzymes are similar to the phagocyte NADPH oxidase in that they are a major source of vascular superoxide production under physiological and pathophysiological conditions [27-29]. However, reports to date maintain that the vascular oxidase is a lower capacity but constitutive producer of ROS in comparison to its phagocyte counterpart [29]. Interestingly, NAD(P)H oxidase-derived ROS has the potential to augment ROS production by other enzymes. For instance, NAD(P)H oxidase-derived ROS can convert xanthine dehydrogenase to xanthine oxidase as well as uncouple NOS via oxidative degradation of NOS cofactor tetrahydrobiopterin, thus leading to a further enhanced ROS generation [30, 31]. Incidentally, it has been reported that NAD(P)H oxidase-derived ROS may decrease the activity of key antioxidant enzymes in the vasculature leading to enhanced oxidative stress upon the production of the same amount of superoxide. Indeed, increased vascular superoxide production and p47phox activity were associated with lower expression of glutathione peroxidase (GPX) -1 and -4, SOD-2 and catalase in high-fat diet fed LDLR-/- mice versus control [32]. Moreover, the decreased expression and activity of SOD-1, -2 and 3 and catalase in calcified regions of stenotic valves were associated with increased superoxide and H2O2 levels [33].
In terms of the expression and activity of vascular NAD(P)H oxidase, both basal and stimulated vascular oxidase activity have been reported in all segments of the vessel wall, i.e. the intima, the media, and the adventitia. Under physiological conditions, basal levels of ROS derived from vascular NAD(P)H oxidase contribute to a wide range of physiological processes, including regulation of vascular tone, cellular signaling, gene expression, angiogenesis, cellular senescence and cell growth [34-39]. The kinetics of vascular NAD(P)H oxidase activation are also unique: superoxide anion is produced within minutes to hours in vascular cells, in contrast to the instantaneous superoxide release of phagocytes [40-42]. Furthermore, over recent years it has become clear that the transmembrane ‘anchoring’ subunit, Nox2 (a.k.a. gp91phox), is only one member of a family of homologous proteins termed Nox (for NADPH oxidase) [43, 44]. The Nox family of oxidases has seven members defined by their anchoring components, namely Nox1, Nox2, Nox3, Nox4, Nox5, Duox1 and Duox2 [45], which differ biochemically in their regulation, expression and activity but retain their function in the electron transfer from NAD(P)H to oxygen. Importantly, functional studies demonstrate that in contrast to Nox1-, Nox2- and Nox3-, Nox4-oxidase (hereafter referred to by the anchoring component) does not require the conventional cytoplasmic subunit p47phox and p67phox or its homologues for activation. This suggests that it may be either regulated by other unrelated cytosolic subunits or that its interaction with p22phox is sufficient for its reported constitutive activity [46]. Moreover, it is important to note that Nox1, Nox2 and Nox4 are likely to play distinct roles in the vasculature due to their different vascular expression and subcellular locations [47, 48]. Evidence suggests that homologues of p47phox (NOXO1, for Nox organizer subunit 1) and p67phox (NOXA1, for Nox activator subunit 1) supplant the need for p47phox and p67phox in vascular cells [49, 50]. p47phox is the canonical cytoplasmic organizer subunit for Nox2, whereas its counterpart NOXO1 appears to play a similar role for Nox1 as well as Nox3 [44, 51, 52]. p67phox is the activator subunit for Nox2, whereas NOXA1 is the Nox1 activator [51]. The dependence of Nox3 on NOXA1 is controversial as some studies show that Nox3 requires NOXA1 for activation in HEK293 cells [44], while other studies demonstrate that Nox3 constitutively produces a substantial amount of superoxide in the absence of activator subunits [53]. The structure, regulation and molecular features of the entire Nox family have been covered extensively in excellent reviews by others [22, 29, 47], and thus will not be discussed at length in this review.
Endothelial cells
Reverse transcriptase polymerase chain reaction (RT-PCR), immunohistochemistry, and Western blot analysis have demonstrated the expression of Nox2 and p22phox in rat coronary and porcine pulmonary endothelial cells [27, 54, 55]. In humans, Nox2 and p22phox expression has been detected in coronary endothelial cells and HUVEC lines by means of Western blot and RT-PCR [40, 56] (Table 1). The expression of cytoplasmic protein subunits p47phox and p67phox has been also demonstrated in porcine and bovine pulmonary artery endothelium [55, 57] and in HUVECs [56]. In the endothelium, the expression of Nox homologues, including Nox1, Nox4 and Nox5, have been reported [47, 49, 58], however to date evidence suggest that Nox4 is the most abundant isoform [59, 60]. In the endothelium, modulation of Nox1-, Nox2-, Nox4- and Nox5-based NAD(P)H oxidase has been shown in response to various stimuli (Please see Table 2). With respect to the new homologues of p47phox and p67phox, immunohistological staining showed low expression of NOXO1 and NOXA1 in cultured basilar artery endothelial cells [49]. It was, however suggested that NOXA1 plays an essential role in generation of ROS in response to oxidized low-density lipoprotein (ox-LDL) [50]. These results demonstrate the expression of distinct anchoring and cytosolic components of NAD(P)H oxidase in the vascular endothelium and the important role of endothelium-derived ROS in vascular remodeling and dysfunction.
Table 1.
Expression of NAD(P)H Oxidase Components in the Vascular Wall
| NAD(P)H oxidase components | Endothelial cells | VSMC from conduit vessels | VSMC from resistanc vessels | Adventitia/Adventitial Fibroblasts |
|---|---|---|---|---|
| p22phox | [27, 54-56] | [61, 178] | [65, 179] | [9, 74] |
| Nox1 | [49] | [43, 67] | [65] | [85] |
| Nox2 | [27, 54-56] | ND [40] or low [67] | [65] | [9, 59] |
| Nox4 | [49, 60] | [67] | [65] | [85] |
| p47phox | [55-57] | [63, 64] | [65] | [9] |
| p67phox | [55, 56] | ND [64, 71] | [65] | [9] |
| NOXO1 | [49] | ? | ? | ? |
| NOXA1 | [49] | [71] | ? | ? |
| Rac | [71, 180] | [71] | ? | ? |
ND, non-detectable; VSMC, vascular smooth muscle cells
Table 2.
Modulation of NAD(P)H Oxidase Subunits
| NAD(P)H oxidase components | Endothelial cells | VSMC | Fibroblasts |
|---|---|---|---|
| Nox1 | BMP4 [181] | AngII [67, 182] | Hypoxia [85] |
| VEGF [183] | PDGF [43, 183] | ||
| PGF2α [184] | |||
| Nox2 | AngII [80] | TNF-α [66] | balloon injury [78] |
| TNF-α [66] | LPS [66] | cuff injury [185] | |
| LPS [66] | IL-1α [66] | AngII [9, 186] | |
| IL-1α [66] | |||
| OS [187] | |||
| Nox4 | AngII [80] | 7-Kchol [188] | hypoxia [85] |
| OS [187] | AngII [182] | TGF-β1 [75] | |
| VEGF [189] | AngII* [67] | PUFAs [190] | |
| balloon injury [78] | H2O2 [191] | ||
| OAG [191] | |||
| AA [191] | |||
| Nox5 | thrombin [58] |
downregulation by AngII
AA, arachidonic acid; AngII, angiotensin II; BMP4, bone morphogenic protein 4; IL-1α, interleukin 1α; 7-Kchol, 7-ketocholesterol; LPS, lipopolysaccharide; OAG, 1-oleoyl-2-acetyl-sn-glycerol; OS, oscillatory shear stress; PDGF, platelet-derived growth factor; PGF2α, prostaglandin F2α; PUFAs, polyunsaturated fatty acids; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
Smooth muscle cells
In contrast to the endothelium, it would appear that the medial milieu is somewhat anomalous. For instance, not all phagocyte-like subunits were detected. Only p22phox and p47phox seem to be consistently expressed in vascular smooth muscle cells. p22phox expression was established in rat thoracic aorta vascular smooth muscle cells by Northern blot analysis [61] and in human coronary artery smooth muscle cells by immunohistochemistry and Western blot [62]. Western blot demonstrated p47phox protein expression in rat [63] and human aortic smooth muscle cells [64]. In contrast, p67phox and Nox2 protein are undetectable in aortic smooth muscle [40, 64] (Table 1). However, smooth muscle cells from human resistance arteries [65] and pig pulmonary arteries expressed Nox2 [66]. Although it was originally thought that medial Nox2 expression was specific for resistance vessels, these later findings in pulmonary arteries appear to dispute that notion.
With regard to the anchoring components of the other Nox enzyme systems, Lassègue et al reported higher expression of Nox4 compared to Nox1 in rat aortic smooth muscle [67]. In contrast, Western blot revealed that Nox1 and Nox4 are similarly expressed in mouse pulmonary and mesenteric arteries [68]. Western blot demonstrates Nox4 protein in human pulmonary artery smooth muscle cells [69]. Moreover, cultured human aortic smooth muscle cells are found to predominantly express Nox4 and several isoforms of Nox5 [70]. RT-PCR has revealed the expression of Nox5 in human pulmonary smooth muscle cells [58]. Finally, RT-PCR reveals Nox1 mRNA in human aortic smooth muscle cells but not in human resistance artery smooth muscle cells [65]. Though the function of endothelial Nox5 is yet to be established, it appears to play an important role in endothelial cell proliferation and capillary formation [58].
Medial NAD(P)H oxidases can also be modulated by a variety of agents (Table 2). In addition, it was reported that NOXA1, expressed in the smooth muscle layer but not in the endothelium or the adventitia of the mouse carotid artery, plays an important role in the activation of NAD(P)H oxidase in response to epidermal growth factor [71]. Overall, these results suggest the distinct expression of Nox homologues in the media and a complex interplay of oxidase subunits in vascular smooth muscle pathology. In the case of Nox4, it is important to point out that AngII may be unique in its action to inhibit this isoform [67].
The adventitia and adventitial fibroblasts
Early studies in our laboratory demonstrated relatively abundant co-localization of Nox2, p22phox, p47phox and p67phox in aortic vascular adventitia and enrichment of p67phox-dependent NAD(P)H oxidase activity in cultured adventitial fibroblasts of the rabbit aorta [9, 72] (Table 1). Immunohistochemical staining showed that these four major components of the Nox2 oxidase were concentrated in rat and mouse aortic adventitia [16, 73]. Fluorescent immunohistochemistry demonstrated that Nox2 is higher in the adventitial layer of human coronary arteries [59], corroborating earlier evidence for relatively higher adventitial p22phox by other groups [62, 74].
With regard to the other Nox isoforms and cytosolic subunits, it has been reported that Nox4 is weakly expressed in adventitial fibroblasts of human coronary arteries [59]. Freshly isolated cultured cardiac fibroblasts express high levels of p22phox, and lower levels of Nox4 > Nox2 > Nox1 [59]. On the other hand, Cucoranu et al demonstrated that Nox5 and Nox4 mRNA are abundantly expressed in cardiac fibroblasts, whereas Nox1 and Nox2 are barely detectable [75]. Using laser scanning microscopy, positive staining for NOXA1 was not observed in the adventitia of the mouse carotid artery [71].
Our laboratory reported constitutive NAD(P)H oxidase superoxide-generating activity in aortic adventitial fibroblasts [9]. In general, fibroblast NAD(P)H oxidase activity can be further enhanced by a wide variety of stimuli, including cytokines, hormones, metabolic factors and mechanical injury [72, 76-79]. Indeed, hypoxia, mechanical injury, polyunsaturated fatty acids (PUFAs), inflammatory cytokines and H2O2 stimulate the release of ROS from fibroblasts (Table 2). In addition, results showing that hyper-insulinemia increases ROS production by human fibroblast NAD(P)H oxidase suggest an important role for fibroblast-derived ROS in the development of oxidative stress associated with type 2 diabetes mellitus [79]. Finally, Szöcs et al reported that 3 days after balloon angioplasty p22phox protein expression and ROS production increases in the adventitial layer of the carotid artery [78].
Taken together, these results demonstrate distinct expression of membrane and cytosolic components of NAD(P)H oxidase in the vascular endothelium, media and adventitia. Moreover, they predict a complex orchestration of ROS-mediated signaling across the vessel wall in response to various disease processes and stimuli. Although further inquiry is required, agents that modulate NAD(P)H oxidase in various vascular cell types and segments appear to differ with regard to the particular stimulus and Nox isoforms. Notably, AngII emerges as the most broadly acting on the Noxes across the vessel wall, be it stimulatory [80] or inhibitory[67]. Intriguingly, it would also appear from the data in Table 2 that fibroblast NAD(P)H oxidases are particularly sensitive to hypoxic conditions although it may be premature to draw that conclusion without careful comparison of hypoxia's effects among a variety of cell types.
4. Adventitial NAD(P)H oxidase-derived ROS in vascular diseases
Diabetes mellitus, hypertension, atherosclerosis, and vascular injury all activate adventitial cells and increase NAD(P)H oxidase-derived ROS production in the adventitial layer [16, 28, 59, 62, 81-83]. For instance, streptozotocin-induced diabetes mellitus increased oxidative stress in porcine coronary arteries largely as a result of augmented adventitial NAD(P)H oxidase activity [82]. The incubation of coronary fibroblasts with advanced glycation end products (AGEs) increased expression of inflammatory mediators, such as IL-6, VCAM-1 and monocyte chemotactic protein-1 (MCP-1) that was inhibited by antioxidants [82]. In AngII-induced hypertension, the NADPH-oxidase derived superoxide production was 5.5-fold higher in the aortic adventitia than in the intima [81]. Indeed, in aortic rings isolated from hypertensive rats nitroblue tetrazolium staining localized superoxide production predominantly to the adventitial layer, while immunohistochemistry showed the expression of p22phox, p47phox and p67phox primarily in the adventitia [81]. In addition, results from our laboratory also suggest that in the AngII-induced hypertension model, the adventitia is a major source of NADPH oxidase-derived superoxide production [16]. Moreover, Shi et al demonstrated that expression of p47phox and p67phox were low in normal coronary arteries, but significantly increased in the adventitia after balloon injury, concomitant with enhanced superoxide production [84]. Szöcs et al demonstrated that superoxide production was increased 3 days after balloon injury in the adventitial and in the innermost medial layer of carotid arteries [78]. Furthermore, in response to hypoxia, Nox4-based NAD(P)H oxidase-derived ROS production increased in pulmonary artery adventitial fibroblasts, suggesting an important role of adventitia-derived ROS in hypoxia-induced vascular remodeling and pulmonary hypertension [85]. Taken together, these results suggest that adventitial NAD(P)H oxidase is activated and adventitia-derived ROS production is increased by a wide variety of conditions ranging from metabolic diseases and mechanical injury to atherosclerotic risk factors. It is therefore not surprising that several studies demonstrated the activation of adventitia positively correlated with the degree of adventitial inflammation and the severity of atherosclerosis [86]. Moreover, findings that in human atherosclerotic coronary arteries the immunoreactivity of p22phox in the adventitia was more pronounced than in non-atherosclerotic arteries appear to suggest that adventitial superoxide plays a role in atherosclerosis [62]. Since ROS from the adventitia are capable of inactivating endothelium-derived NO [73] as well as inducing vascular remodeling in a paracrine manner [87], it is tempting to speculate that activation of adventitial NAD(P)H oxidase may be an important signaling agent in vascular diseases. However, the expectedly complex role of NAD(P)H oxidase-derived ROS in atherosclerosis is not yet clear as some [88] but not all studies [89] show diminished progression of atherosclerosis in ApoE-/- mice lacking an essential component of NAD(P)H oxidase, p47phox or Nox2.
5. Adventitia-derived ROS, and vascular inflammation
Chronic vascular inflammation is a self-perpetuating condition in which oxidative stress-induced phenotypic changes in the vessel wall lead to vascular remodeling and development of vascular disease. Vascular inflammation is known to involve chemotaxis, chemokinesis, monocyte adhesion and transmigration [5]. Less understood, but perhaps equally important, is how adventitial cell proliferation and vasa vasorum neovascularization participate in this process as well as an underlying pivotal role for ROS and lipid peroxidation [90]. Having established adventitial fibroblasts as a robust source of NAD(P)H oxidase-derived superoxide [15-18], we will address the role of adventitia-derived ROS in the initiation and progression of vascular inflammation and address how even subtle increases in ROS and factors derived from these infiltrating cells may affect vascular remodeling.
Using three-dimensional computed tomography, Ritman and coworkers illustrated increased density of the vasa vasorum, small arteries that penetrate the tunica adventitia and media of large vessels as early hallmarks of vascular inflammatory disease [90, 91]. Indeed, in hypertension, hypercholesterolemia and atherosclerosis, a ‘hotbed’ of perivascular hyperplasia results in neovascularization of the adventitia and outer media in the form of a more dense vasa vasorum [11, 90]. The initiating factor for this process purportedly involves decreased oxygen at the core of a thickened media, which elicits expression of critical growth and transcription factors and hypoxia-inducible factor 1α (HIF-1α) [11]. With a denser vasa vasorum and increased adhesion molecule expression on its endothelial lining (vide infra), one would expect a positive feedback loop leading to an early and exaggerated accumulation of leukocytes in the adventitia and outer media. It is our contention therefore that the combined parenchymal cell and inflammatory cell-derived ROS synergize in their local and paracrine influence on vascular dysfunction which includes changes in vascular responsiveness, adventitial and medial cell proliferation and migration, medial hypertrophy, and neointima formation.
The expression of various adhesion molecules, such as VCAM-1, ICAM-1, P- and E-selectins on large vessel arterial lumen endothelial cells is an early and critical step in the inflammatory process that leads to the transmigration of leukocytes to the subendothelium [92, 93]. In addition, increased expression of adhesion molecules on the endothelial surface of the vasa vasorum leads to the accumulation of leukocytes in the adventitia and outer media (Figure 1). Although the precise mechanism underlying this early process is not entirely clear, NAD(P)H oxidase-derived ROS are increasingly being recognized as important players [94, 95]. Recent evidence suggests that ROS up-regulate NF-κB, which plays a pivotal role in the release of pro-inflammatory cytokines, such as IL-1, IL-6 and TNFα, leading to increased expression of endothelial adhesion molecules [96]. The findings that IL-1β- and TNFα-induced VCAM-1 expression is inhibited by antioxidants also suggest that expression of adhesion molecules on vascular endothelium is ROS-dependent [97]. Moreover, Li et al demonstrated that TNFα stimulation failed to induce expression of ICAM-1 in coronary microvascular endothelial cells from p47phox-/- mice suggesting a role of Nox2-based NADPH oxidase-derived ROS in this process [98]. It was also reported that LPS increases the expression of ICAM-1 in human aortic endothelial cells in an NF-κB-dependent manner via the activation of Nox4 [95]. The existence of a feed-forward interaction among leukocyte-derived and adventitial parenchymal cell-derived ROS on NF-κB-dependent inflammatory responses has not yet been confirmed. However, a recent review proposed that ROS-mediated cross-talk between adventitial fibroblasts and leukocytes may have significant implications for general inflammatory responses, leading to a further increased NF-κB activation and downstream adhesion molecule expression in the endothelial layer of the vasa vasorum [99]. As a result of this cross-talk, activated adventitial fibroblasts release a number of potent mediators, such as ET-1, TGF-β, TNF-α and PDGF [99, 100] that may induce IκBα phosphorylation and ubiquitination leading to the release and translocation of NF-κB to the nucleus and subsequent expression of adhesion molecules [101].
Fig.1. The known and proposed mechanisms leading to the initiation and progression of vascular disease from an “outside-in” perspective of vascular inflammation.
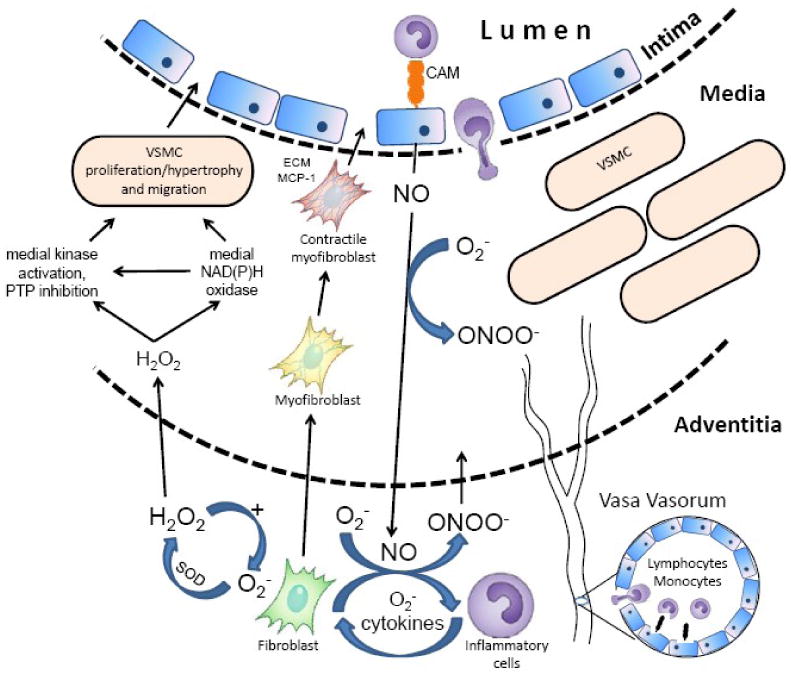
Early in the development of vascular disease, adventitial fibroblasts produce a substantial amount of NAD(P)H oxidase-derived ROS in response to vascular injury, inflammatory cytokines, lipids and various hormones. There is also clear evidence showing that the appearance of microvessels in the form of denser vasa vasorum is an early hallmark of vascular inflammatory disease. Increased levels of adventitial fibroblasts-derived cytokines and ROS would be expected to increase the expression of adhesion molecules on the endothelial lining of an expanded vasa vasorum leading to the transmigration of inflammatory cells to the adventitia and outer media. Invading leukocytes coalesce in the adventitia in response to hormonal stimulants, i.e. AngII, and contribute to the adventitial cytokine and ROS production, thus leading to further increased fibroblast cytokine secretion and a feed-forward propagation of adventitial ROS production. The local cytokine milieu influences the adventitial fibroblasts to undergo a phenotypic switch into myofibroblasts, which migrate to the medial and intimal layer. During neointimal hyperplasia, MCP-1 produced by migratory myofibroblasts is proposed as a chemotactic signal for monocytes leading to their exaggerated transmigration across the vessel wall. In the adventitial layer, resident fibroblasts and inflammatory cells produce superoxide that may be spontaneously or enzymatically dismuted to H2O2, which is relatively stable and cell-permeable and thus likely to act as a paracrine mediator across the vessel wall. H2O2 and perhaps ONOO- are postulated to diffuse to the medial layer, where they directly activate smooth muscle cell kinases. This paracrine oxidative signaling is proposed to mediate medial proliferation and hypertrophy and/or initiate activation of medial NAD(P)H oxidase, which indirectly contributes to medial remodeling.
With this information along with the knowledge of a robust adventitial oxidase activity, it is not difficult to envision a strong positive relationship between adventitial ROS and adhesion molecule expression in the vasa vasorum. Supporting this assumption, AngII infusion for one week increased NAD(P)H oxidase-derived ROS production in the rat aorta and enhanced ICAM-1 expression and subsequent adventitial macrophage infiltration [102]. Similarly, AngII infusion in mice induced a convergence of macrophages to the adventitia [10]. Other models of vascular pathology show a similar association. For example, Zhang et al reported that, in streptozotocin-induced diabetes, increased NAD(P)H oxidase-derived superoxide production in the coronary adventitia was accompanied by up-regulation of VCAM-1 expression [82]. During peri-arterial collar-induced vascular remodeling, adventitial NAD(P)H oxidase-derived superoxide production is elevated concomitant with increased expression of VCAM-1 and ICAM-1 [83]. More direct evidence of a role for ROS promoting an inflammatory role of the vasa vasorum is provided by Okamoto, who reported increased expression of P-selectin mRNA and protein in the vasa vasorum endothelium of balloon-injured porcine coronary arteries accompanied by neutrophil infiltration into the adventitia [103]. These results are not surprising, especially if one considers that the adventitial vasa vasorum is highly enriched in endothelium and is likely to far exceed the number of endothelial cells on the large vessel lumen per unit longitudinal length of the artery. Thus, on closer examination of conduit arteries in cardiovascular diseases, one might expect a specific enrichment of adhesion molecule expression in this segment. In line with this notion, O'Brien et al demonstrated that in human atherosclerotic coronary segments the expression of adhesion molecules on the vasa vasorum (neovasculature) was twice as prevalent as their expression on the large arterial luminal surface [92].
An increasing body of evidence indicates that adventitial superoxide production not only induces increased expression of adhesion molecules, but may also directly contribute to the chemotactic movement of leukocytes and their increased penetration into the vessel wall. Indeed, ROS derived from adventitial fibroblasts are likely to serve as a direct chemotactic signal for leukocytes [102] and increase MCP-1 levels [104]. In fact, as findings by Xu et al. illustrate, adventitial fibroblast MCP-1 levels are increased in apoE-/- mice prior to the appearance of intimal lesions [105]. In vitro measurements showed that scavenging of ROS by SOD and catalase suppressed induction of MCP-1 mRNA accumulation by TNF-alpha [106]. Furthermore, it was demonstrated that following angioplasty, CXCL2, a chemotactic cytokine belonging to the CXC chemokine family, is transiently activated in the adventitial vasa vasorum at day 2 as well as in luminal neointimal cells at day 7 [107]. It stands to reason that once leukocytes invade the adventitia, they produce a large amount of inflammatory cytokines and ROS locally that, in combination with ROS derived from activated adventitial fibroblasts, further exaggerate leukocyte accumulation, ultimately leading to the activation of downstream signaling processes in the adventitia, media and intima. Such a feed-forward interaction among leukocytes, cytokines, ROS and vascular tissue has not been addressed to our knowledge. It is important to note, however, that the same mechanism occurs in the luminal subendothelium in response to cardiovascular risk factors, thus a broader process incorporating both the “outside-in” and the “inside-out” paradigms is likely to operate in the pathophysiology of vascular disease.
6. Adventitial cells and vascular remodeling
In cultured rat aortic smooth muscle cells, it is well established that AngII-stimulated NAD(P)H oxidase-derived ROS mediate the hypertrophic response. For instance, in p22phox-deficient smooth muscle cells AngII-stimulated NAD(P)H oxidase activity decreases, concomitant with reduced hydrogen peroxide production and attenuated medial hypertrophy [61]. These findings indicate that localized ROS acting in an autocrine fashion are capable of inducing medial hypertrophy.
In recent years, evidence has accumulated to support a paracrine effect of adventitial NAD(P)H oxidase-derived ROS in vascular cell proliferation and medial hypertrophy [102, 108, 109]. Wang et al demonstrated that AngII infusion into mice for 6 days increased intimal and adventitial NAD(P)H oxidase-derived superoxide production concomitant with aortic medial thickening [108]. Furthermore, in the same experimental setting, deletion of Nox2 decreased intimal and adventitial superoxide formation and led to reduced aortic medial area, suggesting a paracrine effect of intimal and adventitial Nox2-based NAD(P)H oxidase-derived ROS on medial hypertrophy. Subsequently, our laboratory showed that AngII infusion enhanced adventitial NAD(P)H oxidase activity and increased medial growth independent of changes in blood pressure. Additionally, co-infusion of cell-permeant inhibitor of Nox2-p47phox interaction (nox2ds-tat) attenuated AngII–induced medial hypertrophy [87]. Targeted delivery of this inhibitor to predominantly adventitial fibroblasts using adenovirus attenuated carotid artery medial hypertrophy as well as lipid peroxidation byproduct 4-hydroxynonenal (4-HNE) deposition across the vessel wall [87]. Thus these data are consistent with adventitial NAD(P)H oxidase promoting medial hypertrophy via an oxidizing ROS, the exact nature of which is still unknown. Furthermore, it is important to re-emphasize that in the AngII-induced hypertension model macrophages are abundantly detected in the adventitia [10], and the release of ROS and cytokines from these infiltrating cells together with the activated adventitial cell-derived ROS may synergize their paracrine effect on vascular remodeling and dysfunction.
Having established ROS as important mediators of medial growth in AngII-induced vascular remodeling, it is important to address which ROS is most likely to act as a paracrine mediator across the vessel wall. Baas et al reported that both superoxide and H2O2 are capable of stimulating vascular smooth muscle cell growth via distinct mechanisms [110]. Since superoxide has a diffusion radius of a few microns and an extremely short half-life, it is unlikely to act as a paracrine mediator across the vessel wall but rather as an autocrine or intracellular agent [111]. Indeed, superoxide has been described to activate multiple intracellular proteins and enzymes, including epidermal growth factor receptor, c-Src, p38 mitogen-activated protein kinase (p38 MAPK) and Ras [112]. In contrast, emerging evidence suggests that H2O2 is an important paracrine agent in vascular biology. Given the fact that H2O2 is freely diffusible within and between cells and does not possess an unpaired electron in its outer shell, it would appear that H2O2 is a better candidate as a paracrine agent (Figure 1). In line with this notion, Zhang et al demonstrated that AngII treatment-induced aortic wall hypertrophy was dramatically decreased in transgenic mice with vascular smooth muscle cell-specific overexpression of human catalase, suggesting that H2O2 plays a critical role in the development of vascular hypertrophy in vivo [113]. Human coronary artery adventitial fibroblasts possess NADPH oxidase components, Nox2 and Nox4, and produce superoxide in response to AngII [59]. In the adventitial layer, superoxide can be converted to H2O2 by extracellular SOD [114, 115], which is markedly stimulated by AngII and inflammatory cytokines, such as interferon γ [116, 117]. It is therefore likely that superoxide derived from adventitial fibroblasts and macrophages is rapidly converted to H2O2 in cardiovascular diseases associated with high levels of Ang II and inflammatory cytokines. Interestingly, it has been reported that HEK293 cells stably expressing human Nox4 produce large amounts of H2O2 as a primary metabolite [118], thus raising the possibility that H2O2 derived directly from Nox4 in the adventitial layer can also act as a paracrine mediator.
Under physiological conditions, endothelium-derived NO contributes to paracrine signaling to the media, where it activates soluble guanylate cyclase and elicits relaxation [119-121]. Under pathophysiological conditions, however, superoxide in the adventitia may rapidly inactivate NO and impair endothelium-dependent relaxation. It is not difficult then to envision that in all cases in which adventitial NAD(P)H oxidase is activated, large amounts of the highly reactive ROS, peroxynitrite (ONOO-), are expected. Although the latter has a limited stability and diffusion radius [111, 122], as a potent oxidizing agent, ONOO-, it is expected to cause extensive lipid peroxidation along with its byproducts. One of these byproducts is 4-HNE, which is shown to activate the p38 MAPK pathway, which plays a pivotal role in COX-2 induction [123]. Based on this information, adventitial ONOO- has the potential to either (a) initiate paracrine signaling locally which then spreads across the vessel wall or (b) facilitate the generation of secondary metabolites like 4-HNE which, via diffusion, effect signaling in the vascular media.
7. Paracrine effect of adventitial fibroblast-derived ROS and restenosis
Atherosclerosis, with its major ischemic complications, myocardial infarction and ischemic stroke, is one of the leading causes of death in the Western world [5]. Current therapeutic strategies to restore blood flow in stenotic arteries and thus prevent ischemic complications include transluminal angioplasty and stent placement. During these procedures, however, the endothelial layer and the internal elastic lamina are injured, resulting in endothelial denudation, medial tearing, and attachment of circulating platelets and leukocytes to the exposed subendothelial matrix. Platelets and leukocytes, together with activated vascular cells, produce substantial amounts of inflammatory cytokines, growth factors and ROS leading to phenotypic modulation of medial and adventitial cells [124-126]. In response, smooth muscle cells and adventitial fibroblasts proliferate and migrate to the intima where they secrete extracellular matrix proteins and contribute to the characteristic features of restenosis: neointima formation and constrictive remodeling [12, 127-129]. There is now increasing evidence that ROS play a fundamental role in balloon injury-induced vascular remodeling and that among other potential sources of ROS the membrane-bound NAD(P)H oxidase system is the major source of superoxide in response to vascular injury [78, 130]. In recent years, it has become increasingly clear that in response to balloon injury, NAD(P)H oxidase-derived ROS assume an important role in smooth muscle cell and adventitial fibroblast proliferation and their migration to the neointima [78, 131, 132]. Interestingly, Shi et al demonstrated that adventitial cell proliferation and thickness in the coronary arteries exceeded that in the media at 3 and 7 days after balloon injury [133]. Moreover, in the same post-angioplasty setting, adventitial fibroblasts showed an increased α-smooth muscle (α-SM) actin immunostaining at day 8, indicating the modulation of their phenotype into migratory myofibroblasts in a TGF-β dependent manner [134]. During this process, myofibroblasts acquire different markers of muscle differentiation and migrate to the media as well as neointima, where they may transform into contractile myofibroblasts and contribute to constrictive remodeling (Figure 1). Incidentally, MCP-1 is highly expressed in myofibroblasts and may serve as a chemotactic signal for monocytes from the sub-endothelium leading to monocyte transmigration at later stage of neointimal hyperplasia [107]. Since neointimal proliferation and restenosis after stretch injury coincide with elevated levels of ROS, it is likely that ROS play an important role in activating a variety of medial and adventitial growth-related signaling pathways in both a highly localized and paracrine manner [130, 135].
To test the paracrine role of adventitia-derived ROS in restenosis, Dourron et al employed targeted delivery of an adenoviral vector expressing NAD(P)H oxidase inhibitor nox2ds in the adventitia and investigated its effect on stretch-induced vascular superoxide production and neointima formation [109]. The delivery of a nox2ds-expressing virus to the perivascular space resulted in a localized adventitial expression of nox2ds that suppressed distension-induced superoxide production and significantly attenuated neointima formation. Similarly, inhibition of adventitial p67phox activity reduced angioplasty-induced neointimal thickness, but not to the same degree as did targeting of the p47phox-nox2 interaction by the nox2ds [136]. These results are suggestive of adventitial NAD(P)H oxidase-derived ROS having a paracrine effect on smooth muscle cell proliferation and neointimal growth, without excluding the direct role of fibroblasts in this process. Indeed, it is likely that proliferating adventitial fibroblasts and medial smooth muscle cells directly contribute to neointima formation after balloon injury. However, the demonstrated importance of smooth muscle migration to neointimal growth [137] and the finding that H2O2 increases smooth muscle cell DNA synthesis and cell number (as well as c-myc and c-fos mRNA levels), suggest an important paracrine influence of H2O2 on medial and intimal growth [135].
8. H2O2 as a mediator of medial signal transduction
Numerous studies demonstrate that ROS, especially superoxide and H2O2, are important signaling molecules in the vasculature. Both superoxide and H2O2 have been demonstrated to serve as second messengers activating a variety of signaling cascades involved in smooth muscle cell contraction and hypertrophy, including epidermal growth factor receptor (EGF-R), p21ras, phosphatidylinositol-3 kinase (PI3-K) and Rho-kinase pathways [42, 112, 138]. H2O2 is capable of directly increasing smooth muscle cell DNA synthesis and cell number in association with induction of c-myc and c-fos mRNA [135]. Furthermore, H2O2 induces tyrosine phosphorylation of EGF-R in smooth muscle cells, which forms a complex with Shc (src homology complex), Grb2 (growth factor receptor-bound protein 2) and Sos (son-of-sevenless) leading to the downstream activation of ras and extracellular signal-regulated protein kinases (ERK1/2) [138]. Ushio-Fukai et al reported that AngII-induced transactivation of EGF-R requires ROS in smooth muscle cells and suggested that c-Src is a signaling molecule that links ROS and EGF-R phosphorylation [139]. Interestingly, Abe et al reported that H2O2 directly activates c-Src in mouse fibroblast, which is an essential step for big mitogen-activated protein kinase 1 (BMK1) activation [140]. Additionally, it has been suggested that H2O2 mediates AngII-induced c-Src phosphorylation in smooth muscle cells [112]. Seshiah et al demonstrate that Rac activation by AngII is dependent on PI3-K in smooth muscle cells and required for NAD(P)H oxidase activation [42]. Taken together, H2O2 activates c-Src in smooth muscle cells, which in turn transactivates the EGF-R leading to MAPK activation and stimulation of PI3-K and Rac-dependent NAD(P)H-derived ROS production (Figure 2). The activation of these signaling pathways in smooth muscle cells leads to a further activation of c-Src, a feed-forward propagation of signaling mechanisms and amplification of medial remodeling [112, 139].
Fig.2.
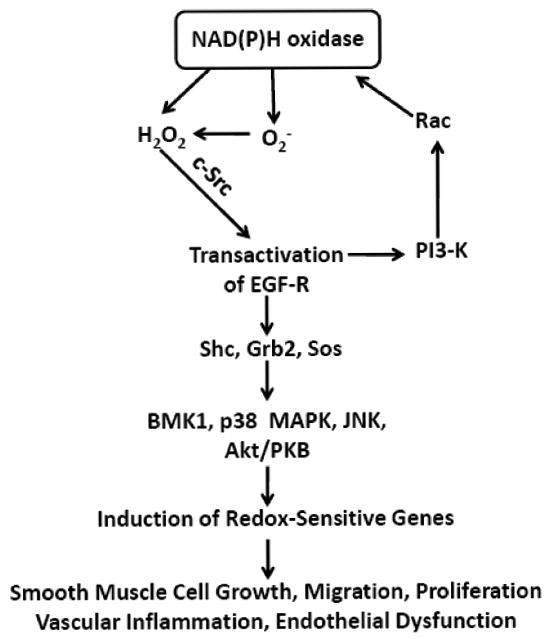
Schematic diagram illustrating the signaling pathways activated by H2O2 in vascular smooth muscle cells. As noted in the text, H2O2 activates numerous signaling molecules and stimulates NAD(P)H oxidase activity in smooth muscle cells, leading to a feed-forward propagation of signaling mechanisms, increased ROS production and amplification of medial remodeling. Akt/PKB, Akt/protein kinase B; BMK1, big mitogen-activated protein kinase 1; EGF-R, epidermal growth factor receptor; Grb2, growth factor receptor-bound protein 2; JNKs; c-Jun N-terminal kinases; p38 MAPK, p38 mitogen-activated protein kinase, PI3-K, phosphatidylinositol-3 kinase; Shc, src homology complex; Sos, son-of-sevenless.
Emerging evidence indicates that the MAPK family, including BMK1, p38 MAPK, c-Jun N-terminal kinases (JNKs) and ERK1/2 regulates vascular growth and migration [141, 142]. In smooth muscle cells, H2O2 has been shown to activate BMK1 [143], p38 MAPK [144] and JNK [145]. In contrast, the effect of H2O2 on ERK1/2 is controversial as some [146], but not all, studies [143, 144] show activation in smooth muscle cells. Moreover, AngII-stimulated p38 MAPK activation was significantly inhibited in smooth muscle cells stably transfected with catalase [144]. Transfection with antisense p22phox oligonucleotides decreased AngII-induced H2O2 and p38 MAPK activity in smooth muscle cells [147]. Ushio-Fukai et al reported that both H2O2 and AngII stimulate Akt/protein kinase B (Akt/PKB) phosphorylation in smooth muscle cells [148]. Diphenylene iodonium, an inhibitor of flavin-containing oxidases, and overexpression of catalase inhibited AngII-induced Akt/PKB phosphorylation in smooth muscle cells. These results suggest a role of NAD(P)H oxidase-derived H2O2 in AngII-induced medial Akt/PKB activation.
A major component of the H2O2-dependent modulation of cell-signaling is the dynamic regulation of protein function by reversible cysteine oxidation [149, 150]. H2O2 is able to oxidize cysteine residues in proteins to cysteine sulfenic acid (Cys-SOH), which can react with another protein thiol (as an intra- or intermolecular interaction) or with a neighboring amide nitrogen to form either disulfides (Cys-S-S-protein) or sulfenamides (Cys-S-N-protein), respectively. These reversibly oxidized forms can be reduced back to the sulfhydryl state (Cys-SH) by reduced glutathione (GSH), a tripeptide that is also regulated by oxidants. Protein-tyrosine phosphatases (PTPs) are particularly attractive candidates for H2O2-induced redox signaling in the vasculature because PTPs contain reactive cysteine residues within their active sites. Indeed, H2O2 has been reported to oxidize catalytic cysteines of PTPs leading to their inactivation in smooth muscle cells and fibroblasts [151, 152]. Interestingly, a previous study demonstrated that H2O2-induced PTP inactivation led to the phosphorylation of PDGF β receptor and subsequent migratory and proliferative responses in smooth muscle cells [152]. Moreover, it has been reported that antioxidants reduce PDGF β receptor phosphorylation, increase PTP activity and inhibited neointima formation [152]. These results emphasize a role for H2O2 in the activation of redox sensitive medial signaling cascades leading to smooth muscle cell migration, proliferation, and vascular inflammation. The likelihood, however, of the non-enzymatic direct oxidation of PTP by H2O2 has been called into question due to the high cellular activity of GPX and catalase and a considerably higher rate of reaction of H2O2 with these enzymes [153]. On the contrary, it is plausible that a high local production of H2O2 close to PTP could account for this oxidation. Alternatively, PTPs may be reversibly glutathionylated or oxidized to a disufide in reactions that are dependent upon H2O2 via a yet-to-be identified enzymatic reaction [153].
9. Mast cells and T-lymphocytes in the adventitia and their role in vascular inflammation
The tunica adventitia is composed primarily of fibroblasts, collagen and elastin fibers, however a variety of inflammatory cells, including macrophages, mast cells and lymphocytes are also present. In addition to macrophages, adventitial mast cells and T- lymphocytes have been demonstrated to play a critical role in the initiation of vascular inflammation and progression of cardiovascular disease [154-157]. Indeed, the number of mast cells was found to increase in the adventitial layer of infarct-related coronary arteries and aortic aneurysm [154, 156], where they release various inflammatory mediators, such as histamine, prostaglandins, cytokines, chemokines, growth factors and proteases [158, 159]. Degranulation of mast cells is therefore postulated to contribute to the recruitment of macrophages and T-lymphocytes as well as to fibroblast stimulation [158, 160, 161]. In addition, immunohistochemical analysis revealed the presence of the angiotensin converting enzyme, chymase, in adventitial mast cells of atherosclerotic aorta and aneurysmal lesions [162], suggesting that AngII released from adventitial mast cells could potentiate vascular ROS formation. Additionally, findings that mast cells synthesize ROS via NADPH oxidase suggest that activated mast cells in the vessel wall may directly contribute to ROS production [163, 164]. Recent studies have demonstrated that stimulation of mast cells through high-affinity IgE receptors induces intracellular production of NAD(P)H oxidase-derived ROS, which appear to be involved in the regulation of calcium signals, degranulation and cytokine release [164].
With regard to the role of adventitial T lymphocytes in vascular inflammation, studies show that the adventitia of atherosclerotic apoE-/- mice is a major site of T-cell accumulation that exceeds that of the intima [165]. In addition, adventitial T-cell infiltration is associated with a marked increase in macrophage inflammatory protein-1α (MIP-1α) transcript in atherosclerotic aorta [165]. Similar to mast cells, T lymphocytes also contain a functional NADPH oxidase [166], which has been implicated in AngII-induced hypertension and increased vascular superoxide production [157]. Notably, it was reported that T-cell infiltration occurs primarily in the peri-adventitial fat [157]. Moreover, in T-cells NADPH oxidase-derived ROS contribute to cytokine production and T cell activation [157, 166].
After vessel injury, but prior to neointimal development, the adventitia and perivascular tissue become highly populated with neutrophils, macrophages, lymphocytes and mast cells [103]. These inflammatory cells produce a large number of inflammatory cytokines and NAD(P)H oxidase-derived ROS that are likely to contribute to the activation and proliferation of fibroblasts leading to a more pronounced adventitial ROS generation and activation of the downstream signaling pathways. These observations suggest a complex interaction among inflammatory cells, cytokines, and NADPH oxidase in a positive feedback cycle of ROS generation and vascular dysfunction.
10. NOX and NOS, synergize in the production of pro-inflammatory oxidants
Over the last few decades NO has emerged as an important cardio- and vasculo-protective molecule via its vasodilator, antioxidant, antithrombotic, antiproliferative and anti-inflammatory actions [167]. Under pathophysiological conditions, however, NO may become deleterious if it reacts with superoxide to form ONOO-, a powerful oxidant, which may cause extensive peroxidation and nitration in the vascular wall. The critical balance between local concentrations of NO and superoxide and the competition between NO and SOD for superoxide is important in understanding NO biology and its deleterious role in the form of ONOO-. The local concentration of NO is determined by the rate of its production and degradation. The balance between the total superoxide-generating capacity of the vessel wall and its degradation via SOD determines local concentration of superoxide in the vasculature. Relevant to this point, the rate of superoxide reacting with NO is fast enough to out-compete endogenous SOD for superoxide [168].
In healthy individuals with preserved vascular NO bioavailability and low superoxide production, limited ONOO- is formed. In contrast, one might expect an increased formation of ONOO- in the vasculature in a variety of pathophysiological conditions associated with an increased rate of superoxide production. In line with this notion, Yasmin et al. demonstrated increased production of dityrosine, which is formed by the reaction of ONOO- with L-tyrosine, was inhibited by L-NMMA, a specific inhibitor of NOS, or by SOD after ischemia-reperfusion injury of the heart [169]. On the contrary, despite the fact that ONOO- is widely recognized as a mediator of NO toxicity, studies also indicate that this compound may induce vasodilatation and inhibit platelet aggregation [170].
11. The adventitia as a ‘staging ground’ for therapeutics delivery
Cardiovascular disorders of large and medium-sized arteries are often treated with pharmacological as well as endovascular interventions. Some of the pitfalls of these procedures in these vessels include a short duration of treatment exposure or the potential for systemic adverse effects from large doses of the pharmacological agents. Delivery of agents or gene vectors into the vascular wall using a periadventitial approach (external stenting, local delivery to the adventitial surface) could be advantageous in providing a sustained, continuous and controlled application of treatment to the diseased vessel.
For instance, local release of chemically-modified heparin from polymeric matrices placed at the adventitial surface inhibited smooth muscle cell proliferation after vascular injury [171]. Moreover, vascular radiation that reduces adventitial cell proliferation prevents intimal lesion development in response to angioplasty [172]. After adenoviral gene transfer of eNOS to the adventitia of carotid arteries, enhanced endothelium-dependent relaxation of atherosclerotic vessels was observed [173]. A variety of other studies demonstrate the effectiveness of this approach and illustrate that delivery of enzymatic and non-enzymatic antioxidants to the adventitia can produce salutary effects across the blood vessel wall [83, 174]. These studies provide proof-of-principle for the use of the adventitia as a therapeutic ‘staging ground’ in vascular disease.
12. Clinical trials
Experimental and observational data support a role for oxidative stress in cardiovascular disease and the benefit of increased antioxidants in the diet. However, a number of high-profile clinical trials examining a protective effect of antioxidants have reported negative results, calling into question the therapeutic advantage of antioxidants to ameliorate human disease (for a thorough review, see [175]. Yet, the solution may be as straightforward as a clear understanding of the role of ROS in cardiovascular disease and what truly constitutes an ‘antioxidant’ from this perspective. That is, many studies to date have used antioxidant vitamins without proper analysis of their ROS-inhibitory potential, duration of treatment, or the cohort of patients studied [176]. Even the possibility that these vitamins, under certain conditions, can have pro-oxidizing effects has been overlooked in many cases. Complementary studies are needed to determine the proper dosing of these therapies and their in vivo effectiveness to reduce oxidative stress in patients. Moreover, it is important to consider the regimen time course and whether treatment has commenced beyond the point of efficacy. That is, antioxidant administration may come “too little, too late”, especially in advanced diseases driven by multiple factors.
Since a significant body of evidence indicates that superoxide and H2O2 are important in their own right as signaling agents involved in vascular homeostasis [112, 113], the nature and delivery of such inhibitors must be such that they only target deleterious oxidase activity. Moreover, since some therapeutics and vitamins can “redox cycle,” emphasis should be placed on developing agents that either inhibit the production or scavenge these other biologically important ROS without expediting the production of other oxidants. For instance, singular scavenging of superoxide with SOD or its mimetics could actually increase H2O2 and thus promote oxidation either directly or via conversion of H2O2 to hydroxyl anion in the presence of reduced metals [177]. Thus, clinical studies should be carried out with careful consideration of these factors as well as a clear understanding of the complex redox chemistry involved in these pathways. One way to avoid this problem may be by inhibiting the source of superoxide in tissue. Since superoxide is the precursor to multiple ROS, inhibition of superoxide-generating oxidases could avert the problems discussed above. Perhaps even more important and relevant to the current discussion, strategies targeting oxidase isozymes in distinct cell types or in blood vessel segments, i.e. the adventitia, could provide the advantage of selective inhibition of deleterious vs. salubrious oxidant production.
13. Summary
Vascular inflammation is traditionally considered an “inside-out” response in which the inflammatory process is initiated at the luminal surface and progresses toward the outer layers of the vessel wall. In recent years, however, evidence has accumulated showing early activation of adventitial cells in response to various stimuli and cardiovascular risk factors, preceding more ostensible indicators of vascular disease such as endothelial dysfunction and inflammation. It is proposed that early in the development of vascular disease, adventitial fibroblasts produce a significant amount of NAD(P)H oxidase-derived ROS, resulting in increased expression of adhesion molecules on the endothelial surface of the vasa vasorum and ultimately leading to adventitial leukocyte infiltration. Transmigration of leukocytes to the adventitia through the expanded vasa vasorum and activation of resident mast cells could contribute to the adventitial cytokine and ROS production, leading to further increased fibroblast cytokine production, feed-forward activation of Nox isoforms and increased adventitial ROS formation. This positive feedback loop is expected to occur across vessel segments and suggests synergy in this regard between the adventitial, medial and endothelial layers. During this process, the adventitial layer is expected to produce a large amount of H2O2, which is stable and cell permeable and therefore likely to act as a paracrine mediator of oxidative stress across the vessel wall. Moreover, NAD(P)H oxidase-derived superoxide in the adventitia may rapidly inactivate endothelium-derived NO leading to decreased NO bioavailability, impairment of endothelium-dependent relaxation and generation of highly reactive ONOO-. Adventitial H2O2, and perhaps ONOO- are postulated to diffuse to the medial layer, where they may activate medial signaling cascades, leading to medial proliferation, growth and migration. In fact, under some circumstances low levels of ROS in one segment of the blood vessel may be protective, ie. involved in cell turnover and angiogenesis, which then may be surpassed by exaggerated production in others. Thus, more sophisticated approaches aimed at understanding the complex interplay of ROS signaling across the vasculature are necessary. Taken together, substantial evidence exists demonstrating the importance of the vascular adventitia and its ROS in vascular disease. The traditional “inside-out” and the newer “outside-in” paradigms necessarily overlap in promoting vascular disease and a greater appreciation of the latter's importance in this process will lead to significantly improved therapies targeting disease progression.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Padwal R, Straus SE, McAlister FA. Evidence based management of hypertension. Cardiovascular risk factors and their effects on the decision to treat hypertension: evidence based review. BMJ. 2001;322:977–980. doi: 10.1136/bmj.322.7292.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell SC, Moffatt RJ, Stamford BA. Smoking and smoking cessation-The relationship between cardiovascular disease and lipoprotein metabolism: A review. Atherosclerosis. 2008 doi: 10.1016/j.atherosclerosis.2008.04.046. [DOI] [PubMed] [Google Scholar]
- 3.Ishigaki Y, Katagiri H, Gao J, Yamada T, Imai J, Uno K, Hasegawa Y, Kaneko K, Ogihara T, Ishihara H, Sato Y, Takikawa K, Nishimichi N, Matsuda H, Sawamura T, Oka Y. Impact of plasma oxidized low-density lipoprotein removal on atherosclerosis. Circulation. 2008;118:75–83. doi: 10.1161/CIRCULATIONAHA.107.745174. [DOI] [PubMed] [Google Scholar]
- 4.Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. The aging endothelium, cardiovascular risk and disease. Exp Physiol. 2008 doi: 10.1113/expphysiol.2008.043356. [DOI] [PubMed] [Google Scholar]
- 5.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross R. Atherosclerosis -- an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 7.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovascular Research. 2007;75:640–648. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capers QI, Alexander RW, Lou P, de Leon H, Wilcox JN, Ishizaka N, Howard AB, Taylor WR. Monocyte chemoattractant protein-1 expression in aortic tissues of hypertensive rats. Hypertension. 1997;30:1397–1402. doi: 10.1161/01.hyp.30.6.1397. [DOI] [PubMed] [Google Scholar]
- 9.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proceedings of the National Academy of Sciences. 1997;94:14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush E, Maeda N, Kuziel WA, Dawson TC, Wilcox JN, DeLeon H, Taylor WR. CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II-induced hypertension. Hypertension. 2000;36:360–363. doi: 10.1161/01.hyp.36.3.360. [DOI] [PubMed] [Google Scholar]
- 11.Kuwahara F, Kai H, Tokuda K, Shibata R, Kusaba K, Tahara N, Niiyama H, Nagata T, Imaizumi T. Hypoxia-inducible factor-1alpha/vascular endothelial growth factor pathway for adventitial vasa vasorum formation in hypertensive rat aorta. Hypertension. 2002;39:46–50. doi: 10.1161/hy1201.097200. [DOI] [PubMed] [Google Scholar]
- 12.Scott NA, Cipolla GD, Ross CE, Dunn B, Martin FH, Simonet L, Wilcox JN. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation. 1996;93:2178–2187. doi: 10.1161/01.cir.93.12.2178. [DOI] [PubMed] [Google Scholar]
- 13.Arribas SM, Gonzalez C, Graham D, Dominiczak AF, McGrath JC. Cellular changes induced by chronic nitric oxide inhibition in intact rat basilar arteries revealed by confocal microscopy. J Hypertens. 1997;15:1685–1693. doi: 10.1097/00004872-199715120-00073. [DOI] [PubMed] [Google Scholar]
- 14.Kantachuvesiri S, Fleming S, Peters J, Peters B, Brooker G, Lammie AG, McGrath I, Kotelevtsev Y, Mullins JJ. Controlled hypertension, a transgenic toggle switch reveals differential mechanisms underlying vascular disease. J Biol Chem. 2001;276:36727–36733. doi: 10.1074/jbc.M103296200. [DOI] [PubMed] [Google Scholar]
- 15.Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA. An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol. 1995;268:H2274–H2280. doi: 10.1152/ajpheart.1995.268.6.H2274. [DOI] [PubMed] [Google Scholar]
- 16.Cifuentes ME, Rey FE, Carretero OA, Pagano PJ. Upregulation of p67 phox and gp91 phox in aortas from angiotensin II-infused mice. Am J Physiol Heart Circ Physiol. 2000;279:H2234–H2240. doi: 10.1152/ajpheart.2000.279.5.H2234. [DOI] [PubMed] [Google Scholar]
- 17.Rey FE, Pagano PJ. The reactive adventitia: fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol. 2002;22:1962–1971. doi: 10.1161/01.atv.0000043452.30772.18. [DOI] [PubMed] [Google Scholar]
- 18.Chamseddine AH, Miller FJ., Jr gp91 phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H2284–H2289. doi: 10.1152/ajpheart.00459.2003. [DOI] [PubMed] [Google Scholar]
- 19.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR, Quinn MT. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. J Leukoc Biol. 2007;82:729–741. doi: 10.1189/jlb.1206735. [DOI] [PubMed] [Google Scholar]
- 20.Cross AR, Segal AW. The NADPH oxidase of professional phagocytes--prototype of the NOX electron transport chain systems. Biochim Biophys Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol. 2004;76:760–781. doi: 10.1189/jlb.0404216. [DOI] [PubMed] [Google Scholar]
- 22.Geiszt M. NADPH oxidases: new kids on the block. Cardiovascular Research. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 24.Knaus UG, Morris S, Dong HJ, Chernoff J, Bokoch GM. Regulation of human leukocyte p21-activated kinases through G protein-coupled receptors. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- 25.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 26.Cross AR. p40 phox participates in the activation of NADPH oxidase by increasing the affinity of p47 phox for flavocytochrome b 558. Biochem J. 2000;349:113–117. doi: 10.1042/0264-6021:3490113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayraktutan U, Draper N, Lang D, Shah AM. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovascular Research. 1998;38:256–262. doi: 10.1016/s0008-6363(98)00003-0. [DOI] [PubMed] [Google Scholar]
- 28.Paravicini TM, Gulluyan LM, Dusting GJ, Drummond GR. Increased NADPH oxidase activity, gp91phox expression, and endothelium-dependent vasorelaxation during neointima formation in rabbits. Circ Res. 2002;91:54–61. doi: 10.1161/01.res.0000024106.81401.95. [DOI] [PubMed] [Google Scholar]
- 29.Lassègue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 30.McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, Harrison DG. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285:H2290–H2297. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- 31.Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114:1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- 32.Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Pratico D, Harrison DG, Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–54. doi: 10.1161/CIRCRESAHA.108.188771. [DOI] [PubMed] [Google Scholar]
- 33.Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, Imajoh-Ohmi S, Igarashi K, Shibata Y, Sueishi K, Sumimoto H. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells. 2005;10:1139–1151. doi: 10.1111/j.1365-2443.2005.00907.x. [DOI] [PubMed] [Google Scholar]
- 35.Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond) 2005;109:217–226. doi: 10.1042/CS20050067. [DOI] [PubMed] [Google Scholar]
- 36.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 37.Frey RS, Ushio-Fukai M, Malik A. NADPH Oxidase-Dependent Signaling in Endothelial Cells: Role in Physiology and Pathophysiology. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imanishi T, Kobayashi K, Kuroi A, Ikejima H, Akasaka T. Pioglitazone inhibits angiotensin II-induced senescence of endothelial progenitor cell. Hypertens Res. 2008;31:757–765. doi: 10.1291/hypres.31.757. [DOI] [PubMed] [Google Scholar]
- 39.Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res. 2000;87:26–32. doi: 10.1161/01.res.87.1.26. [DOI] [PubMed] [Google Scholar]
- 41.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 42.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity. Upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 43.Suh YA, Arnold RS, Lassègue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1 [letter] Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 44.Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 45.Van Heerebeek L, Meischl C, Stooker W, Meijer CJ, Niessen HW, Roos D. NADPH oxidase(s): new source(s) of reactive oxygen species in the vascular system? J Clin Pathol. 2002;55:561–568. doi: 10.1136/jcp.55.8.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ambasta RK, Kumar P, Griendling KK, Schmidt HHHW, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 47.Griendling KK. Novel NAD(P)H oxidases in the cardiovascular system. Heart. 2004;90:491–493. doi: 10.1136/hrt.2003.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;2006:re8. doi: 10.1126/stke.3492006re8. [DOI] [PubMed] [Google Scholar]
- 49.Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, Wakisaka M, Kawahara T, Rokutan K, Ibayashi S, Iida M. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 50.Honjo T, Otsui K, Shiraki R, Kawashima S, Sawamura T, Yokoyama M, Inoue N. Essential role of NOXA1 in generation of reactive oxygen species induced by oxidized low-density lipoprotein in human vascular endothelial cells. Endothelium. 2008;15:137–141. doi: 10.1080/10623320802125433. [DOI] [PubMed] [Google Scholar]
- 51.Geiszt M, Lekstrom K, Witta J, Leto TL. Proteins homologous to p47 phox and p67 phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J Biol Chem. 2003;278:20006–20012. doi: 10.1074/jbc.M301289200. [DOI] [PubMed] [Google Scholar]
- 52.Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJH, Verpy E, B nfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol. 2006;16:208–213. doi: 10.1016/j.cub.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 53.Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- 54.Bayraktutan U, Blayney L, Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91- phox and p22- phox in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:1903–1911. doi: 10.1161/01.atv.20.8.1903. [DOI] [PubMed] [Google Scholar]
- 55.Hohler B, Holzapfel B, Kummer W. NADPH oxidase subunits and superoxide production in porcine pulmonary artery endothelial cells. Histochem Cell Biol. 2000;114:29–37. doi: 10.1007/s004180000160. [DOI] [PubMed] [Google Scholar]
- 56.Jones SA, O'Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OTG. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271:H1626–H1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 57.Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K + Circ Res. 1998;83:730–737. doi: 10.1161/01.res.83.7.730. [DOI] [PubMed] [Google Scholar]
- 58.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. NOX5 variants are functionally active in endothelial cells. Free Radic Biol Med. 2007;42:446–459. doi: 10.1016/j.freeradbiomed.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 59.Sorescu D, Weiss D, Lassègue B, Clempus RE, Szöcs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 60.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 61.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. 1996;271:23317–23321. doi: 10.1074/jbc.271.38.23317. [DOI] [PubMed] [Google Scholar]
- 62.Azumi H, Inoue N, Takeshita S, Rikitake Y, Kawashima S, Hayashi Y, Itoh H, Yokoyama M. Expression of NADH/NADPH oxidase p22 phox in human coronary arteries. Circulation. 1999;100:1494–1498. doi: 10.1161/01.cir.100.14.1494. [DOI] [PubMed] [Google Scholar]
- 63.Liu S, Ma X, Gong M, Shi L, Lincoln T, Wang S. Glucose down-regulation of cGMP-dependent protein kinase I expression in vascular smooth muscle cells involves NAD(P)H oxidase-derived reactive oxygen species. Free Radic Biol Med. 2007;42:852–863. doi: 10.1016/j.freeradbiomed.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 64.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47 phox may participate in forming this oxidase in vitro and in vivo. J Biol Chem. 1999;274:19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 65.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries. Regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 66.Muzaffar S, Shukla N, Angelini G, Jeremy JY. Nitroaspirins and morpholinosydnonimine but not aspirin inhibit the formation of superoxide and the expression of gp91phox induced by endotoxin and cytokines in pig pulmonary artery vascular smooth muscle cells and endothelial cells. Circulation. 2004;110:1140–1147. doi: 10.1161/01.CIR.0000139851.50067.E4. [DOI] [PubMed] [Google Scholar]
- 67.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91 phox homologues in vascular smooth muscle cells. Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 68.Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. 2008;45:1223–1231. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Djordjevic T, BelAiba RS, Bonello S, Pfeilschifter J, Hess J, Gorlach A. Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler Thromb Vasc Biol. 2005;25:519–525. doi: 10.1161/01.ATV.0000154279.98244.eb. [DOI] [PubMed] [Google Scholar]
- 70.Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassègue B, Griendling KK. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. 2008;45:329–335. doi: 10.1016/j.freeradbiomed.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ambasta RK, Schreiber JG, Janiszewski M, Busse R, Brandes RP. Noxa1 is a central component of the smooth muscle NADPH oxidase in mice. Free Radic Biol Med. 2006;41:193–201. doi: 10.1016/j.freeradbiomed.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 72.Pagano PJ, Chanock SJ, Siwik DA, Colucci WS, Clark JK. Angiotensin II induces p67 phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension. 1998;32:331–337. doi: 10.1161/01.hyp.32.2.331. [DOI] [PubMed] [Google Scholar]
- 73.Wang HD, Pagano PJ, Du Y, Cayatte AJ, Quinn MT, Brecher P, Cohen RA. Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ Res. 1998;82:810–818. doi: 10.1161/01.res.82.7.810. [DOI] [PubMed] [Google Scholar]
- 74.Yokoyama M, Inoue N, Kawashima S. Role of the vascular NADH/NADPH oxidase system in atherosclerosis. Ann N Y Acad Sci. 2000;902:241–247. 247–248. doi: 10.1111/j.1749-6632.2000.tb06319.x. discussion. [DOI] [PubMed] [Google Scholar]
- 75.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 76.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, Habermehl GG. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem J. 1989;263:539–545. doi: 10.1042/bj2630539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thannickal VJ, Fanburg BL. Activation of an H 2 O 2 -generating NADH oxidase in human lung fibroblasts by transforming growth factor á1. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 78.Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, Wilcox JN, Quinn MT, Lambeth JD, Griendling KK. Upregulation of nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. 2002;22:21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 79.Ceolotto G, Bevilacqua M, Papparella I, Baritono E, Franco L, Corvaja C, Mazzoni M, Semplicini A, Avogaro A. Insulin generates free radicals by an NAD(P)H, phosphatidylinositol 3′-kinase-dependent mechanism in human skin fibroblasts ex vivo. Diabetes. 2004;53:1344–1351. doi: 10.2337/diabetes.53.5.1344. [DOI] [PubMed] [Google Scholar]
- 80.Yamagishi S, Nakamura K, Ueda S, Kato S, Imaizumi T. Pigment epithelium-derived factor (PEDF) blocks angiotensin II signaling in endothelial cells via suppression of NADPH oxidase: a novel anti-oxidative mechanism of PEDF. Cell Tissue Res. 2005;320:437–445. doi: 10.1007/s00441-005-1094-8. [DOI] [PubMed] [Google Scholar]
- 81.Wang HD, Hope S, Du Y, Quinn MT, Cayatte A, Pagano PJ, Cohen RA. Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension. 1999;33:1225–1232. doi: 10.1161/01.hyp.33.5.1225. [DOI] [PubMed] [Google Scholar]
- 82.Zhang L, Zalewski A, Liu Y, Mazurek T, Cowan S, Martin JL, Hofmann SM, Vlassara H, Shi Y. Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation. 2003;108:472–478. doi: 10.1161/01.CIR.0000080378.96063.23. [DOI] [PubMed] [Google Scholar]
- 83.Chan EC, Datla SR, Dilley R, Hickey H, Drummond GR, Dusting GJ. Adventitial application of the NADPH oxidase inhibitor apocynin in vivo reduces neointima formation and endothelial dysfunction in rabbits. Cardiovasc Res. 2007;75:710–718. doi: 10.1016/j.cardiores.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 84.Shi Y, Niculescu R, Wang D, Patel S, Davenpeck KL, Zalewski A. Increased NAD(P)H oxidase and reactive oxygen species in coronary arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2001;21:739–745. doi: 10.1161/01.atv.21.5.739. [DOI] [PubMed] [Google Scholar]
- 85.Li S, Tabar SS, Malec V, Eul BG, Klepetko W, Weissmann N, Grimminger F, Seeger W, Rose F, Hanze J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid Redox Signal. 2008;10:1687–1698. doi: 10.1089/ars.2008.2035. [DOI] [PubMed] [Google Scholar]
- 86.Schwartz CJ, Mitchell JRA. Cellular infiltration of the human arterial adventitia associated with atheromatous plaques. Circulation. 1962;26:73–78. doi: 10.1161/01.cir.26.1.73. [DOI] [PubMed] [Google Scholar]
- 87.Liu J, Ormsby A, Oja-Tebbe N, Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res. 2004;95:587–594. doi: 10.1161/01.RES.0000142317.88591.e6. [DOI] [PubMed] [Google Scholar]
- 88.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ETH, Runge MS. p47 phox is required for atherosclerotic lesion progression in ApoE -/- mice. J Clin Invest. 2001;108:1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kirk EA, Dinauer MC, Rosen H, Chait A, Heinecke JW, LeBoeuf RC. Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2000;20:1529–1535. doi: 10.1161/01.atv.20.6.1529. [DOI] [PubMed] [Google Scholar]
- 90.Herrmann J, Lerman LO, Rodriguez-Porcel M, Holmes DR, Jr, Richardson DM, Ritman EL, Lerman A. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc Res. 2001;51:762–766. doi: 10.1016/s0008-6363(01)00347-9. [DOI] [PubMed] [Google Scholar]
- 91.Ritman EL, Lerman A. The dynamic vasa vasorum. Cardiovascular Research. 2007;75:649–658. doi: 10.1016/j.cardiores.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O'Brien KD, McDonald TO, Chait A, Allen MD, Alpers CE. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation. 1996;93:672–682. doi: 10.1161/01.cir.93.4.672. [DOI] [PubMed] [Google Scholar]
- 93.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 94.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 95.Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72:447–455. doi: 10.1016/j.cardiores.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 96.Sarada S, Himadri P, Mishra C, Geetali P, Ram MS, Ilavazhagan G. Role of oxidative stress and NFkB in hypoxia-induced pulmonary edema. Exp Biol Med (Maywood) 2008;233:1088–1098. doi: 10.3181/0712-RM-337. [DOI] [PubMed] [Google Scholar]
- 97.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25:2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006;21:134–145. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 100.Shivakumar K, Sollott SJ, Sangeetha M, Sapna S, Ziman B, Wang S, Lakatta EG. Paracrine effects of hypoxic fibroblast-derived factors on the MPT-ROS threshold and viability of adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2653–2658. doi: 10.1152/ajpheart.91443.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Browatzki M, Schmidt J, Kubler W, Kranzhofer R. Endothelin-1 induces interleukin-6 release via activation of the transcription factor NF-kappaB in human vascular smooth muscle cells. Basic Res Cardiol. 2000;95:98–105. doi: 10.1007/s003950050170. [DOI] [PubMed] [Google Scholar]
- 102.Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23:776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- 103.Okamoto E, Couse T, de Leon H, Vinten-Johansen J, Goodman RB, Scott NA, Wilcox JN. Perivascular inflammation after balloon angioplasty of porcine coronary arteries. Circulation. 2001;104:2228–2235. doi: 10.1161/hc4301.097195. [DOI] [PubMed] [Google Scholar]
- 104.Wung BS, Cheng JJ, Hsieh HJ, Shyy YJ, Wang DL. Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ Res. 1997;81:1–7. doi: 10.1161/01.res.81.1.1. [DOI] [PubMed] [Google Scholar]
- 105.Xu F, Ji J, Li L, Chen R, Hu WC. Adventitial fibroblasts are activated in the early stages of atherosclerosis in the apolipoprotein E knockout mouse. Biochem Biophys Res Commun. 2007;352:681–688. doi: 10.1016/j.bbrc.2006.11.073. [DOI] [PubMed] [Google Scholar]
- 106.Chen XL, Zhang Q, Zhao R, Medford RM. Superoxide, H2O2, and iron are required for TNF-alpha-induced MCP-1 gene expression in endothelial cells: role of Rac1 and NADPH oxidase. Am J Physiol Heart Circ Physiol. 2004;286:H1001–1007. doi: 10.1152/ajpheart.00716.2003. [DOI] [PubMed] [Google Scholar]
- 107.Jabs A, Okamoto E, Vinten-Johansen J, Bauriedel G, Wilcox JN. Sequential patterns of chemokine- and chemokine receptor-synthesis following vessel wall injury in porcine coronary arteries. Atherosclerosis. 2007;192:75–84. doi: 10.1016/j.atherosclerosis.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 108.Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, Cohen RA. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res. 2001;88:947–953. doi: 10.1161/hh0901.089987. [DOI] [PubMed] [Google Scholar]
- 109.Dourron HM, Jacobson GM, Park JL, Liu J, Reddy DJ, Scheel ML, Pagano PJ. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am J Physiol Heart Circ Physiol. 2005;288:H946–H953. doi: 10.1152/ajpheart.00413.2004. [DOI] [PubMed] [Google Scholar]
- 110.Baas AS, Berk BC. Differential activation of mitogen-activated protein kinases by H2O2 and O2- in vascular smooth muscle cells. Circ Res. 1995;77:29–36. doi: 10.1161/01.res.77.1.29. [DOI] [PubMed] [Google Scholar]
- 111.Saran M, Bors W. Signalling by O2-. and NO.: how far can either radical, or any specific reaction product, transmit a message under in vivo conditions? Chem Biol Interact. 1994;90:35–45. doi: 10.1016/0009-2797(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 112.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension. 2005;46:732–737. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]
- 114.Strålin P, Karlsson K, Johansson BO, Marklund SL. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler Thromb Vasc Biol. 1995;15:2032–2036. doi: 10.1161/01.atv.15.11.2032. [DOI] [PubMed] [Google Scholar]
- 115.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability. In vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. 2003;93:622–629. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 116.Fukai T, Siegfried MR, Ushio-Fukai M, Griendling KK, Harrison DG. Modulation of extracellular superoxide dismutase expression by angiotensin II and hypertension. Circ Res. 1999;85:23–28. doi: 10.1161/01.res.85.1.23. [DOI] [PubMed] [Google Scholar]
- 117.Strålin P, Marklund SL. Multiple cytokines regulate the expression of extracellular superoxide dismutase in human vascular smooth muscle cells. Atherosclerosis. 2000;151:433–441. doi: 10.1016/s0021-9150(99)00427-x. [DOI] [PubMed] [Google Scholar]
- 118.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 119.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 120.Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 121.Waldman SA, Murad F. Biochemical mechanisms underlying vascular smooth muscle relaxation: the guanylate cyclase-cyclic GMP system. J Cardiovasc Pharmacol. 1988;12 5:S115–118. [PubMed] [Google Scholar]
- 122.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, Uchida K. A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. J Biol Chem. 2004;279:48389–48396. doi: 10.1074/jbc.M409935200. [DOI] [PubMed] [Google Scholar]
- 124.Murrell GAC, Francis MJO, Bromley L. Modulation of fibroblast proliferation by oxygen free radicals. Biochem J. 1990;265:659–665. doi: 10.1042/bj2650659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest. 1992;89:507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Strålin P, Marklund SL. Vasoactive factors and growth factors alter vascular smooth muscle cell EC-SOD expression. Am J Physiol Heart Circ Physiol. 2001;281:H1621–H1629. doi: 10.1152/ajpheart.2001.281.4.H1621. [DOI] [PubMed] [Google Scholar]
- 127.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 128.Li G, Chen SJ, Oparil S, Chen YF, Thompson JA. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101:1362–1365. doi: 10.1161/01.cir.101.12.1362. [DOI] [PubMed] [Google Scholar]
- 129.Zargham R. Preventing restenosis after angioplasty: a multistage approach. Clin Sci (Lond) 2008;114:257–264. doi: 10.1042/CS20070228. [DOI] [PubMed] [Google Scholar]
- 130.Souza HP, Souza LC, Anastacio VM, Pereira AC, Junqueira MdL, Krieger JE, da Luz PL, Augusto O, Laurindo FRM. Vascular oxidant stress early after balloon injury: evidence for increased NAD(P)H oxidoreductase activity. Free Radic Biol Med. 2000;28:1232–1242. doi: 10.1016/s0891-5849(00)00240-9. [DOI] [PubMed] [Google Scholar]
- 131.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 132.Kanellakis P, Nestel P, Bobik A. Angioplasty-induced superoxide anions and neointimal hyperplasia in the rabbit carotid artery: suppression by the isoflavone trans-tetrahydrodaidzein. Atherosclerosis. 2004;176:63–72. doi: 10.1016/j.atherosclerosis.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 133.Shi Y, Pieniek M, Fard A, O'Brien J, Mannion JD, Zalewski A. Adventitial remodeling after coronary arterial injury. Circulation. 1996;93:340–348. doi: 10.1161/01.cir.93.2.340. [DOI] [PubMed] [Google Scholar]
- 134.Shi Y, O'Brien JE, Jr, Fard A, Zalewski A. Transforming growth factor-á1 expression and myofibroblast formation during arterial repair. Arterioscler Thromb Vasc Biol. 1996;16:1298–1305. doi: 10.1161/01.atv.16.10.1298. [DOI] [PubMed] [Google Scholar]
- 135.Rao GN, Berk BC. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ Res. 1992;70:593–599. doi: 10.1161/01.res.70.3.593. [DOI] [PubMed] [Google Scholar]
- 136.Weaver M, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol. 2006;290:H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 137.Clowes AW, Schwartz SM. Significance of quiescent smooth muscle migration in the injured rat carotid artery. Circ Res. 1985;56:139–145. doi: 10.1161/01.res.56.1.139. [DOI] [PubMed] [Google Scholar]
- 138.Rao GN. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates Ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene. 1996;13:713–719. [PubMed] [Google Scholar]
- 139.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 140.Abe J, Takahashi M, Ishida M, Lee JD, Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J Biol Chem. 1997;272:20389–20394. doi: 10.1074/jbc.272.33.20389. [DOI] [PubMed] [Google Scholar]
- 141.Lee HM, Lee CK, Lee SH, Roh HY, Bae YM, Lee KY, Lim J, Park PJ, Park TK, Lee YL, Won KJ, Kim B. p38 mitogen-activated protein kinase contributes to angiotensin II-stimulated migration of rat aortic smooth muscle cells. J Pharmacol Sci. 2007;105:74–81. doi: 10.1254/jphs.fp0070770. [DOI] [PubMed] [Google Scholar]
- 142.Richard MN, Deniset JF, Kneesh AL, Blackwood D, Pierce GN. Mechanical stretching stimulates smooth muscle cell growth, nuclear protein import, and nuclear pore expression through mitogen-activated protein kinase activation. J Biol Chem. 2007;282:23081–23088. doi: 10.1074/jbc.M703602200. [DOI] [PubMed] [Google Scholar]
- 143.Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- 144.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 145.Yoshizumi M, Abe J, Haendeler J, Huang Q, Berk BC. Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. J Biol Chem. 2000;275:11706–11712. doi: 10.1074/jbc.275.16.11706. [DOI] [PubMed] [Google Scholar]
- 146.Zhang J, Jin N, Liu Y, Rhoades RA. Hydrogen peroxide stimulates extracellular signal-regulated protein kinases in pulmonary arterial smooth muscle cells. Am J Respir Cell Mol Biol. 1998;19:324–332. doi: 10.1165/ajrcmb.19.2.3209. [DOI] [PubMed] [Google Scholar]
- 147.Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kübler W, Kreuzer J. Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II. Involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol. 2000;20:940–948. doi: 10.1161/01.atv.20.4.940. [DOI] [PubMed] [Google Scholar]
- 148.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 149.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 150.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 152.Kappert K, Sparwel J, Sandin A, Seiler A, Siebolts U, Leppanen O, Rosenkranz S, Ostman A. Antioxidants relieve phosphatase inhibition and reduce PDGF signaling in cultured VSMCs and in restenosis. Arterioscler Thromb Vasc Biol. 2006;26:2644–2651. doi: 10.1161/01.ATV.0000246777.30819.85. [DOI] [PubMed] [Google Scholar]
- 153.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Laine P, Kaartinen M, Penttil A, Panula P, Paavonen T, Kovanen PT. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation. 1999;99:361–369. doi: 10.1161/01.cir.99.3.361. [DOI] [PubMed] [Google Scholar]
- 155.Zhao L, Moos MP, Grabner R, Pedrono F, Fan J, Kaiser B, John N, Schmidt S, Spanbroek R, Lotzer K, Huang L, Cui J, Rader DJ, Evans JF, Habenicht AJ, Funk CD. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat Med. 2004;10:966–973. doi: 10.1038/nm1099. [DOI] [PubMed] [Google Scholar]
- 156.Tsuruda T, Kato J, Hatakeyama K, Kojima K, Yano M, Yano Y, Nakamura K, Nakamura-Uchiyama F, Matsushima Y, Imamura T, Onitsuka T, Asada Y, Nawa Y, Eto T, Kitamura K. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ Res. 2008;102:1368–1377. doi: 10.1161/CIRCRESAHA.108.173682. [DOI] [PubMed] [Google Scholar]
- 157.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shakoory B, Fitzgerald SM, Lee SA, Chi DS, Krishnaswamy G. The role of human mast cell-derived cytokines in eosinophil biology. J Interferon Cytokine Res. 2004;24:271–281. doi: 10.1089/107999004323065057. [DOI] [PubMed] [Google Scholar]
- 159.Gulliksson M, Palmberg L, Nilsson G, Ahlstedt S, Kumlin M. Release of prostaglandin D2 and leukotriene C4 in response to hyperosmolar stimulation of mast cells. Allergy. 2006;61:1473–1479. doi: 10.1111/j.1398-9995.2006.01213.x. [DOI] [PubMed] [Google Scholar]
- 160.Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, Schwartz LB, Korn JH. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol. 1997;158:2310–2317. [PubMed] [Google Scholar]
- 161.Wang Y, Thorlacius H. Mast cell-derived tumour necrosis factor-alpha mediates macrophage inflammatory protein-2-induced recruitment of neutrophils in mice. Br J Pharmacol. 2005;145:1062–1068. doi: 10.1038/sj.bjp.0706274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ihara M, Urata H, Kinoshita A, Suzumiya J, Sasaguri M, Kikuchi M, Ideishi M, Arakawa K. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension. 1999;33:1399–1405. doi: 10.1161/01.hyp.33.6.1399. [DOI] [PubMed] [Google Scholar]
- 163.Fukuishi N, Sakaguchi M, Matsuura S, Nakagawa C, Akagi R, Akagi M. The mechanisms of compound 48/80-induced superoxide generation mediated by A-kinase in rat peritoneal mast cells. Biochem Mol Med. 1997;61:107–113. doi: 10.1006/bmme.1997.2594. [DOI] [PubMed] [Google Scholar]
- 164.Suzuki Y, Yoshimaru T, Matsui T, Inoue T, Niide O, Nunomura S, Ra C. Fc epsilon RI signaling of mast cells activates intracellular production of hydrogen peroxide: role in the regulation of calcium signals. J Immunol. 2003;171:6119–6127. doi: 10.4049/jimmunol.171.11.6119. [DOI] [PubMed] [Google Scholar]
- 165.Moos MP, John N, Grabner R, Nossmann S, Gunther B, Vollandt R, Funk CD, Kaiser B, Habenicht AJ. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2386–2391. doi: 10.1161/01.ATV.0000187470.31662.fe. [DOI] [PubMed] [Google Scholar]
- 166.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5:818–827. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 167.Walford G, Loscalzo J. Nitric oxide in vascular biology. J Thromb Haemost. 2003;1:2112–2118. doi: 10.1046/j.1538-7836.2003.00345.x. [DOI] [PubMed] [Google Scholar]
- 168.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovasc Res. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- 170.Mayer B, Schrammel A, Klatt P, Koesling D, Schmidt K. Peroxynitrite-induced accumulation of cyclic GMP in endothelial cells and stimulation of purified soluble guanylyl cyclase. Dependence on glutathione and possible role of S-nitrosation. J Biol Chem. 1995;270:17355–17360. doi: 10.1074/jbc.270.29.17355. [DOI] [PubMed] [Google Scholar]
- 171.Edelman ER, Adams DH, Karnovsky MJ. Effect of controlled adventitial heparin delivery on smooth muscle cell proliferation following endothelial injury. Proc Natl Acad Sci U S A. 1990;87:3773–3777. doi: 10.1073/pnas.87.10.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wilcox JN, Waksman R, King SB, Scott NA. The role of the adventitia in the arterial response to angioplasty: the effect of intravascular radiation. Int J Radiat Oncol Biol Phys. 1996;36:789–796. doi: 10.1016/s0360-3016(96)00299-4. [DOI] [PubMed] [Google Scholar]
- 173.Ooboshi H, Toyoda K, Faraci FM, Lang MG, Heistad DD. Improvement of relaxation in an atherosclerotic artery by gene transfer of endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol. 1998;18:1752–1758. doi: 10.1161/01.atv.18.11.1752. [DOI] [PubMed] [Google Scholar]
- 174.Kullo IJ, Mozes G, Schwartz RS, Gloviczki P, Crotty TB, Katusic ZS, O'Brien T. Adventitial gene transfer of recombinant endothelial nitric oxide synthase to rabbit carotid arteries alters vascular reactivity [abstract] J Am Coll Cardiol. 1997;29(Suppl A):228A–229A. doi: 10.1161/01.cir.96.7.2254. [DOI] [PubMed] [Google Scholar]
- 175.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 176.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 177.Sato K, Akaike T, Kohno M, Ando M, Maeda H. Hydroxyl radical production by H2O2 plus Cu,Zn-superoxide dismutase reflects the activity of free copper released from the oxidatively damaged enzyme. J Biol Chem. 1992;267:25371–25377. [PubMed] [Google Scholar]
- 178.Fukui T, Lassègue B, Kai H, Alexander RW, Griendling KK. Cytochrome b -558 α-subunit cloning and expression in rat aortic smooth muscle cells. Biochim Biophys Acta. 1995;1231:215–219. doi: 10.1016/0005-2728(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 179.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 180.Kovacic HN, Irani K, Goldschmidt-Clermont PJ. Redox regulation of human Rac1 stability by the proteasome in human aortic endothelial cells. J Biol Chem. 2001;276:45856–45861. doi: 10.1074/jbc.M107925200. [DOI] [PubMed] [Google Scholar]
- 181.Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, Boyd NL, Platt MO, Lassègue B, Griendling KK, Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a Nox1-based NADPH oxidase. Circ Res. 2004;95:773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- 182.Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med. 2001;31:1456–1464. doi: 10.1016/s0891-5849(01)00727-4. [DOI] [PubMed] [Google Scholar]
- 183.Kobayashi S, Nojima Y, Shibuya M, Maru Y. Nox1 regulates apoptosis and potentially stimulates branching morphogenesis in sinusoidal endothelial cells. Exp Cell Res. 2004;300:455–462. doi: 10.1016/j.yexcr.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 184.Katsuyama M, Fan C, Yabe-Nishimura C. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: induction of NOX1 by PGF2alpha. J Biol Chem. 2002;277:13438–13442. doi: 10.1074/jbc.M111634200. [DOI] [PubMed] [Google Scholar]
- 185.Kawai J, Ando K, Tojo A, Shimosawa T, Takahashi K, Onozato ML, Yamasaki M, Ogita T, Nakaoka T, Fujita T. Endogenous adrenomedullin protects against vascular response to injury in mice. Circulation. 2004;109:1147–1153. doi: 10.1161/01.CIR.0000117231.40057.6D. [DOI] [PubMed] [Google Scholar]
- 186.Rey FE, Li XC, Carretero OA, Garvin JL, Pagano PJ. Perivascular superoxide anion contributes to impairment of endothelium-dependent relaxation: role of gp91(phox) Circulation. 2002;106:2497–2502. doi: 10.1161/01.cir.0000038108.71560.70. [DOI] [PubMed] [Google Scholar]
- 187.Hwang J, Ing MH, Salazar A, Lassègue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression. Implication for native LDL oxidation. Circ Res. 2003;93:1225–1232. doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, O'Dowd Y, Bens M, Vandewalle A, Gougerot-Pocidalo MA, Lizard G, Ogier-Denis E. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Xu H, Goettsch C, Xia N, Horke S, Morawietz H, Forstermann U, Li H. Differential roles of PKCalpha and PKCepsilon in controlling the gene expression of Nox4 in human endothelial cells. Free Radic Biol Med. 2008;44:1656–1667. doi: 10.1016/j.freeradbiomed.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 190.Rossary A, Arab K, Steghens JP. Polyunsaturated fatty acids modulate NOX 4 anion superoxide production in human fibroblasts. Biochem J. 2007;406:77–83. doi: 10.1042/BJ20061009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Colston JT, de la Rosa SD, Strader JR, Anderson MA, Freeman GL. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett. 2005;579:2533–2540. doi: 10.1016/j.febslet.2005.03.057. [DOI] [PubMed] [Google Scholar]