

Abstract
Organization of G protein-coupled receptors and cognate signaling partners at the plasma membrane has been proposed to occur via multiple mechanisms, including membrane microdomains, receptor oligomerization, and protein scaffolding. Here, we investigate the organization of six types of Gi/o-coupled receptors endogenously expressed in SH-SY5Y cells. The most abundant receptor in these cells was the μ-opioid receptor (MOR), the activation of which occluded acute inhibition of adenylyl cyclase (AC) by agonists to δ-opioid (DOR), nociceptin/orphanin FQ peptide (NOPr), α2-adrenergic (α2AR), cannabinoid 1, and serotonin 1A receptors. We further demonstrate that all receptor pairs share a common pool of AC. The MOR agonist [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) also occluded the ability of DOR agonist to stimulate G proteins. However, at lower agonist concentrations and at shorter incubation times when G proteins were not limiting, the relationship between MOR and DOR agonists was additive. The additive relationship was confirmed by isobolographic analysis. Long-term coadministration of MOR and DOR agonists caused cAMP overshoot that was not additive, suggesting that sensitization of AC mediated by these two receptors occurs by a common pathway. Furthermore, heterologous inhibition of AC by agonists to DOR, NOPr, and α2AR reduced the expression of cAMP overshoot in DAMGO-dependent cells. However, this cross-talk did not lead to heterologous tolerance. These results indicate that multiple receptors could be tethered into complexes with cognate signaling proteins and that access to shared AC by multiple receptor types may provide a means to prevent opioid withdrawal.
Introduction
Opioid receptors are members of the G protein-coupled receptor (GPCR) family and signal via activation of adenylyl cyclase (AC)-inhibitory (Gi/o) GTP-binding proteins. It has been suggested that the probability of opioid receptor/G protein interaction is enhanced by compartmentalization in the membrane (Alt et al., 2001), allowing rapidity of GPCR signal propagation (Hur and Kim, 2002). Various modes of organization in the plasma membrane have been proposed to describe these compartments, including dimerization of receptors (George et al., 2000; Jordan et al., 2003; Gomes et al., 2004; Rios et al., 2004, 2006; Wang et al., 2005), membrane microdomains (Allen et al., 2007), or protein scaffolds (Hall and Lefkowitz, 2002). However, mathematical modeling of experimental findings supporting compartmentalization has claimed that these data can be explained by a collision coupling model (Tolkovsky and Levitzki, 1978; Stickle and Barber, 1992) without the need to invoke compartments (Brinkerhoff et al., 2008).
Compartments also prevent interactions between two proteins by constraining cross-talk and/or sharing of effector molecules, thus leading to signaling specificity. In NG108-15 cells, muscarinic receptors and δ-opioid receptors (DOR) did not share G proteins with α2-adrenergic receptors (α2AR), as measured by agonist binding (Graeser and Neubig, 1993). In this scenario, coadministration of agonists for separately compartmentalized receptors would result in an additive response as each receptor type activated its own pool of effectors. Thus, in N18TG2 neuroblastoma cells, agonists to endogenous DOR and cannabinoid (CB1) receptors activated G proteins in an additive manner (Shapira et al., 2000). On the other hand, in SH-SY5Y cells, coadministration of a μ-opioid receptor (MOR) agonist and a DOR agonist produced the same level of G protein activation as the MOR agonist alone, indicating that MOR and DOR activate the same G proteins (Alt et al., 2002). Likewise, DOR and CB1 receptors cotransfected in COS-7 cells shared G proteins (Shapira et al., 2000), and MOR and α2AR endogenously expressed in SH-SY5Y cells were observed to access the same AC enzymes (Lameh et al., 1992).
The conflicting data on DOR and CB1 receptor competition in N18TG2 and COS-7 cells can potentially be explained by differences in the level of expression of receptors. At high density, receptors compete for a limiting pool of G proteins, whereas at low receptor concentrations, G proteins are in excess, and agonists for two receptor types activate G proteins in an additive manner regardless of compartmentalization (Brinkerhoff et al., 2008). However, at low receptor levels, artificially reducing G protein number [using pertussis toxin (PTX)] did not increase competition (Graeser and Neubig, 1993; Shapira et al., 2000), suggesting that receptor number is more predictive of competition than G protein number (Brinkerhoff et al., 2008).
Competition between only two GPCR types would be observed if the receptors were able to freely diffuse along the cell membrane to access all available G proteins or if they were corralled together (i.e., in a membrane microdomain, by scaffolding proteins, or by dimerization). By considering competition between multiple receptor types, the chance of all receptors sharing the same compartment decreases, and it should therefore be easier to differentiate between receptors that are somehow constrained together and those that are not. The goal of the experiments presented here was to determine the degree of competition or effector sharing between multiple inhibitory GPCRs endogenously expressed in SH-SY5Y cells and the consequences of this competition for signaling to AC. We show that agonist-occupied MOR can access all ACs available to these other Gi/o-coupled GPCRs, suggesting a lack of compartmentalization and/or the presence of complexes containing multiple receptors. Moreover, depending on the level of receptor expression, agonists at non-MOR GPCRs are able to attenuate the cAMP overshoot observed after withdrawal from exposure to a long-term MOR agonist, thus suggesting a mechanism for the prevention of opioid withdrawal.
Materials and Methods
Materials.
[3H]5-(1,1-Dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-phenol (CP 55,940), [3H][d-Ala2, N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO), [3H][d-Pen2,5]-enkephalin (DPDPE), [3H]diprenorphine, [3H]nociceptin/OFQ, [3H]5-bromo-6-(2-imidazolin-2-ylamino)quinoxaline (UK14,304), [3H]yohimbine, and guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) were obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA). (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC80) was obtained from the Narcotic Drug and Opioid Peptide Basic Research Center at the University of Michigan (Ann Arbor, MI). DAMGO, DPDPE, naloxone, d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP), nociceptin/orphanin FQ (nociceptin/OFQ), UK14,304, clonidine, forskolin, and 3-isobutyl-1-methylxanthine (IBMX) were from Sigma-Aldrich (St. Louis, MO). CP 55,940 and WIN 55212-2 were from Cayman Chemical (Ann Arbor, MI). N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH (ICI 174,864) and 1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (J113397) were from Tocris Bioscience (Ellisville, MO). Retinoic acid was obtained from Calbiochem (La Jolla, CA). PTX was from List Biological Laboratories (Campbell, CA). Tissue culture media, fetal bovine serum, and trypsin were from Invitrogen (Carlsbad, CA). All other chemicals were obtained from Sigma-Aldrich and were of analytical grade.
Cell Culture.
Human neuroblastoma SH-SY5Y cells, a subclone of SK-N-SH cells, were obtained from the American Type Culture Collection (Manassas, VA), grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C in 5% CO2, and used within passages 34 to 44 from subcloning to maintain consistent neuroblast properties between experiments. All experiments were performed in SH-SY5Y cells differentiated with 10 μM retinoic acid for 4 to 7 days before assay.
Radioligand Binding Assays.
Membranes were prepared from retinoic acid-differentiated SH-SY5Y cells. Cells were rinsed with phosphate-buffered saline (150 mM NaCl, 0.61 mM Na2HPO4, and 0.38 mM KH2PO4, pH 7.4), resuspended in warm harvesting buffer (150 mM NaCl, 20 mM HEPES, and 0.68 mM EDTA, pH 7.4), and centrifuged at 500g in an IEC Centra CL2 centrifuge (Thermo Fisher Scientific, Waltham, MA) with a swinging bucket rotor. Cells were resuspended in ice-cold 50 mM Tris-HCl buffer, pH 7.4, homogenized with a Tissue Tearor (Biospec, Inc; Bartlesville, OK) for 20 s at setting 4, and centrifuged at 27,000g in a Beckman Coulter (Fullerton, CA) centrifuge. The crude membrane pellet was then resuspended in Tris buffer, homogenized for 10 s at setting 2, and centrifuged as above. Final membrane pellets were resuspended in 50 mM Tris-HCl buffer, pH 7.4, separated into aliquots, and stored at −80°C. Protein concentration was measured using the Bradford assay.
Receptor density was determined by incubating membranes (50 μg) for 60 min at 25°C with shaking in 50 mM Tris-HCl, pH 7.4, buffer containing saturating concentrations of radiolabeled ligand as follows: 12 nM [3H]DAMGO or 4 nM [3H]diprenorphine in the presence of 1 μM ICI 174,864 for MOR, 16 nM [3H]DPDPE or 1 nM [3H]naltrindole for DOR, 1 nM [3H]nociceptin/OFQ for nociceptin/orphanin FQ peptide receptor (NOPr), 15 nM [3H]UK14,304 or 10 nM [3H]yohimbine for α2AR or 6 nM [3H]CP 55,940 for CB1. Nonspecific binding was determined with unlabeled naloxone (MOR and DOR), J113397 (NOPr), UK14,304 (α2AR), or WIN 55212-2 (CB1). All plasticware was precoated with Sigma Cote (Sigma-Aldrich), and 0.1% bovine serum albumin was included for [3H]CP 55,940 binding. Assays were stopped by rapid filtration through GF/C filters presoaked in 0.1% polyethylenimine using a harvester (Brandel Inc., Gaithersburg, MD) and rinsed three times with ice-cold 50 mM Tris-HCl wash buffer, pH 7.4. Dried filters were saturated with EcoLume liquid scintillation cocktail (MP Biomedicals, Solon, OH), and radioactivity was counted in a Wallac 1450 MicroBeta (PerkinElmer Life and Analytical Sciences).
Stimulation of [35S]GTPγS Binding.
Membranes were prepared from retinoic acid-differentiated SH-SY5Y cells as described under Radioligand Binding Assays. In some experiments, cells were treated overnight with agonist (SNC80 or DAMGO) or for 24 h with PTX (100 ng/ml) before membrane preparation.
Membranes (50 μg of protein) were incubated with 0.1 nM [35S]GTPγS for 60 min (unless otherwise indicated) at 25°C with or without various concentrations of SNC80 and/or DAMGO in [35S]GTPγS binding buffer (50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol, and 30 μM GDP). Membranes with bound [35S]GTPγS were collected on GF/C filters (Whatman, Maidstone, UK) using a Brandel harvester and rinsed three times with cold wash buffer (50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, and 100 mM NaCl). Bound radioactivity was determined by liquid scintillation counting as described under Radioligand Binding Assays.
cAMP Accumulation Assays.
For inhibition of AC, SH-SY5Y cells were plated in 24-well plates (5 × 105 cells/well) and differentiated with 10 μM retinoic acid 4 days before assay. Cells were incubated with 1 μM concentration of the indicated agonist(s) in the presence of 5 μM forskolin and 1 mM IBMX in DMEM/10% FBS for 10 min at 37°C. The assay was stopped by replacing the media with 1 ml of ice-cold 3% perchloric acid. After at least 30 min at 4°C, a 400-μl aliquot of sample was neutralized with 2.5 M KHCO3 and centrifuged at 13,000g. cAMP was measured from the supernatant using a [3H]cAMP assay system (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) following the manufacturer's instructions. Inhibition of cAMP formation was calculated as the percentage of inhibition of forskolin-stimulated cAMP accumulation in the absence of opioid agonist.
For AC sensitization experiments, differentiated SH-SY5Y cells were incubated overnight in the presence or absence of DAMGO and/or DPDPE in DMEM/10% FBS at 37°C. Drug-containing media were replaced with media containing 5 μM forskolin, 1 mM IBMX, and 1 μM MOR antagonist CTAP for DAMGO-treated cells; 30 μM forskolin, 1 mM IBMX, and 1 μM DOR antagonist ICI 174,864 for DPDPE-treated cells; or 5 μM forskolin, 1 mM IBMX, and 100 μM opioid antagonist naloxone for long-term DAMGO- and DPDPE-treated cells, to precipitate cAMP overshoot. In some experiments, DAMGO-containing media were replaced with media containing 5 μM forskolin, 1 mM IBMX, 1 μM MOR antagonist CTAP, and 1 μM Gi/o-coupled receptor agonist. After 10 min at 37°C, the assay was stopped with ice-cold 3% perchloric acid, and cAMP accumulation was quantified as described above. Overshoot was calculated as either the percentage of cAMP overshoot or as a percentage of forskolin-stimulated cAMP accumulation in the absence of opioid agonist.
Isobologram Analysis.
An isobologram for agonists (SNC80 and DAMGO) with different maxima and therefore a variable potency ratio was constructed based on the following equation (Tallarida, 2006):
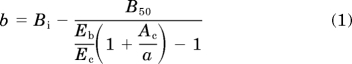 |
The parameters for the equation were based on values from the individual concentration-effect curves for DAMGO and SNC80. As the more efficacious drug in these experiments, DAMGO was assigned as drug B and SNC80 as drug A (Tallarida, 2006). B50 and Ac represent the EC50 of DAMGO and SNC80, respectively. Eb and Ec represent the maximal effect (in femtomoles of [35S]GTPγS bound per milligram) produced by DAMGO and SNC80, respectively. The equation was solved for either a or b (concentration of SNC80 or DAMGO, in nanomoles, respectively) at the concentration of DAMGO that produced 50% of its maximal effect (Bi). Therefore, in this case, Bi = B50. The derived (a, b) coordinates were fit to exponential one-phase decay in Prism version 5 (GraphPad Software Inc., San Diego, CA) to produce the line of additivity. Concentration-effect curves for DAMGO were then obtained in the presence of set concentrations of SNC80 (1, 5, 10, 20, or 30 nM). The concentration of DAMGO, when in combination with SNC80, needed to produce 50% of its maximum effect was determined and plotted on the isobologram as mean ± S.E.M. for three separate experiments. Because the error for all points overlaps the line of additivity, they were assumed to not be statistically different from the line, and no further statistical analysis was conducted.
Western Blot.
SH-SY5Y membranes (40 μg) were mixed with sample buffer (63 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.008% bromphenol blue, and 50 mM dithiothreitol), separated by SDS-polyacrylamide gel electrophoresis on a 10% polyacrylamide gel, and transferred to nitrocellulose membranes (Pierce Chemical, Rockford, IL) for Western blotting. G proteins were detected with rabbit anti-Gαo (Santa Cruz Biotechnology, Santa Cruz, CA) and mouse anti-Gαi2 (Millipore Corporation, Billerica, MA). As a loading control, tubulin was detected with mouse anti-α-tubulin (Sigma-Aldrich). Above antibodies were diluted 1:1000 in 5% nonfat dry milk (Gαo and tubulin) or 1% bovine serum albumin (Gαi2) prepared with Tris-buffered saline/0.05% Tween 20. Secondary antibodies used were goat anti-mouse horseradish peroxidase or goat anti-rabbit horseradish peroxidase (Santa Cruz Biotechnology) diluted 1:10,000 in 5% nonfat dry milk in Tris-buffered saline/0.05% Tween 20. SuperSignal West Pico chemiluminescent substrate (Pierce Chemical) was used to detect immunoreactivity. Immunoreactive band densities were quantified using NIH Image software (http://rsbweb.nih.gov/ij/), normalized to tubulin loading control, and presented as fold over vehicle (mean ± S.E.M. for three separate experiments).
Statistical Analysis.
All data were analyzed using GraphPad Prism version 5 software. Data points represent at least three separate experiments in duplicate and are presented as mean ± S.E.M. unless otherwise noted. The addition of single agonist concentrations was analyzed by one-way ANOVA with Bonferroni's post hoc test. The percentage competition values were compared with 100 by one-sample t test. Effects on agonist responses at various concentrations were analyzed by two-way ANOVA with Bonferroni's post hoc test. EC50 values were calculated from individual concentration-effect curves using nonlinear three parameter log [agonist]-response curve-fit analysis in GraphPad Prism and compared for statistical significance by unpaired, two-tailed Student's t test. For all tests, significance was set at p < 0.05.
Results
Gi/o-Coupled Receptors Expressed in SH-SY5Y Cells.
Human neuroblastoma SH-SY5Y cells were differentiated with retinoic acid (10 μM for 4–7 days) to produce a neuronal-like phenotype. Differentiation increased MOR density from 232 ± 33 to 305 ± 42 fmol/mg protein, as identified by the specific MOR agonist [3H]DAMGO, and increased the level of AC inhibition by DAMGO, as reported previously (Zadina et al., 1994). In differentiated SH-SY5Y cells, agonists for the following receptors were shown to inhibit AC: MOR, DOR, NOPr, α2AR, CB1, and 5-HT1A (Fig. 1A). However, the ability of a maximal concentration (1 μM) of these agonists to inhibit AC was not equal. The most effective agonist was the MOR agonist DAMGO, followed closely by the DOR agonist SNC80 and the NOPr agonist nociceptin/OFQ. The following agonists had similar activity but caused significantly less inhibition than DAMGO: UK14,304 (α2AR), clonidine (α2AR), CP 55,9140 (CB1), and 8-OH-DPAT (5-HT1A). All of the agonists used are commonly regarded as full agonists, except for clonidine and 8-OH-DPAT, which display partial agonist activity in certain assays. However, in this assay, clonidine caused the same degree of cAMP inhibition as the full α2AR agonist UK14,304.
Fig. 1.

Gi/o-coupled receptors endogenously expressed in SH-SY5Y cells share a common pool of AC. A, short-term inhibition of 5 μM forskolin-stimulated AC by 1 μM concentration of the indicated agonist alone. Agonist (receptor): DAMGO (MOR), SNC80 (DOR), OFQ (NOPr), UK14,304 (α2AR), clonidine (α2AR), CP 55,940 (CB1), and 8-OH-DPAT (5-HT1A). ***, p < 0.001 compared with DAMGO by one-way ANOVA with Bonferroni's post-test. B, coincubation with 1 μM DAMGO occludes inhibition by 1 μM concentration of all indicated agonists. All bars are not statistically different from 1 μM DAMGO alone (p > 0.05 by one-way ANOVA with Bonferroni's post-test). C, lower efficacy agonists (1 μM) were also not additive when coadministered in the indicated pairs (p > 0.05 for all pairs compared with the most efficacious agonist of the pair by one-way ANOVA with Bonferroni's post test). Data are presented as mean ± S.E.M. (n = 4, in duplicate) of the percentage of cAMP inhibition, where stimulation by 5 μM forskolin alone is represented as 0%.
The endogenous expression level of the above identified receptors was determined in membranes from differentiated SH-SY5Y cells using maximal concentrations of selective agonist radioligands for each receptor. Receptor densities are listed in Table 1 and follow a similar rank order as the ability of selective agonists for each receptor to inhibit AC. Although the agonist radioligands will preferentially recognize receptors in a high-affinity conformation, the binding experiments were performed in the absence of guanine nucleotides and Na+ ions to shift the equilibrium toward high-affinity receptor conformations. To confirm this, we compared antagonist binding at MOR, DOR, and α2AR (Table 1). At MOR and DOR similar values were obtained, suggesting that all receptors were in the high-affinity state in the absence of Na+ ions and GTPγS. For the α2AR receptors, there was a difference in agonist and antagonist binding, indicating that approximately 20% of the receptors were in a low-affinity state. Agonist binding at CB1, and NOPr was reduced by 89 and 90%, respectively (n = 2), in the presence of Na+ ions and GTPγS, confirming that these ligands were recognizing mostly high-affinity sites.
TABLE 1.
Receptor density in differentiated SH-SY5Y cells
Receptor numbers in cell membranes were determined using selective agonist radioligands at a maximal concentration as described under Materials and Methods. 5-HT1A was not tested. Binding of maximal concentrations of antagonists for MOR ([3H]diprenorphine in the presence of the δ-selective ICI 174,864), DOR ([3H]naltrindole), and α2AR ([3H]yohimbine) was 103 ± 8.0, 98 ± 11, and 79 ± 6% of the agonist binding, respectively. Results are presented as mean ± S.E.M. (n = 3–6).
| Receptor Type | Radioligand | Receptor Density |
|---|---|---|
| fmol/mg protein | ||
| MOR | [3H]DAMGO | 305 ± 42 |
| DOR | [3H]DPDPE | 191 ± 21 |
| CB1 | [3H]CP 55,940 | 60 ± 25 |
| α2AR | [3H]UK 14304 | 35 ± 1 |
| NOPr | [3H]Nociceptin/OFQ | 30 ± 11 |
Gi/o-Coupled Receptors Compete for AC.
To evaluate the level of interaction between MOR and other Gi/o-coupled receptors in SH-SY5Y cells, maximal concentrations of Gi/o-coupled receptor agonists were combined with a maximal concentration of DAMGO. When added to DAMGO, none of the agonists was able to inhibit AC to a greater degree than DAMGO alone (Fig. 1B), indicating that MORs are able to access and inhibit the same AC enzymes as other Gi/o-coupled receptors. We next addressed whether agonists to α2AR, CB1, or 5-HT1A receptors, which produced lower maximal inhibition than DAMGO, would compete with each other for AC. Even these less efficacious agonists were not able to inhibit AC to any greater extent in combination than when applied alone (Fig. 1C).
The extent of competition between receptors for AC can be calculated using the following equation (Brinkerhoff et al., 2008):
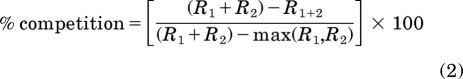 |
where R1 and R2 are two different receptor types giving a theoretical maximum response (R1 + R2), which is compared with the experimentally determined effect of R1 + 2 and the effect of the most efficacious agonist alone [max(R1,R2)]. If two receptors do not compete, the theoretical additive and experimental additive will be equivalent, so there will be 0% competition. When there is complete (100%) competition between two receptors, the experimental addition of both agonists does not increase the response over the most efficacious agonist alone. Using the data from experiments in SH-SY5Y cells described above, complete competition was observed for all receptor pairs (Table 2). In addition, the DOR agonist SNC80 or the NOPr agonist nociceptin/OFQ occluded responses from the other, less effective agonists, resulting in competition that was not significantly different from 100% (Table 3). Therefore, at maximal agonist concentrations, Gi/o-coupled receptors, including the opioid receptors, compete for a shared pool of AC.
TABLE 2.
Competition between indicated agonists (1 μM) for short-term inhibition of AC
Percentage competition was calculated as described under Results (Brinkerhoff et al., 2008) using experimental data shown in Fig. 1. The percentage of competition from three individual experiments was compiled and is presented as mean ± S.E.M.
| Agonist R1 (Receptor) | Agonist R2 (Receptor) | Competition |
|---|---|---|
| % | ||
| DAMGO (MOR) | SNC80 (DOR) | 78 ± 8 |
| DAMGO (MOR) | Nociceptin/OFQ (NOPr) | 97 ± 12 |
| DAMGO (MOR) | UK14,304 (α2AR) | 115 ± 10 |
| DAMGO (MOR) | Clonidine (α2AR) | 106 ± 22 |
| DAMGO (MOR) | CP 55,9140 (CB1) | 121 ± 24 |
| DAMGO (MOR) | 8-OH-DPAT (5-HT1A) | 97 ± 13 |
| UK 14301 (α2AR) | CP 55,9140 (CB1) | 95 ± 19 |
| 8-OH-DPAT (5-HT1A) | Clonidine (α2AR) | 95 ± 16 |
| 8-OH-DPAT (5-HT1A) | CP 55,9140 (CB1) | 83 ± 6 |
TABLE 3.
Competition between 1 μM SNC80 or nociceptin/OFQ and other Gi/o-coupled receptor agonists for short-term inhibition of AC
Inhibition of AC by 1 μM of agonist(s) alone or in combination was performed as described for Fig. 1 and under Materials and Methods. Percentage competition was calculated from three individual experiments, in duplicate, as described for Table 2.
| Agonist R1 (Receptor) | Agonist R2 (Receptor) | Competition |
|---|---|---|
| % | ||
| SNC80 (DOR) | Nociceptin/OFQ (NOPr) | 79 ± 18 |
| SNC80 (DOR) | UK14,304 (α2AR) | 84 ± 30 |
| SNC80 (DOR) | Clonidine (α2AR) | 72 ± 21 |
| SNC80 (DOR) | CP 55,9140 (CB1) | 88 ± 32 |
| SNC80 (DOR) | 8-OH-DPAT (5-HT1A) | 119 ± 12 |
| Nociceptin/OFQ (NOPr) | UK14,304 (α2AR) | 108 ± 15 |
| Nociceptin/OFQ (NOPr) | Clonidine (α2AR) | 119 ± 11 |
| Nociceptin/OFQ (NOPr) | CP 55,9140 (CB1) | 119 ± 7 |
| Nociceptin/OFQ (NOPr) | 8-OH-DPAT (5-HT1A) | 124 ± 10 |
MOR and DOR Competition for G Proteins.
Interactions between receptors could occur at the level of AC or G protein. To evaluate this, we studied the interaction between MOR and DOR for G proteins as measured by binding of [35S]GTPγS. Maximum concentrations of DAMGO stimulated a greater degree of [35S]GTPγS binding than SNC80, consistent with their relative degrees of AC inhibition and the greater expression of MOR compared with DOR in these cells (Fig. 2A). When added together, DAMGO and SNC80 stimulation of [35S]GTPγS binding was similar to binding stimulated by DAMGO alone (p > 0.05 comparing DAMGO to DAMGO/SNC80 by one-way ANOVA with Bonferroni's post-test) and significantly less than the theoretical additive (p < 0.01 comparing R1 + R2 to DAMGO/SNC80 by one-way ANOVA with Bonferroni's post-test) (Fig. 2A), giving a percentage of competition between DAMGO and SNC80 of 88 ± 2%, similar to the level of competition between these two agonists for AC. These results indicate significant sharing of G proteins between MOR and DOR in differentiated SH-SY5Y cells.
Fig. 2.

MOR and DOR share pertussis toxin-sensitive G proteins. A, stimulation of [35S]GTPγS binding in membranes from SH-SY5Y cells after 60-min incubation with 1 μM DAMGO or SNC80 alone or in combination (DAMGO/SNC80). Incubation with DAMGO and SNC80 in combination (DAMGO/SNC80) did not significantly increase [35S]GTPγS binding more than DAMGO alone (p > 0.05 by one-way ANOVA with Bonferroni's post test) and stimulated significantly less [35S]GTPγS binding than the theoretical additive of the individual responses (R1 + R2) (**, p < 0.01 by one-way ANOVA with Bonferroni's post test). B, pretreatment of SH-SY5Y cells with PTX (100 ng/ml) for 24 h before membrane preparation blocked the stimulation of [35S]GTPγS binding by 1 μM DAMGO, SNC80, or the combination (DAMGO/SNC80) (***, p < 0.001 by two-way ANOVA with Bonferroni's post test). Pertussis toxin treatment did not alter spontaneous [35S]GTPγS binding in the absence of agonist (p > 0.05 by two-way ANOVA with Bonferroni's post test). Data are presented as mean ± S.E.M. (n = 3, in triplicate); n.s., not significant.
It has been proposed that MOR and DOR heterodimerize and that these oligomers can activate PTX-resistant G proteins (George et al., 2000). However, in the differentiated SH-SY5Y cells used in our experiments, [35S]GTPγS binding stimulated by the combination of DAMGO and SNC80 was completely eliminated by PTX treatment (Fig. 2B; PTX 100 ng/ml, 24 h), indicating that the combination of agonists still signals through PTX-sensitive Gαi/o proteins. In addition, PTX did not alter spontaneous [35S]GTPγS binding (Fig. 2B). Likewise, AC inhibition by the combination of DAMGO and SNC80 was also blocked by 24-h pretreatment with PTX (data not shown).
Isobolographic Analysis of Interactions between MOR and DOR Agonists.
Combinations of maximally effective concentrations of agonists resulted in less than additive effects and predict competition for a common effector; however, at lower agonist concentrations, it should be possible to identify additive, subadditive, and synergistic interactions. To this end, the concentration-dependence of DAMGO to stimulate [35S]GTPγS binding was determined in the presence of a submaximal concentration of SNC80. The addition of 30 nM SNC80 with DAMGO did not significantly change the potency of DAMGO to stimulate [35S]GTPγS binding (Fig. 3A; EC50 of DAMGO alone, 121 ± 32 nM; EC50 of DAMGO + 30 nM SNC80, 64 ± 12 nM; p > 0.05), and at maximal concentrations of DAMGO, the level of [35S]GTPγS binding was similar in the presence or absence of SNC80 (at 1 and 10 μM DAMGO, p > 0.05 by two-way ANOVA with Bonferroni's post-test) and significantly less than the theoretical additive (at 10 μM DAMGO, p < 0.05 by two-way ANOVA with Bonferroni's post-test; Fig. 3A). However, at lower concentrations of DAMGO, combination with 30 nM SNC80 was similar to the theoretical additive (Fig. 3A), indicating an additive interaction between MOR and DOR agonists until G proteins become limiting, at which point DAMGO is occlusive.
Fig. 3.

DAMGO and SNC80 activation of G protein is additive at concentrations or time points when G protein is not limiting. A, concentration-dependent stimulation of [35S]GTPγS binding in SH-SY5Y membranes after 60-min incubation with various concentrations of DAMGO alone (■) or DAMGO with 30 nM SNC80 (□). The EC50 of DAMGO is not significantly altered by the addition of 30 nM SNC80 (DAMGO alone, 121 ± 32 nM, DAMGO + 30 nM SNC80, 64 ± 12 nM; p = 0.14 by two-tailed Student's t test). Coincubation of DAMGO with 30 nM SNC80 produces additive [35S]GTPγS binding similar to the theoretical additive curve (●), which diverges only when DAMGO becomes occlusive at maximal concentrations (*, p < 0.05 at 10 μM for DAMGO + 30 nM SNC80 compared with the “theoretical addition” by two-way ANOVA with Bonferroni's post test; n = 4, in duplicate). B, isobologram for agonists with a variable potency ratio calculated as described under Materials and Methods. Stimulation of [35S]GTPγS binding by DAMGO was conducted in the presence of indicated concentrations of SNC80. Concentration combinations that produced 50% of the maximum effect of DAMGO alone are plotted from three separate experiments as mean ± S.E.M. Points on the line indicate additivity between DAMGO and SNC80. C, stimulation of [35S]GTPγS binding in SH-SY5Y membranes after incubation with 1 μM DAMGO or SNC80 alone or in combination for 10 or 20 min, before the incubation reaches steady state. At both time points, coincubation with DAMGO and SNC80 (DAMGO/SNC80) is greater than DAMGO alone (*, p < 0.05 by one-way ANOVA with Bonferroni's post test) and similar to the theoretical additive (R1 + R2) (p > 0.05 by one-way ANOVA with Bonferroni's post test; n = 2, in triplicate); n.s., not significant.
Additive actions of agonists or deviations from additivity can be observed graphically using an isobologram (Tallarida, 2006). An isobologram for combinations of DAMGO and SNC80 that produced 50% of the maximal DAMGO effect was constructed based on values from the individual concentration-effect curves for DAMGO and SNC80 using eq. 1 (Tallarida, 2006). The line of additivity is not linear because DAMGO and SNC80 have different maxima and therefore a variable potency ratio. [35S]GTPγS concentration-effect curves for DAMGO were performed in the presence of low concentrations of SNC80. The concentration combination that was required to produce 50% of the maximal DAMGO effect was plotted on the isobologram. At these low concentrations, SNC80 produced an additive interaction when combined with DAMGO, as indicated by points falling on the line of additivity (Fig. 3B). This agrees with results above and indicates that MOR and DOR share a common set of G proteins that are activated additively at low concentrations but subadditively when G proteins become limiting.
The binding of [35S]GTPγS is time-dependent with a t1/2 of approximately 20 min for DAMGO or SNC80-stimulated binding. Therefore, at time points shorter than 20 min, G proteins should not be limiting. When [35S]GTPγS binding was measured after a 10- or 20-min incubation, the combination of maximal concentrations of DAMGO and SNC80 was similar to the theoretical additive (Fig. 3C) with competition between receptors only 12 ± 8.6% after 10 min, increasing to 33 ± 11% after 20 min. These results are in agreement with an additive interaction of these agonists at less than saturating concentrations, when G proteins are not limiting.
Heterologous Inhibition of AC Prevents Opioid Receptor-Mediated cAMP Overshoot.
Long-term administration of opioid agonists and other Gi/o-coupled receptor agonists causes a homeostatic sensitization of AC resulting in an overshoot of cAMP production upon the addition of a competitive antagonist (Watts, 2002). To determine whether MOR and DOR accessed the same systems responsible for AC sensitization during long-term treatment, cells were treated overnight with the peptidic DOR agonist DPDPE (10 μM) in the presence or absence of DAMGO. DPDPE alone produced an overshoot response, which was enhanced in the presence of 10 but not 100 nM DAMGO (Fig. 4).
Fig. 4.

MOR and DOR share AC during long-term agonist administration. SH-SY5Y cells were treated overnight with vehicle (□) or 10 μM DPDPE ( ) in the presence or absence of the MOR agonist DAMGO (10 or 100 nM) to induce dependence. Withdrawal was precipitated with the opioid antagonist naloxone (100 μM) in the presence of 5 μM forskolin. Data are presented as mean ± S.E.M. (n = 4, in duplicate) of the percentage of forskolin-stimulated cAMP, where forskolin alone is 100% and is indicated by the dashed line. Overnight incubation with DPDPE produced overshoot on its own and enhanced the overshoot produced by 10 nM but not 100 nM DAMGO. ***, p < 0.001 compared with the vehicle with the same concentration of DAMGO by two-way ANOVA and Bonferroni's post test.
) in the presence or absence of the MOR agonist DAMGO (10 or 100 nM) to induce dependence. Withdrawal was precipitated with the opioid antagonist naloxone (100 μM) in the presence of 5 μM forskolin. Data are presented as mean ± S.E.M. (n = 4, in duplicate) of the percentage of forskolin-stimulated cAMP, where forskolin alone is 100% and is indicated by the dashed line. Overnight incubation with DPDPE produced overshoot on its own and enhanced the overshoot produced by 10 nM but not 100 nM DAMGO. ***, p < 0.001 compared with the vehicle with the same concentration of DAMGO by two-way ANOVA and Bonferroni's post test.
Because MOR and DOR accessed the same pool of AC in both the naive and dependent opioid states we hypothesized that overshoot of cAMP occurring upon precipitation of withdrawal from long-term MOR agonist treatment would be prevented by the short-term addition of a DOR agonist. To test this hypothesis, differentiated SH-SY5Y cells were treated overnight with DAMGO (100 nM), and withdrawal was precipitated with the MOR antagonist CTAP in the presence or absence of 1 μM SNC80. The addition of SNC80 attenuated the AC overshoot response (Fig. 5). Moreover, this attenuation was also observed with the addition of the NOPr agonist nociceptin/OFQ and the α2AR agonists UK14,304 and clonidine (Fig. 5). Agonists that gave a reduced short-term inhibition of cAMP (CP 55,9140 and 8-OH-DPAT) were unable to attenuate MOR-mediated cAMP overshoot.
Fig. 5.

DAMGO-mediated cAMP overshoot is reduced by heterologous inhibition of shared AC by agonist to DOR, NOPr, or α2AR. AC sensitization was developed by incubating SH-SY5Y cells overnight with 100 nM DAMGO. To precipitate withdrawal, DAMGO-containing media were replaced with media containing 5 μM forskolin, 1 mM IBMX, and 1 μM CTAP in the presence or absence of 1 μM concentration of a non-MOR agonist, as indicated, for 10 min. Data are presented as mean ± S.E.M. of the percentage of cAMP overshoot, where stimulation by forskolin alone is represented as 0%. Three of six experiments, in duplicate, were compiled that produced >100% DAMGO overshoot in the absence of a non-MOR agonist. **, p < 0.01; ***, p < 0.001 compared with DAMGO overshoot without non-MOR agonist by one-way ANOVA with Bonferroni's post test.
To determine the concentration-relationship of this effect, DAMGO-mediated cAMP overshoot was precipitated by CTAP in the presence or absence of varying concentrations of SNC80. The addition of SNC80 reduced DAMGO-mediated overshoot in a concentration-dependent manner (Fig. 6A). Furthermore, SNC80 inhibited cAMP production with a similar potency in vehicle or DAMGO-treated cells (vehicle, 14.6 ± 7.8 nM; DAMGO-treated, 13.8 ± 7.2 nM). The effect of SNC80 was via DOR because MOR was blocked by the selective antagonist CTAP in both vehicle and DAMGO-treated cells, and in separate experiments, 1 μM CTAP did not affect AC inhibition by 100 nM SNC80 (100 nM SNC80, 59 ± 3% cAMP inhibition; 100 nM SNC80 + 1 μM CTAP, 55 ± 2% cAMP inhibition; n = 6, p > 0.05).
Fig. 6.

Inhibition of cAMP by MOR or DOR agonists is similar for sensitized or nonsensitized AC. A, SH-SY5Y cells were incubated with vehicle (■) or the MOR agonist DAMGO (100 nM, □) overnight to induce dependence. Withdrawal was precipitated with the MOR antagonist CTAP (1 μM) in the presence of 5 μM forskolin. Short-term cAMP production was inhibited by including various concentrations of the DOR agonist SNC80 in the precipitating media. The concentration-response of SNC80 to inhibit cAMP was similar in control and DAMGO-dependent cells (EC50: vehicle-treated = 14.6 ± 7.8 nM, DAMGO-treated, 13.8 ± 7.2 nM; p > 0.05 by two-tailed student's t test). B, cells were incubated with vehicle (■) or the DOR agonist DPDPE (10 μM, □) overnight to induce dependence. Receptor-specific withdrawal was precipitated with the DOR antagonist ICI 174,864 (1 μM) in the presence of 30 μM forskolin. Various concentrations of the MOR agonist DAMGO were included in the precipitating media. The concentration-response of DAMGO to inhibit cAMP production was similar in control and DPDPE-withdrawn cells (EC50: vehicle-treated, 32.2 ± 12.5 nM, DPDPE-treated, 29.0 ± 7.1 nM; p > 0.05 by two-tailed Student's t test). Data are presented as mean picomoles of cAMP per milligram of protein ± S.E.M. (n = 3 or 4, in duplicate). cAMP produced by forskolin alone is indicated by the dashed line.
Likewise, DOR-mediated cAMP overshoot was attenuated in a concentration-dependent manner by DAMGO (Fig. 6B). In these experiments, cells were treated overnight with DPDPE, and specific DOR-mediated cAMP overshoot was precipitated using the selective DOR antagonist ICI 174,864 (1 μM) in the absence or presence of increasing concentrations of DAMGO. In addition to preventing overshoot, DAMGO inhibited AC with a similar potency in vehicle or DPDPE-treated cells (EC50: vehicle-treated, 32.2 ± 12.5 nM; DPDPE-treated, 29.0 ± 7.1 nM). 1 μM ICI 174,864 did not shift the ability of DAMGO to stimulate [35S]GTPγS binding in SH-SY5Y cells (EC50: control, 263 ± 30 nM; with ICI 174,864, 283 ± 35 nM; p = 0.70, n = 2).
Lack of Heterologous Tolerance between MOR and DOR.
The similar EC50 values for AC inhibition in opioid-treated and naive cells for both MOR and DOR agonists suggest a lack of cross-tolerance between these two receptors in SH-SY5Y cells. To confirm this, agonist-stimulated [35S]GTPγS binding was measured in membranes from SH-SY5Y cells that were treated overnight with vehicle, DAMGO, or SNC80. DAMGO stimulated [35S]GTPγS binding in vehicle-treated SH-SY5Y membranes in a concentration-dependent manner (Fig. 7A, EC50, 86 ± 16 nM). Both the maximum effect (vehicle-treated, 121 ± 3.7% stimulation; DAMGO-treated, 29 ± 2.9% stimulation; p < 0.0001) and EC50 (vehicle-treated, 86 ± 16 nM; DAMGO-treated, 260 ± 62 nM; p < 0.05) of DAMGO-stimulated [35S]GTPγS binding was significantly attenuated in membranes from cells treated overnight with 1 μM DAMGO, indicating the development of tolerance (Fig. 7A). In comparison, SNC80-stimulated [35S]GTPγS binding was similar in membranes from vehicle or DAMGO-treated cells (Fig. 7B; vehicle-treated EC50, 29.8 ± 12.5 nM; DAMGO-treated EC50, 34.6 ± 10.8 nM; vehicle-treated maximum, 53.1 ± 4.7% stimulation; DAMGO-treated maximum, 56.5 ± 3.5% stimulation). The reverse-treatment paradigm produced similar results. Treatment of SH-SY5Y cells overnight with 1 μM SNC80 produced marked homologous tolerance, indicated by a significant reduction in [35S]GTPγS binding by maximal concentrations of SNC80 (Fig. 7C; vehicle-treated, 57.2 ± 3.4% stimulation; SNC80-treated, 14.6 ± 4.8% stimulation; p < 0.0001). However, the potency and efficacy for DAMGO to stimulate [35S]GTPγS binding was not affected by SNC80 pretreatment (Fig. 7d; vehicle-treated EC50, 69 ± 8.5 nM; SNC80-treated EC50, 79 ± 1.6 nM; vehicle-treated maximum, 106.7 ± 7.5%; SNC80-treated maximum, 100.7 ± 5.9%, p > 0.05).
Fig. 7.

Lack of heterologous tolerance between MOR and DOR. SH-SY5Y cells were incubated with 1 μM concentration of the MOR agonist DAMGO (A and B) or the DOR agonist SNC80 (C and D) for 24 h before membrane preparation. [35S]GTPγS binding in membranes from treated cells was stimulated by incubation for 60 min with various concentrations of DAMGO (A and D) or SNC80 (B and C). Long-term treatment with 1 μM DAMGO reduced the maximal effect and potency of DAMGO (EC50: vehicle-treated, 86 ± 16 nM; DAMGO-treated, 260 ± 62 nM, p = 0.04) but not SNC80 (EC50: vehicle-treated, 29.8 ± 12.5 nM; DAMGO-treated EC50, 34.6 ± 10.8 nM; p > 0.05). Likewise, long-term treatment with 1 μM SNC80 almost completely abolished SNC80-mediated [35S]GTPγS binding but did not alter the potency of DAMGO-mediated [35S]GTPγS binding (EC50: vehicle-treated, 69 ± 8.5 nM; SNC80-treated, 79 ± 1.6 nM, p > 0.05). EC50 statistical comparisons were made by two-tailed Student's t test. E, Gαo and Gαi2 were detected in membranes from cells treated overnight with vehicle (DMEM, V), 1 μM DAMGO (D), or 1 μM SNC80 (S). Immunoreactive density was quantified, normalized to tubulin loading control, and compared with vehicle-treated cells (n = 3). There was no difference in G protein levels after agonist treatment (p > 0.05 by one-way ANOVA with Bonferroni's post test).
Basal binding of [35S]GTPγS in the absence of agonist was similar in vehicle, DAMGO, or SNC80-pretreated cells (vehicle-treated, 9.8 ± 0.6 fmol/mg; DAMGO-treated, 11.6 ± 1.4 fmol/mg; SNC80-treated, 11.4 ± 0.6 fmol/mg, p > 0.05 one-way ANOVA with Bonferroni's post-test), indicating that G proteins were unchanged by overnight agonist treatment. To confirm this, Gαo and Gαi2 were identified by Western blot in the membrane samples used in the above experiments. Gαo and Gαi2 protein levels were not changed after overnight treatment with DAMGO or SNC80 (Fig. 7E). Together, these results confirm a lack of cross-tolerance between MOR and DOR in differentiated SH-SY5Y cells, similar to previous reports in undifferentiated cells (Zadina et al., 1994; Alt et al., 2002).
Discussion
In this study, we have shown that AC inhibition by agonists to DOR, NOPr, α2AR, CB1, and 5-HT1A receptors in differentiated SH-SY5Y cells was occluded by a maximal concentration of the MOR agonist DAMGO, suggesting that all of these receptors compete for and inhibit the same AC enzymes. The competition, as shown by MOR and DOR, began at the G protein, was additive when G proteins were not limiting, and reached an occlusive ceiling at maximal agonist concentrations. Similar competition occurred during long-term agonist exposure such that short-term administration of agonists to DOR, NOPr, and α2AR reduced the expression of AC sensitization after long-term DAMGO treatment. However, the cross-talk between MOR and DOR did not lead to heterologous tolerance.
The rank order of AC inhibition by a maximum concentration of full agonists acting at Gi/o-coupled receptors in SH-SY5Y cells was MOR > DOR ≥ NOPr > α2AR ≥ CB1 = 5-HT1A. This order is mostly determined by the relative receptor expression as measured by 3H-agonist binding, which follows a similar pattern. One exception was NOPr, which was expressed at relatively low levels (30 ± 11 fmol/mg protein), yet the NOPr agonist nociceptin/OFQ inhibited AC as well as the DOR agonist SNC80 or the MOR agonist DAMGO. This suggests that NOPr is efficiently coupled to Gαi/o proteins in the cells, which is consistent with evidence that NOPr displays agonist-independent constitutive activity (Beedle et al., 2004). Thus, all of these Gi/o-coupled receptors shared a common pool of AC, but the proportion of the AC pool used by each receptor was determined by the agonist-driven activity of each receptor to inhibit AC (Fig. 8). The most active agonist, the MOR agonist DAMGO, had access to the most AC and at a maximal concentration occluded effects by agonists to DOR, NOPr, α2AR, CB1, and 5-HT1A receptors. Agonists to α2AR, CB1 and 5-HT1A receptors inhibited AC the least, and even these receptors were not additive with each other, suggesting that the pool is always limiting, even for receptors with lower expression levels.
Fig. 8.

Schematic depicting the accessibility of Gi/o-coupled receptors to portions of the total AC pool. The amount of the AC pool used by each receptor type is related to the agonist-mediated activity of each receptor. The most active and most highly expressed receptor, MOR, shares AC with all other receptor types. The other receptor types share AC in a manner predicted by receptor density and/or the ability of agonist to inhibit AC such that three receptor groups exist: one that contains MOR only; one that contains MOR, DOR, and NOPr; and one that contains MOR, DOR, NOPr, α2AR, CB1, and 5-HT1A.
The ability of all receptors to share AC indicates that barriers to prevent free diffusion in the membrane, such as receptor oligomers, membrane microdomains, and protein scaffolds (George et al., 2000; Hall and Lefkowitz, 2002; Gomes et al., 2004; Allen et al., 2007), do not segregate these receptors from the common pool of AC. Two alternative hypotheses could explain these results. First, free access of receptors to all G proteins and AC as predicted by the collision-coupling model would allow receptors to share a common pool of AC and has recently been discussed as an alternative explanation to negative cooperativity data that were attributed to dimerization (Chabre et al., 2009). However, such a scenario would seem unlikely given the evidence that MOR diffusion is restricted to submicrometer domains (Saulière et al., 2006). Second, there could be complexes of multiple receptors isolated with signaling molecules. Most of the Gi/o-coupled receptors expressed in SH-SY5Y cells have been reported to heterodimerize with MOR, including DOR (George et al., 2000; Gomes et al., 2004), NOPr (Wang et al., 2005), α2AR (Jordan et al., 2003), and CB1 (Rios et al., 2006), whereas DOR has been shown to form heterodimers with α2AR (Rios et al., 2004). In addition, preformed signaling complexes containing GPCR and multiple effectors have been identified (Davare et al., 2001). In SH-SY5Y cells, such complexes could include MOR and at least five other Gi/o-coupled receptor types.
Agonists for DOR, NOPr, and α2AR were able to inhibit AC in the naive and opioid-dependent state. The rank order of the effectiveness of agonists to inhibit AC remained the same in control and DAMGO-dependent cells, so that the most efficacious agonists (SNC80 and nociceptin/OFQ) significantly prevented DAMGO-mediated cAMP overshoot. One exception was the α2AR agonist UK14,304, which was equally effective as SNC80 and nociceptin/OFQ at preventing DAMGO-mediated overshoot, but considerably less efficacious at AC inhibition in the naive cell. An increase in α2AR density after long-term DAMGO exposure could explain the enhanced UK14,304 response. However, in rats, α2AR density in various brain regions was either decreased or unchanged after long-term morphine treatment (Smith et al., 1989). On the other hand, the α2AR signaling system may become more efficient after long-term MOR agonist treatment possibly through opioid-induced changes in the activity of regulators of G protein signaling proteins (Traynor, 2010).
AC sensitization occurs after long-term MOR occupation and is believed to be important for the manifestation of withdrawal (Watts, 2002). Specifically, up-regulation of the cAMP/AC/protein kinase A pathway in the locus ceruleus (LC) has been identified as a mediator of opioid dependence and withdrawal, most recently by Zachariou et al., 2008. Thus, drugs that inhibit the cAMP pathway and can counter AC sensitization, such as the Gi/o-coupled receptor agonists presented here, would have therapeutic potential in the treatment of opioid withdrawal. For instance, the clinical utility of the α2AR agonist clonidine in opioid withdrawal has been known for some time (Gold et al., 1978), and α2AR agonists are often used “off label” to treat or prevent opioid withdrawal. Furthermore, the α2AR agonist lofexidine could become the first nonopiate U.S. Food and Drug Administration-approved treatment of opioid withdrawal (Yu et al., 2008).
It is believed that clonidine prevents opioid withdrawal symptoms by reversing hyperactivity of noradrenergic neurons in the LC (Aghajanian, 1978). There are two proposed mechanisms for withdrawal-induced hyperactivity of LC neurons in opioid-dependence. The first is an enhanced input of excitatory glutamate into the LC (Aston-Jones et al., 1997). The second is an intracellular sensitization mediated by up-regulation of the cAMP pathway and is supported by data that in vitro, withdrawal-induced hyperactivity is suppressed by inhibitors of protein kinase A and is enhanced by forskolin or an active cAMP analog (Ivanov and Aston-Jones, 2001). Thus, our findings that clonidine can heterologously inhibit cAMP and prevent DAMGO-mediated cAMP overshoot provides a mechanism for clonidine in opioid withdrawal.
NOP and CB1 receptors are also coexpressed with MOR in LC neurons, in which NOP receptors were found to activate the same population of K+ channels as MOR and α2AR (Connor et al., 1996; Scavone et al., 2010). Furthermore, intracerebroventricular injection of nociceptin/OFQ prevented naloxone-precipitated withdrawal symptoms in morphine-dependent rats and the nonpeptidic NOPr agonist (1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one (Ro 64-6198) reduced the expression of morphine-withdrawal jumping in mice when administered just before precipitation of withdrawal (Kotlinska et al., 2000, 2003). This is again consistent with our findings that administration of nociceptin/OFQ during antagonist-precipitated withdrawal reduced cAMP overshoot and supports an intracellular mechanism of competitive inhibition of shared AC. Although the CB1 agonist CP 55,940 did not prevent MOR-induced cAMP overshoot in SH-SY5Y cells, this may be due to low expression levels. This is pertinent because levels of CB1 in the brain are generally high, and short-term administration of cannabinoid agonists to morphine-dependent rodents prevented withdrawal symptoms including jumping, weight loss, wet dog shakes, and diarrhea, although the mechanism or site of action was not determined (Hine et al., 1975).
In contrast to α2AR, NOPr, and CB1 receptors, the role of DOR in attenuating MOR-mediated withdrawal may be less relevant in vivo. Although we have shown that DOR shares AC with MOR, and the DOR agonist SNC80 inhibits DAMGO-mediated cAMP overshoot in SH-SY5Y cells, an in vivo intracellular mechanism between MOR and DOR to prevent morphine-withdrawal will depend on coexpression in a single neuron. DOR is not expressed in the LC, and although other brain regions are undoubtedly important in withdrawal (Christie et al., 1997), the coexpression of MOR and DOR on neurons in other regions is debatable (Scherrer et al., 2009). Furthermore, administration of the DOR agonist (±)-[1(S*),2α,5β]-4-[[2,5-dimethyl-4-(2-propenyl)-1-piperazinyl](3-hydroxyphenyl)methyl]-N,N-diethylbenzamide dihydrobromide (BW373U86) just before naloxone-precipitated withdrawal in morphine-dependent rats did not reduce withdrawal signs (Lee et al., 1993).
In conclusion, these studies have shown that all identified Gi/o-coupled receptors endogenously expressed in differentiated SH-SY5Y cells shared a common pool of AC. The interaction probably begins at the G-protein level for all receptors, as shown for MOR and DOR. At this stage, we cannot distinguish between a lack of compartmentalization and the presence of signalosomes that contain several receptor types and signaling proteins. However, biophysical data on MOR membrane diffusion would tend to support a model in which there is organization of receptors (Saulière et al., 2006). Regardless of the model, heterologous inhibition of shared AC by DOR, NOPr, and α2AR agonists prevented the expression of cAMP overshoot in MOR agonist-dependent cells. Thus, these studies support an intracellular mechanism for the prevention of morphine-withdrawal symptoms by short-term administration of α2AR, NOPr, or CB1 receptor agonists.
Acknowledgments
We thank Anthony McClafferty for technical assistance.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants R01-DA04087, F31-DA023339, T32-DA07267]; and the National Institutes of Health National Institute of General Medical Sciences [Grant T32-GM07767].
Parts of this work were previously presented at the Society for Neuroscience Annual Meeting, 2009 Oct 17–21, Chicago, IL, and at Experimental Biology, 2010 Apr 25–28, Anaheim, CA.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.064816.
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- SNC80
- (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide
- DPDPE
- [d-Pen2,5]-enkephalin
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- OFQ
- orphanin FQ
- 8-OH-DPAT
- 8-hydroxy-2-(di-n-propylamino)tetralin hydrochloride
- CTAP
- d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2
- ICI 174,864
- N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH
- J113397
- 1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one
- [35S]GTPγS
- guanosine-5′-O-(3-[35S]thio)triphosphate
- MOR
- μ-opioid receptor
- DOR
- δ-opioid receptor
- NOPr
- nociceptin/orphanin FQ peptide receptor
- α2AR
- α2-adrenergic receptor
- CB1
- cannabinoid receptor 1
- 5-HT1A
- 5-hydroxytryptamine-1A receptor
- AC
- adenylyl cyclase
- PTX
- pertussis toxin
- IBMX
- 3-isobutyl-1-methylxanthine
- ANOVA
- analysis of variance
- LC
- locus ceruleus
- UK14,304
- 5-bromo-6-(2-imidazolin-2-ylamino)quinoxaline
- CP 55,940
- 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-phenol
- WIN 55212-2
- (2,3-dihydro-5-methyl-3-((4-morpholinyl)methyl)pyrrolo-(1,2,3-de)-1,4-benzoxazin-6-yl)(1-naphthalenyl)methanone monomethanesulfonate
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- Ro 64-6198
- (1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
- BW373U86
- (±)-[1(S*),2α,5β]-4-[[2,5-dimethyl-4-(2-propenyl)-1-piperazinyl](3-hydroxyphenyl)methyl]-N,N-diethylbenzamide dihydrobromide.
Authorship Contributions
Participated in research design: Levitt and Traynor.
Conducted experiments: Levitt and Purington.
Performed data analysis Levitt, Purington, and Traynor.
Wrote or contributed to the writing of the manuscript: Levitt and Traynor.
References
- Aghajanian GK. (1978) Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature 276:186–188 [DOI] [PubMed] [Google Scholar]
- Allen JA, Halverson-Tamboli RA, Rasenick MM. (2007) Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8:128–140 [DOI] [PubMed] [Google Scholar]
- Alt A, Clark MJ, Woods JH, Traynor JR. (2002) Mu and Delta opioid receptors activate the same G proteins in human neuroblastoma SH-SY5Y cells. Br J Pharmacol 135:217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt A, McFadyen IJ, Fan CD, Woods JH, Traynor JR. (2001) Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding in digitonin-permeabilized C6 rat glioma cells: evidence for an organized association of mu-opioid receptors and G protein. J Pharmacol Exp Ther 298:116–121 [PubMed] [Google Scholar]
- Aston-Jones G, Hirata H, Akaoka H. (1997) Local opiate withdrawal in locus coeruleus in vivo. Brain Res 765:331–336 [DOI] [PubMed] [Google Scholar]
- Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. (2004) Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci 7:118–125 [DOI] [PubMed] [Google Scholar]
- Brinkerhoff CJ, Traynor JR, Linderman JJ. (2008) Collision coupling, crosstalk, and compartmentalization in G-protein coupled receptor systems: can a single model explain disparate results? J Theor Biol 255:278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabre M, Deterre P, Antonny B. (2009) The apparent cooperativity of some GPCRs does not necessarily imply dimerization. Trends Pharmacol Sci 30:182–187 [DOI] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, Osborne PB, Bellchambers CE. (1997) Where is the locus in opioid withdrawal? Trends Pharmacol Sci 18:134–140 [DOI] [PubMed] [Google Scholar]
- Connor M, Vaughan CW, Chieng B, Christie MJ. (1996) Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus neurones in vitro. Br J Pharmacol 119:1614–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. (2001) A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98–101 [DOI] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, O'Dowd BF. (2000) Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem 275:26128–26135 [DOI] [PubMed] [Google Scholar]
- Gold MS, Redmond DE, Jr, Kleber HD. (1978) Clonidine blocks acute opiate-withdrawal symptoms. Lancet 2:599–602 [DOI] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. (2004) A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 101: 5135–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeser D, Neubig RR. (1993) Compartmentation of receptors and guanine nucleotide-binding proteins in NG108–15 cells: lack of cross-talk in agonist binding among the alpha 2-adrenergic, muscarinic, and opiate receptors. Mol Pharmacol 43:434–443 [PubMed] [Google Scholar]
- Hall RA, Lefkowitz RJ. (2002) Regulation of G protein-coupled receptor signaling by scaffold proteins. Circ Res 91:672–680 [DOI] [PubMed] [Google Scholar]
- Hine B, Friedman E, Torrelio M, Gershon S. (1975) Morphine-dependent rats: blockade of precipitated abstinence by tetrahydrocannabinol. Science 187: 443–445 [DOI] [PubMed] [Google Scholar]
- Hur EM, Kim KT. (2002) G protein-coupled receptor signalling and cross-talk: achieving rapidity and specificity. Cell Signal 14:397–405 [DOI] [PubMed] [Google Scholar]
- Ivanov A, Aston-Jones G. (2001) Local opiate withdrawal in locus coeruleus neurons in vitro. J Neurophysiol 85:2388–2397 [DOI] [PubMed] [Google Scholar]
- Jordan BA, Gomes I, Rios C, Filipovska J, Devi LA. (2003) Functional interactions between mu opioid and alpha 2A-adrenergic receptors. Mol Pharmacol 64:1317–1324 [DOI] [PubMed] [Google Scholar]
- Kotlińska J, Suder P, Legowska A, Rolka K, Silberring J. (2000) Orphanin FQ/nociceptin inhibits morphine withdrawal. Life Sci 66:PL119–PL123 [DOI] [PubMed] [Google Scholar]
- Kotlinska J, Wichmann J, Rafalski P, Talarek S, Dylag T, Silberring J. (2003) Non-peptidergic OP4 receptor agonist inhibits morphine antinociception but does not influence morphine dependence. Neuroreport 14:601–604 [DOI] [PubMed] [Google Scholar]
- Lameh J, Eiger S, Sadée W. (1992) Interaction among mu-opioid receptors and alpha 2-adrenoceptors on SH-SY5Y human neuroblastoma cells. Eur J Pharmacol 227:19–24 [DOI] [PubMed] [Google Scholar]
- Lee PH, McNutt RW, Chang KJ. (1993) A nonpeptidic delta opioid receptor agonist, BW373U86, attenuates the development and expression of morphine abstinence precipitated by naloxone in rat. J Pharmacol Exp Ther 267:883–887 [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA. (2004) Interactions between delta opioid receptors and alpha-adrenoceptors. Clin Exp Pharmacol Physiol 31:833–836 [DOI] [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA. (2006) mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol 148:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulière A, Gaibelet G, Millot C, Mazères S, Lopez A, Salomé L. (2006) Diffusion of the mu opioid receptor at the surface of human neuroblastoma SH-SY5Y cells is restricted to permeable domains. FEBS Lett 580:5227–5231 [DOI] [PubMed] [Google Scholar]
- Scavone JL, Mackie K, Van Bockstaele EJ. (2010) Characterization of cannabinoid-1 receptors in the locus coeruleus: relationship with mu-opioid receptors. Brain Res 1312:18–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI. (2009) Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137:1148–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Vogel Z, Sarne Y. (2000) Opioid and cannabinoid receptors share a common pool of GTP-binding proteins in cotransfected cells, but not in cells which endogenously coexpress the receptors. Cell Mol Neurobiol 20:291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CB, Moises HC, Spengler RN, Hollingsworth PJ. (1989) Changes in alpha 2-adrenoceptor number and function in brains of morphine-dependent rats. Eur J Pharmacol 161: 111–119 [DOI] [PubMed] [Google Scholar]
- Stickle D, Barber R. (1992) The encounter coupling model for beta-adrenergic receptor/GTP-binding protein interaction in the S49 cell. Calculation of the encounter frequency. Biochem Pharmacol 43:2015–2028 [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. (2006) An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther 319:1–7 [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM, Levitzki A. (1978) Mode of coupling between the beta-adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry 17:3795. [DOI] [PubMed] [Google Scholar]
- Traynor J. (2010) Regulator of G protein-signaling proteins and addictive drugs. Ann NY Acad Sci 1187:341–352 [DOI] [PubMed] [Google Scholar]
- Wang HL, Hsu CY, Huang PC, Kuo YL, Li AH, Yeh TH, Tso AS, Chen YL. (2005) Heterodimerization of opioid receptor-like 1 and mu-opioid receptors impairs the potency of mu receptor agonist. J Neurochem 92:1285–1294 [DOI] [PubMed] [Google Scholar]
- Watts VJ. (2002) Molecular mechanisms for heterologous sensitization of adenylate cyclase. J Pharmacol Exp Ther 302:1–7 [DOI] [PubMed] [Google Scholar]
- Yu E, Miotto K, Akerele E, Montgomery A, Elkashef A, Walsh R, Montoya I, Fischman MW, Collins J, McSherry F, et al. (2008) A phase 3 placebo-controlled, double-blind, multi-site trial of the alpha-2-adrenergic agonist, lofexidine, for opioid withdrawal. Drug Alcohol Depend 97:158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Liu R, LaPlant Q, Xiao G, Renthal W, Chan GC, Storm DR, Aghajanian G, Nestler EJ. (2008) Distinct roles of adenylyl cyclases 1 and 8 in opiate dependence: behavioral, electrophysiological, and molecular studies. Biol Psychiatry 63:1013–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadina JE, Harrison LM, Ge LJ, Kastin AJ, Chang SL. (1994) Differential regulation of mu and delta opiate receptors by morphine, selective agonists and antagonists and differentiating agents in SH-SY5Y human neuroblastoma cells. J Pharmacol Exp Ther 270:1086–1096 [PubMed] [Google Scholar]