

Abstract
Background & Aims
Corticosteroids are now widely accepted as a treatment for autoimmune pancreatitis (AIP). However, the molecular mechanism by which steroid treatment improves AIP remains largely unknown. The aim of this study was to elucidate cellular mechanisms by which corticosteroids improve both pancreatic exocrine function and histopathology in AIP.
Methods
Pancreatic exocrine function was evaluated by the secretin-stimulated function test and pancreatic biopsy specimens were processed for histologic analysis at the time of diagnosis and 3 months after initiation of steroid treatment. Expression and localization of proteins was assayed by immunohistochemistry. Analysis of immunoglobulin (Ig)G4-positive plasma cells was used to verify inflammation in AIP.
Results
The number of IgG4-positive plasma cells in pancreatic sections was decreased by steroid treatment, indicating reduced inflammation. Fluid, bicarbonate (HCO3−), and digestive enzyme secretions all were impaired in most patients with AIP. Corticosteroids improved both HCO3− and digestive enzyme secretion. A large fraction of the cystic fibrosis transmembrane conductance regulator (CFTR), which plays a central role in pancreatic duct HCO3− secretion, was mislocalized to the cytoplasm of duct cells before treatment. Corticosteroids corrected the localization of CFTR to the apical membrane, accounting for the improved HCO3− secretion. Steroid treatment resulted in regeneration of acinar cells, accounting for restored digestive enzyme secretion.
Conclusions
Corticosteroids reduce inflammation and restore both digestive enzyme and HCO3− secretion in patients with AIP by regenerating acinar cells and correcting CFTR localization in pancreatic duct cells. Mislocalization of CFTR may explain aberrant HCO3− secretion in other forms of pancreatitis.
Keywords: Exocrine Function Test, CFTR, Aquaporin Water Channel, CD133
Autoimmune pancreatitis (AIP) is a relatively new entity in chronic pancreatitis and is characterized by diffuse swelling of the pancreas and a high serum immunoglobulin (Ig)G4 concentration.1 Histologic examination reveals a replacement of acinar cells by inflammatory cells and fibrotic tissue and an intense inflammatory cell infiltration in periductal lesions, suggesting that duct cells are the principal target of the immunologic responses in these patients.2 Oral corticosteroids recently have become established as a treatment of AIP, not only for the swelling of the gland but also for improved histology.3 However, despite recent advances in establishing the clinical characteristics of the disease and treatment of AIP, it remains largely unsolved how steroids affect the inflamed pancreas, decrease tissue damage, and improve pancreatic exocrine function.
The exocrine pancreas is composed of 2 main cell types: acinar and duct cells. Acinar cells secrete digestive enzymes and duct cells secrete bicarbonate (HCO3−)-rich fluid. Ductal fluid and HCO3− secretion are a critical function of the pancreas for inactivation of digestive enzymes and alkalinization of the pancreatic juice. Pancreatic ductal function can be evaluated only by the secretin-stimulated pancreatic function test—a direct analysis of pancreatic juice collected in the duodenum. Ductal fluid and HCO3− secretion is governed by the cystic fibrosis transmembrane conductance regulator (CFTR),4 a cyclic adenosine monophosphate–regulated chloride channel expressed exclusively in the apical plasma membrane of pancreatic duct cells. CFTR regulates ductal HCO3− secretion by either regulating chloride (Cl−)/HCO3− exchangers, the SLC26 transporters family,5 or directly permeating HCO3− flux across the epithelia when luminal Cl− becomes low.6,7 Indeed, cystic fibrosis (CF)-associated mutations in CFTR that impair either channel function8 or targeting of CFTR to the plasma membrane cause aberrant HCO3− secretion by the pancreatic duct.9
By using n-benzoyl-l-tyrosyl-p-aminobenzoic acid excretion tests, it has been documented that steroid therapy restores pancreatic exocrine function by partially regenerating acinar cells that are replaced by fibrotic tissue.10,11 However, the time course of regeneration of acinar cells by treatment with corticosteroids and its cellular mechanism remain unknown.
In the present study, we evaluated both ductal and acinar cell function at the same time in patients with autoimmune pancreatitis using the classic secretin test. Exocrine function was severely impaired in AIP and a 3-month steroid treatment (short-term) improved HCO3− and digestive enzyme secretion, indicating that steroids improve both ductal and acinar cell function. Moreover, 12-month steroid therapy (long-term) further improved digestive enzyme secretion compared with the short-term treatment. Histologic examination of endoscopic ultrasound–guided trucut biopsies revealed that a large fraction of CFTR is mislocalized to the cytoplasm of pancreatic duct cells in AIP. Most notably, steroids restored mislocalized CFTR to the luminal membrane. Importantly, sequencing the entire open reading frame and flanking intronic regions in CFTR of AIP patients revealed no major mutations. Hence, aberrant HCO3− secretion in AIP patients is not caused by mutations in CFTR, but rather by mislocalization of CFTR that was corrected with corticosteroids. An increase in the number of acinar cells in pancreatic tissues observed after steroid treatment indicate that recovery of digestive enzyme secretion was owing mostly to regeneration of acinar cells. These novel findings suggest that steroids affect both duct and acinar cells in AIP, repair cellular damage, and improve pancreatic exocrine function. It will be of particular interest to examine the effect of steroids on similar parameters in other forms of acute and chronic pancreatitis.
Material and Methods
Patients
We studied 21 patients with definite AIP who came to Aichi Cancer Center Hospital or Nagoya University Hospital during the period from 1992 to 2008. Patients met the 2006 revised Japanese clinical diagnostic criteria for AIP12 as follows: diffuse swelling of the pancreas, irregular narrowing of the main pancreatic duct, and a positive test for autoantibodies or a high IgG (≥1800 mg/dL)/IgG4 concentration (≥135 mg/dL).12 Patients who did not fulfill clinical diagnostic criteria were diagnosed by pancreatic biopsy with a full spectrum of lymphoplasmacytic sclerosing pancreatitis, such as a dense lymphoplasmacytic infiltrate, storiform fibrosis, acinar atrophy, and obliterative phlebitis.13,14 All pancreatic tissues obtained had no granulocytic epithelial lesion.2 Patients were examined functionally by the secretin test and histologically before and after initiation of corticosteroids when patients agreed to examinations and tests.
A standard protocol for oral corticosteroids was used accordingly: prednisolone at 30 mg/day for a week as an initial dose, 20 mg/day for a second week, 10 mg/day for 4 additional weeks, and 5 mg/day as a maintenance dose throughout the observation period.
Chronic Pancreatitis With Other Etiologies
Surgical sections of 6 patients with alcoholic pancreatitis (>60 g of daily ethanol consumption for more than 10 years) and 2 patients with obstructive pancreatitis distal to a pancreatic carcinoma were used for histologic examination. Three TCB samples without either full spectrum of pathologic features as lymphoplasmacytic sclerosing pancreatitis or as granulocytic epithelial lesions were used as idiopathic chronic pancreatitis. Four surgically resected pancreatic tissues from patients with other than pancreatic diseases were used as control.
Evaluation of Pancreatic Exocrine Function
Pancreatic exocrine function was evaluated by the secretin test as before.15 Detailed procedures are described in the Supplementary Materials and Methods section.
Pancreatic Biopsy
Written informed consent was obtained from each patient before the procedure. To exclude malignancy, all pancreatic biopsies were performed at Aichi Cancer Center Hospital and the procedure was approved by the ethics committee of the hospital. Under visual guidance of endoscopic ultrasonography (GF-UCT240; Olympus, Tokyo, Japan),14 pancreatic tissues were obtained from the body of the pancreas using a 19-gauge trucut biopsy (TCB) needle (Quick-core, Cook Medical Inc, Bloomington, IN). In some cases, when enough tissue material could not be obtained by a TCB needle, fine-needle aspiration biopsies using a 22-gauge needle were performed. Pancreatic biopsies were repeated at 3 months after the first biopsy to further exclude malignancy.
Immunohistochemistry and Histologic Scoring
Biopsy samples or surgically resected tissues were fixed in 10% formalin and embedded in paraffin. Sections were deparaffinized, permeabilized, and used for immunohistochemistry.16 Detailed procedures for immunohistochemistry and histologic scoring are described in the Supplementary Materials and Methods section.
Statistical Analysis
Statistical analysis was performed with the Student's paired t test. Differences with a P value of less than .05 were considered statistically significant. All values are expressed as mean ± standard error of the mean.
Results
Corticosteroids Reverse Impaired HCO3− Secretion
Table 1 summarizes the serologic characteristics of 21 patients with AIP. Nineteen (90.5%) patients showed positive IgG4 values of more than 135 mg/dL.1 Figure 1A shows the exocrine function evaluated by the secretin test. The secreted volume and maximum HCO3− concentration in response to secretin injection report ductal function, whereas total amylase output reports acinar cell function. In 17 patients examined, the degree of exocrine dysfunction differed significantly among patients with AIP. Fourteen (82%) patients showed reduced pancreatic exocrine function in at least 2 parameters before treatment, including maximum HCO3− concentration, which is consistent with definite chronic pancreatitis.17
Table 1. Serologic Characteristics of the 21 Patients With AIP in This Study.
| Case no. | Age/sex | IgG, mg/dL | IgG4, mg/dL | Auto antibodies | Other organ involvement | Tissue (n = 19) |
Second biopsy (n = 7) |
Exocrine function tests | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-treatment | Post-treatment | Pre-treatment | Post-treatment | Pre-treatment (n = 17) |
3 mo (n = 8) |
1 y (n = 3) |
||||||
| 1 | 44/M | 2770 | 1840 | 741 | 432 | (−) | DU | Resection | + | |||
| 2 | 71/M | 2304 | N/A | 177 | N/A | ANA | SA | Resection | + | |||
| 3 | 61/M | 2820 | 1350 | 1590 | 710 | (−) | Hilar LN | N/A | + | + | + | |
| 4 | 70/M | 1300 | N/A | 183 | N/A | (−) | FNA | + | ||||
| 5 | 59/M | N/A | N/A | 185 | N/A | (−) | Resection | |||||
| 6 | 68/M | 1455 | N/A | 351 | N/A | (−) | SC | FNA | + | + | ||
| 7 | 72/M | 1823 | 1180 | 366 | 185 | RF | SC | TCB | TCB | + | + | + |
| 8 | 76/M | 1604 | 1407 | 227 | 165 | RF | TCB | TCB | + | + | ||
| 9 | 68/M | 2346 | 1663 | 640 | 277 | (−) | Mediastinal LN | TCB | TCB | + | + | + |
| 10 | 70/M | 3180 | 1767 | 419 | 223 | ANA | SC | TCB | + | |||
| 11 | 75/M | 1274 | 988 | 79 | 56 | RF | SC | N/A | + | |||
| 12 | 61/M | 1669 | 1251 | 399 | 266 | (−) | TCB | + | + | |||
| 13 | 66/M | 2060 | 1445 | 342 | 214 | (−) | SA, R fibrosis | TCB | TCB | + | + | |
| 14 | 60/M | 3650 | 1627 | 1550 | 630 | RF | Prostatitis | TCB | + | |||
| 15 | 58/M | 1355 | 1128 | 201 | 136 | (−) | SC | TCB | TCB | |||
| 16 | 55/F | 1404 | 1007 | 127 | 50.5 | (−) | TCB | TCB | ||||
| 17 | 75/M | 4091 | 1632 | 1070 | 348 | ANA | SC | TCB | + | + | ||
| 18 | 41/F | 1545 | 823 | 414 | 218 | RF | TCB | TCB | ||||
| 19 | 67/M | 1874 | 1289 | 181 | 32.7 | ANA | R fibrosis | FNA | + | |||
| 20 | 61/M | 1778 | 1497 | 259 | N/A | ANA | TCB | + | ||||
| 21 | 55/M | 1551 | N/A | 549 | N/A | (−) | SC | FNA | + | |||
NOTE. Total serum IgG and IgG4 were examined at the time of diagnosis (Pre-treatment) and 3 months after initiation of oral corticosteroids (Post-treatment).
ANA, antinuclear antibody; DU, duodenal ulcer; FNA, fine-needle aspiration biopsy; LN, lymph node swelling; N/A, not available; R fibrosis, retroperitoneal fibrosis; RF, rheumatoid factor; SA, sialoadenitis; SC, sclerosing cholangitis.
Figure 1.
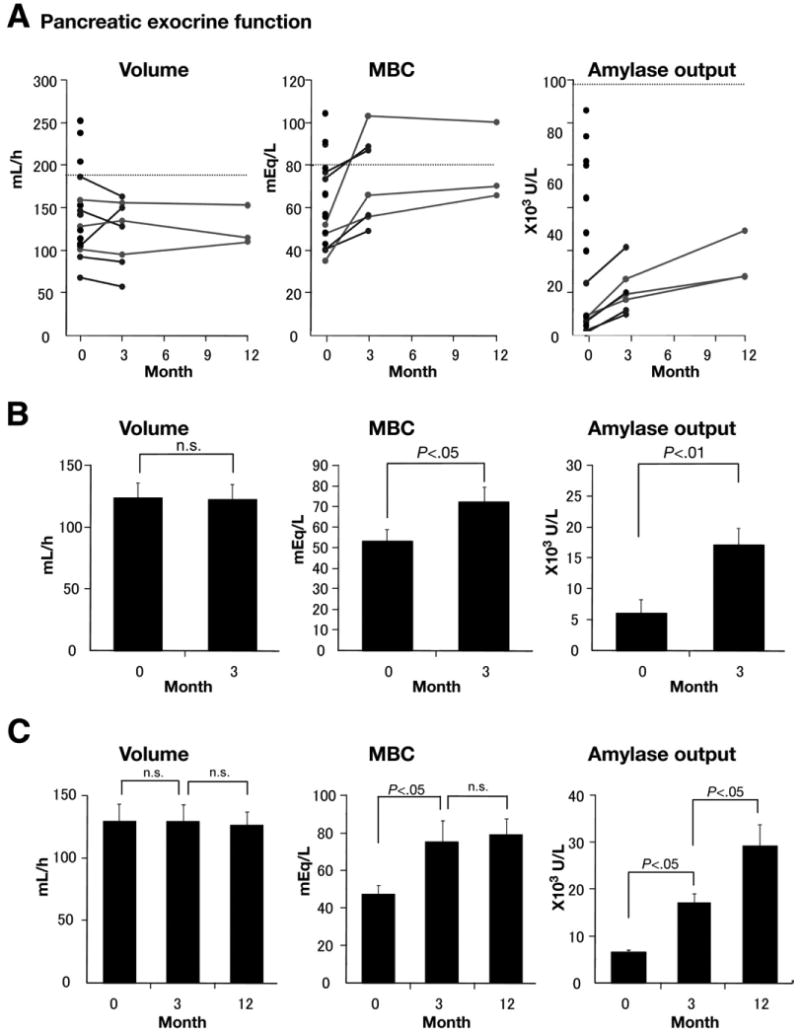
(A) Pancreatic exocrine function in AIP was examined by the secretin test. Tests were performed at the time of diagnosis (0 months), 3 months, and 12 months after initiation of corticosteroids. Lower limits of normal are marked by dashed lines. Black lines indicate change of exocrine function in patients whose function was evaluated at 0 and 3 months after initiation of corticosteroids. Gray lines indicate changes of exocrine function in patients whose exocrine function was evaluated at 0, 3, and 12 months. (a) Total secreted volume for an hour (normal, ≥183 mL/h). (b) Maximum HCO3− concentration (mbc) (normal, ≥80 mEq/L). (c) Total amylase output for an hour (normal, ≥99,000 U/h). (B) Change in pancreatic exocrine functions caused by short-term treatment. (a) Secreted volume was not altered by steroids (n = 8, mean ± standard error of the mean; at 0 months, 123.5 ± 12.9 mL/h; at 3 months, 122.1 ± 12.7 mL/h). (b) MBC (n = 7; at 0 months, 53.3 ± 5.8 mEq/L; at 3 months, 72.5 ± 7.2 mEq/L), and (c) total amylase output (n = 8; at 0 months, 6138 ± 2107 U/h; at 3 months, 17,143 ± 2710 U/h) were improved significantly by treatment. (C) Change of pancreatic exocrine functions at 0, 3, and 12 months after initiation of corticosteroids (n = 3). (a) Secretory volume and (b) MBC were not altered by prolonging treatment up to 12 months. (c) Amylase output is improved by short-term treatment (at 0 months, 6683 ± 444 U/h; at 3 months, 17,200 ± 1968 U/h) and further by long-term treatment (at 12 months, 28,962 ± 4894 U/h).
To examine whether corticosteroids affect pancreatic exocrine function in AIP, secretin tests were repeated before and 3 months after initiation of treatment. Unlike a previous report,18 steroids did not restore secreted volume (Figure 1B; n = 8; NS). However, a 3-month administration of steroids to patients with AIP significantly increased the maximum HCO3− concentration (n = 7; P < .05) and total amylase output in the secreted fluid (n = 8; P < .01), suggesting that treatment improves both ductal and acinar cell function.
Pancreatic exocrine function in 3 patients with AIP was evaluated 3 times: before, 3 months (short-term) after initiation of treatment, and 12 months (long-term) after initiation of treatment (Figure 1C), to test whether long-term corticosteroids further improve pancreatic exocrine function. Long-term corticosteroids had no additional effect on both secreted volume (NS) and maximum HCO3− concentration (NS), but further improved amylase secretion when compared with short-term treatment (P < .05).
These results indicate that short-term corticosteroids are sufficient for recovery of HCO3− secretion by pancreatic ducts. However, functional recovery for digestive enzyme secretion by acinar cells requires steroid administration for a longer period of time.
Regeneration of Acini and Disappearance of Fibrosis by Corticosteroids
To define cellular mechanisms of the restored HCO3− secretion by duct cells and recovery of amylase secretion by acinar cells, we first examined pancreatic tissues by H&E and Masson's trichrome staining. In AIP, extensive disappearance of acini and their replacement by fibrotic tissue were evident (Figure 2B, D, and E) when compared with normal subjects (Figure 2A and C). A 3-month steroid treatment repaired the tissue damage and resulted in partial regeneration of acinar cells and disappearance of fibrosis (Figure 2F). Regeneration of exocrine cells can account for increased amylase output owing to steroid administration. Most of the nuclei lining the basal membrane of duct cells appeared to be deformed before treatment (insets in Figure 2B and E), and the shape of duct cells and their nuclei was improved by treatment (inset in Figure 2F). However, these histologic findings could not explain aberrant fluid and HCO3− secretion by pancreatic ducts before treatment and improvement of HCO3− secretion in pancreatic juice by corticosteroids.
Figure 2.
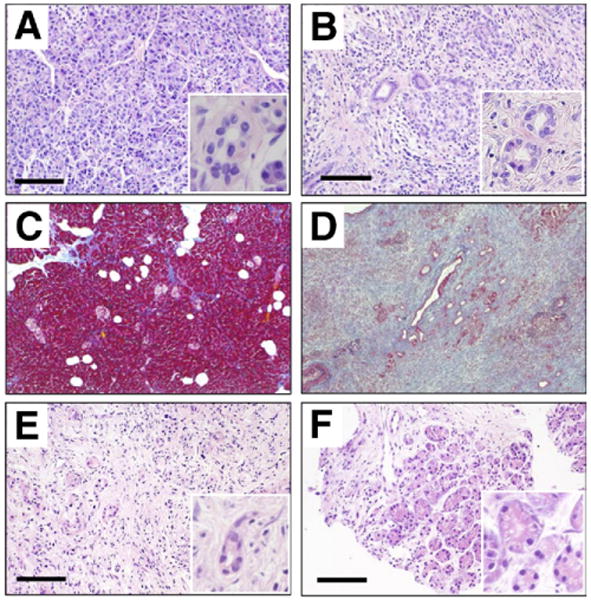
Pancreatic sections were stained with H&E. (A) Normal subject. (B) AIP, surgically resected tissue. Masson's trichrome staining of (C) normal pancreas and (D) AIP. H&E staining of specimen (E) before and (F) 3 months after initiation of steroid treatment. (E and F) Sections were obtained from the same patient. Bars, 100 μm. Insets show images at a higher magnification.
Up-regulation But no Effect of Corticosteroids on Aquaporin 1 Expression and Localization in Pancreatic Ducts
CFTR regulates overall pancreatic ductal fluid and HCO3− secretion5–7 and fluid secretion requires aquaporin 1 (AQP1),19 which is the dominant water channel in the human pancreatic duct.20 Therefore, to elucidate the underlying mechanism of aberrant fluid secretion in AIP, we first examined the subcellular localization of AQP1 water channel in pancreatic ducts.
In normal subjects, AQP1 immunolabeling was found mainly in the apical and lateral plasma membranes, but to some extent also in basal membranes from centroacinar cells to large interlobular ducts (Figure 3A). AQP1 also was detected in capillaries and other small vessels. In contrast to what is expected from the impaired fluid secretion (Figure 1A), expression of AQP1 was increased markedly in AIP (Figure 3B and C). AQP1 immunoreactivity appeared not only to be up-regulated on the plasma membrane but also in the cytoplasm of pancreatic duct cells. Short-term treatment (Figure 3D) reversed neither reduced fluid secretion (Figure 1B) nor increased AQP1 expression in pancreatic ducts (Figure 3C). The increased expression of AQP1 likely is compensatory in response to reduced fluid secretion.
Figure 3.
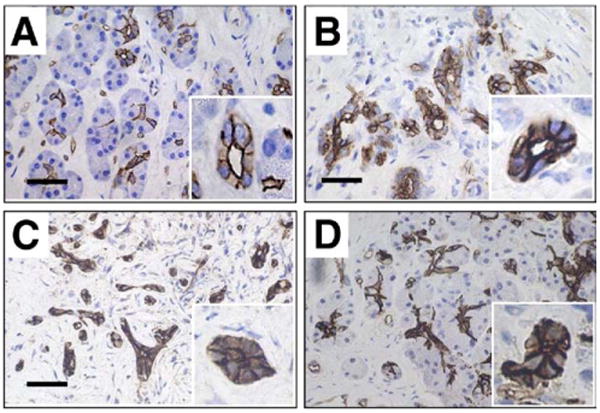
Immunolocalization of AQP1 in the pancreas. (A) Control subject. (B) AIP, surgically resected. Biopsy specimen (C) before treatment and (D) after treatment. (C and D) Sections were obtained from the same patient. The pathologic changes were seen in all sections examined. Bars, 20 μm. Insets show images at a higher magnification.
Cytoplasmic Mislocalization of CFTR and Relocation to the Apical Plasma Membrane by Corticosteroids
CFTR governs overall ion transport across most epithelia and plays a central role in pancreatic ductal HCO3− secretion. Therefore, we next examined the subcellular localization of CFTR with the anti-CFTR antibody, M3A7. Remarkably, a large fraction of CFTR was found in the cytoplasm of pancreatic ducts of AIP patients, probably owing to partial mistargeting of protein to the plasma membrane domain. In normal subjects, CFTR immunolabeling is confined exclusively to the apical membrane of intercalated, intralobular, and small interlobular ducts (Figure 4A). By contrast, in patients with AIP a large fraction of the CFTR immunolabeling was found in the cytoplasm of pancreatic ducts and apical labeling appeared less intense compared with the labeling in normal pancreatic ducts (Figure 4B and C). Strikingly, this mislocalization largely was corrected by corticosteroid treatment. Most of the protein again was confined to the apical plasma membrane after 3 months of treatment (Figure 4D). To quantify these changes, slides were scored by a person who was blinded to the source of tissue (see Materials and Methods section). Figure 4E shows that the histologic score was improved markedly by corticosteroids. These findings indicate that restoration of CFTR localization by the anti-inflammatory treatment most likely accounts for improved pancreatic HCO3− secretion (Figure 1).
Figure 4.
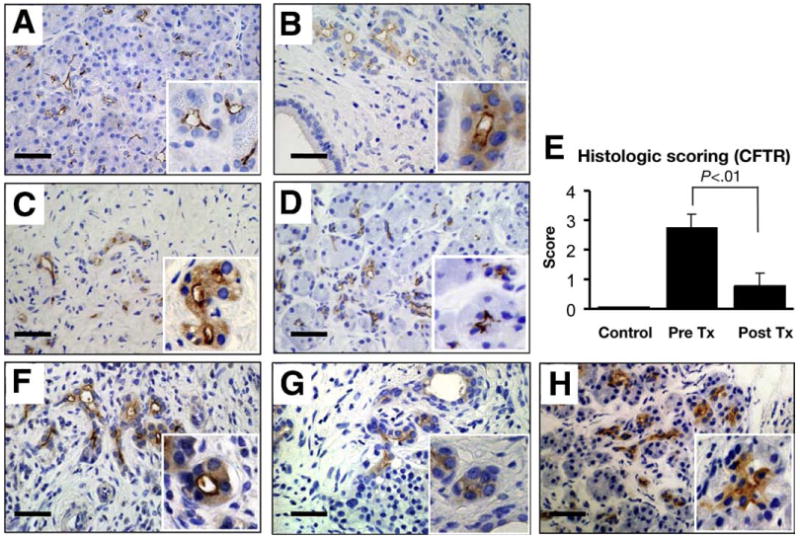
Immunolocalization of CFTR in the pancreas. (A) Normal subject. (B) AIP, surgically resected. Biopsy specimen (C) before and (D) after treatment. (C and D) Sections are from the same patient. (E) Pancreatic sections obtained from normal subjects (n = 4), from AIP patients at 0 (n = 7) or 3 months (n = 7) treatment were scored at 0 (none), 1 (slight), 2 (moderate), and 3 (severe) for the degree of cytoplasmic staining of CFTR in pancreatic ducts. CFTR localization also was examined in the pancreas from the patients with (F) alcoholic, (G) obstructive, and (H) idiopathic chronic pancreatitis. Each panel represents 6 alcoholic, 2 obstructive, and 3 idiopathic cases of pancreatitis, respectively. Bars, 20 μm. Insets show images at a higher magnification.
CFTR Mislocalization in Other Forms of Chronic Pancreatitis
Impaired HCO3− secretion is a hallmark of pancreatic exocrine dysfunction in all forms of chronic pancreatitis.17 To investigate whether partial mislocalization of CFTR to the cytoplasm also can be found in other forms of chronic pancreatitis, we examined the expression and subcellular localization of CFTR in 6 patients with alcoholic pancreatitis (Figure 4F), 2 patients with obstructive pancreatitis (Figure 4G), and 3 patients with idiopathic chronic pancreatitis (Figure 4H). Intriguingly, mislocalization of CFTR was found in all forms of chronic pancreatitis examined, similar to the findings in AIP.
Numbers of IgG4-Positive Plasma Cells Are Decreased by Corticosteroids
In previous studies, increased numbers of IgG4-positive plasma cells in the pancreas have been established as a marker for the diagnosis of AIP. Because serum IgG4 levels in patients with AIP were decreased markedly by corticosteroids, IgG4 immunostains using biopsy specimens obtained before and after corticosteroids were performed. IgG4-positive cells were abundant in the tissues before treatment (Figure 5A). In contrast, the number of IgG4-positive cells was reduced remarkably in response to corticosteroid treatment (Figure 5B). The decrease in the numbers of IgG4-positive cells was in good agreement with the improved CFTR localization (Figures 5C and 4E). These findings suggest a relation between reduced inflammation and restored localization of CFTR to the luminal membrane.
Figure 5.
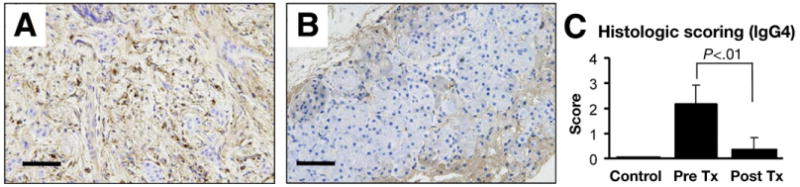
Immunohistochemical staining for IgG4. (A) Marked IgG4-positive plasma cell infiltrates are present in tissue before treatment. (B) The number of IgG4-positive plasma cells was decreased significantly after treatment. (C) Pancreatic sections obtained from normal subjects (n = 4), and AIP patients at 0 (n = 7) or 3 months (n = 7) treatment were scored at 0 (none), 1 (slight), 2 (moderate), and 3 (severe) for extent of IgG4-positive plasma cell infiltration.
CD133 Expression in the Human Pancreas
CD133, a hematopoietic stem/progenitor cell marker known as prominine-1, is reported to be a good candidate gene for the identification of pancreatic stem/progenitor cells.21,22 CD133 is expressed in the ducts of fetal and adult pancreas and in pancreatic cancers.23 CD133-positive pancreatic duct cells isolated from fetal pancreas can be differentiated into insulin-containing endocrine or amylase-containing exocrine cells, suggesting that CD133-positive duct cells could be the source of progenitor cells in the development of the fetal organ. The finding in the present study of regeneration of acinar cells by steroids in AIP raised the question of whether acinar cell regeneration is associated with CD133-positive ductal cells. To address this question, we used anti–prominine-1 antibody and examined CD133 expression and localization in normal pancreas and AIP. Figure 6 shows the CD133 expression in adult human pancreas. In normal subjects, CD133 expression was confined to the apical membrane domain of all cells from intercalated to small interlobular ducts (Figure 6A). CD133 also was detected in the apical membrane of small interlobular ducts in tissues with AIP where most of the acini had disappeared and were replaced by fibrotic tissue (Figure 6B). By contrast, with the localization of CFTR, CD133 immunolabeling was detected exclusively in the apical plasma membrane of residual ductal trees and no cytoplasmic staining was found (Figure 6B). Short-term treatment caused clustered regeneration of acinar cells and CD133 immunolabeling again was detected in the apical membrane in most of the ductal structures surrounded by regenerated acinar cells (Figure 6C). Significantly, no CD133 labeling was detected in the residual fibrotic area even after treatment, where no acinar cell regeneration was found (Figure 6D). These findings are in line with the hypothesis that CD133 is a good marker for pancreatic cell differentiation and regeneration.
Figure 6.
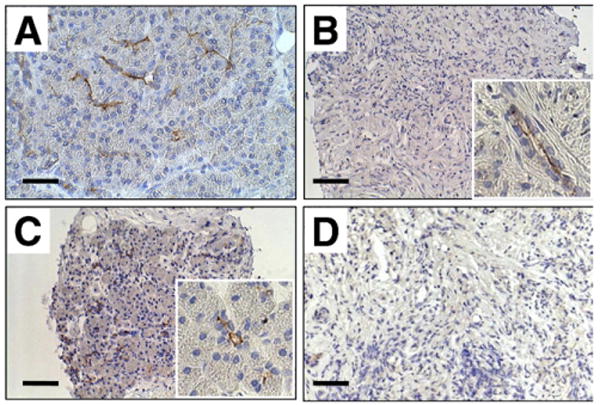
Immunoperoxidase labeling of CD133 (prominine-1) in the pancreas. (A) Normal subject. (B) Absence of acinar cells and massive fibrosis are evident before treatment. CD133 was detected in the apical membrane of residual pancreatic ducts. (C) CD133 and fibrosis after short-term steroid treatment. (D) CD133-positive cells are absent and acinar cell regeneration did not occur. Bars, 20 μm. Insets show images at a higher magnification.
Discussion
The present study shows that pancreatic fluid and HCO3− secretion by the duct and digestive enzyme secretion by acinar cells are severely compromised in patients with AIP. Treatment of these patients with corticosteroids repaired the pancreatic parenchymal damage, improved ductal HCO3− secretion, and improved digestive enzyme secretion by acinar cells. The most remarkable finding of the present study was that the aberrant pancreatic ductal function in AIP and its reversal by corticosteroids could be traced to partial mislocalization of CFTR to the cytoplasm and correction of its targeting to the duct cells apical membranes.
CFTR is an epithelial Cl− channel expressed in the apical plasma membrane of intercalated and small interlobular pancreatic ducts. The role of CFTR in HCO3− secretion is evident from the lack of pancreatic HCO3− secretion in CF.9 In fact, CFTR governs the overall ductal HCO3− secretory process either by virtue of its regulation of major HCO3− transporters in the apical membrane of the pancreatic duct and other epithelia5 or by directly conducting HCO3− when luminal Cl− is low.6,7
Recent work showed that knockdown of CFTR by RNA interference markedly reduced fluid secretion by the mouse pancreatic duct.24 Therefore, we postulated that reduction in CFTR function may underlie the reduced HCO3− secretion observed in patients with chronic pancreatitis. Indeed, we discovered that CFTR is mislocalized significantly in pancreatic duct cells in all forms of chronic pancreatitis examined. Hence, reduction of the amount of CFTR protein expressed in the apical membrane can explain the aberrant HCO3− secretion by the pancreatic duct in these patients. Importantly, reversal of CFTR mislocalization and restoration of HCO3− secretion by steroid therapy in AIP establish a strong connection between the membrane localization of CFTR, aberrant HCO3− secretion in vivo, and the severity of exocrine dysfunction.
A somewhat unexpected finding was that the increased ductal HCO3− secretion by steroid treatment was not accompanied by increased volume (Figure 1), in particular in view of the improved acinar function that should supply the duct with more NaCl-rich fluid. There are several potential explanations for these findings. Paracellular Na+ flux is vital for ductal fluid secretion. It is possible that, as in other forms of pancreatitis,25 tight junction function is compromised in AIP and was not corrected by steroid treatment to restore fluid secretion. Another possibility is that the inflamed pancreas in AIP may secrete more fluid than expected from the damage caused by the pancreatitis, and correction of the disease restored a more normal fluid secretion, but the sum of the effects resulted in no apparent change in total volume.
The present findings indicate that CFTR plays a more prominent role in pancreatic ductal function in patients with chronic pancreatitis than previously appreciated. In previous studies, a link between CFTR mutations and pancreatitis has been established by showing a higher frequency of CFTR mutations in idiopathic pancreatitis than in normal subjects in the Caucasian population.26,27 Furthermore, the risk for pancreatitis is higher in CF carriers than in subjects without apparent mutations in CFTR.28 However, the previous studies could not establish a link between CFTR mistargeting and pancreatic ductal dysfunction because to the extent examined the mutations have a minor, if any, effect on CFTR Cl− channel activity. Thus, the present study examined the role of CFTR in pancreatic HCO3− secretion irrespective of mutations in CFTR, and implicated a role of CFTR in a disease (pancreatitis) that is not the result of mutations in CFTR, but rather is mediated by deranged handling of CFTR by epithelial cells. The mislocalization of CFTR in all forms of chronic pancreatitis examined likely was caused by inflammation of the pancreas. These findings should promote further studies to establish roles of the mistargeting of membrane proteins such as SLC26 transporters5,29 in secretory or absorbing defects in other inflammatory diseases in epithelia.
Because of the central role of CFTR in pancreatic HCO3− secretion and regulation of HCO3− transporters by CFTR, mistargeting of CFTR is expected to have multiple effects on ductal HCO3− secretory process. Hence, mistargeting of CFTR will result in the loss of apical membrane Cl− conductance and Cl− recirculation across the apical membrane,30 loss of CFTR-mediated HCO3− conductance at low luminal Cl−,6,7 loss of Cl−/HCO3− exchange by the apical SLC26 transporters that are activated by CFTR,5,29 and potentially loss of regulation of other apical membrane transporters regulated by CFTR and participating in HCO3− secretion.31 Hence, it is likely that the activity and perhaps localization of other apical membrane transporters are affected by mistargeting of CFTR. Nevertheless, mistargeting of CFTR in chronic pancreatitis reported here is the principal event leading to aberrant HCO3− secretion in the disease, whereas potential changes in the activity of other transporters are likely to be secondary to the change in mistargeting of CFTR.
In AIP, not only pancreatic ductal function was impaired, but also the ability to secrete digestive enzymes was compromised severely. This digestive enzyme malsecretion can be explained by the absence of acinar cells and their replacement by fibrotic tissue in the pancreas of AIP. Here, we have found that even short-term (3-month) steroid treatment results in partial regeneration of acinar cells and thereby recovery of digestive enzyme secretion. Moreover, although the number of patients examined was small (n = 3), when prednisolone at 5 mg was given for 12 months, further improvement in digestive enzyme secretion was found. This probably reflects the time required for acinar cell regeneration, which appears to occur at a slower rate than correction of CFTR localization. Whether a treatment longer than 1 year will result in further improvement of digestive enzyme secretion is being examined.
Chronic pancreatitis with any etiology is a progressive disease that damages pancreatic parenchyma, resulting in tissue damage that is thought to be irreversible by any known treatment.32 All treatments available for chronic pancreatitis are aimed at alleviating symptoms of the disease and none is aimed at either halting the progressive inflammation, or correcting tissue damage. The effectiveness of short-term steroid treatment in correcting mislocalized CFTR and in promoting regeneration of acinar cells in AIP raise the prospect of careful administration of steroids as a new treatment to reverse dysfunction and tissue damage to other forms of chronic pancreatitis.
Another potential implication of our findings is that mislocalization of CFTR can be a contributing factor to the severity of secretory or absorbing defects seen in other inflammatory diseases of epithelia. In Sjögren syndrome, an inflammation of salivary and lacrimal glands leads to reduced fluid secretion and altered electrolyte composition of the secreted fluid,33 suggesting abnormal ductal function. In inflammatory bowel diseases, such as Crohn's disease and ulcerative colitis, diarrhea and malnutrition are the primary symptoms, suggesting abnormal secretory and absorbing functions of the small and large intestinal epithelium.34 In CF much heterogeneity exists between genotype and phenotype, in particular in CF lung disease. Inflammation caused by bacterial or viral infection determines disease severity in the CF lung.35 Consequently, anti-inflammatory drugs such as corticosteroids or non-steroidal anti-inflammatory drugs increasingly are prescribed to patients with CF, although it has not been established whether anti-inflammatory drugs reduce the disease severity.36 If inflammation causes partial mislocalization of CFTR in the lung epithelium, similar to that found in pancreatic ducts in chronic pancreatitis, a treatment with anti-inflammatory drugs may affect the membrane localization of CFTR, in particular CFTR mutations that reduce channel activity but reside in the plasma membrane. Hence, our findings suggest that an anti-inflammatory regimen may be beneficial in delaying the onset of diseases associated with aberrant HCO3− secretion, such as chronic pancreatitis, Sjögren syndrome, and CF.
Supplementary Material
Supplementary Table 1. Primers Used in this Study to Amplify CFTR Gene Fragments
Supplementary Table 2. CFTR Genotypes in Japanese Patients With Autoimmune Pancreatitis
Acknowledgments
The authors are deeply indebted to Mr K. Ishida (Aichi Cancer Center Hospital) for his expertise in immunohistochemistry, Mrs M Yamazaki (Nagoya university) for secretarial support, and Mrs M. Nakakuki (Nagoya University) for measuring the pancreatic juice samples.
Funding: This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology, the Ministry of Health, Labor and Welfare for intractable pancreatic diseases, Pancreas Research Foundation of Japan (S.B.H.K. and N.M.), Aichi D.R.G Foundation (S.B.H.K.), and The Japanese Foundation for Research and Promotion of Endoscopy (N.M.).
Abbreviations used in this paper
- AIP
autoimmune pancreatitis
- AQP1
aquaporin 1
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- TCB
trucut biopsy
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Supplementary Material: Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2010.01.001.
References
- 1.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 2.Kloppel G, Luttges J, Lohr M, et al. Autoimmune pancreatitis: pathological, clinical, and immunological features. Pancreas. 2003;27:14–19. doi: 10.1097/00006676-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Finkelberg DL, Sahani D, Deshpande V, et al. Autoimmune pancreatitis. N Engl J Med. 2006;355:2670–2676. doi: 10.1056/NEJMra061200. [DOI] [PubMed] [Google Scholar]
- 4.Choi JY, Muallem D, Kiselyov K, et al. Aberrant CFTR-dependent HCO3− transport in mutations associated with cystic fibrosis. Nature. 2001;410:94–97. doi: 10.1038/35065099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ko SB, Shcheynikov N, Choi JY, et al. A molecular mechanism for aberrant CFTR-dependent HCO3− transport in cystic fibrosis. EMBO J. 2002;21:5662–5672. doi: 10.1093/emboj/cdf580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishiguro H, Steward MC, Naruse S, et al. CFTR functions as a bicarbonate channel in pancreatic duct cells. J Gen Physiol. 2009;133:315–326. doi: 10.1085/jgp.200810122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shcheynikov N, Kim KH, Kim KM, et al. Dynamic control of cystic fibrosis transmembrane conductance regulator Cl−/HCO3− selectivity by external Cl−. J Biol Chem. 2004;279:21857–21865. doi: 10.1074/jbc.M313323200. [DOI] [PubMed] [Google Scholar]
- 8.Sheppard DN, Rich DP, Ostedgaard LS, et al. Mutations in CFTR associated with mild-disease-form Cl− channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- 9.Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999;79:S3–S22. doi: 10.1152/physrev.1999.79.1.S3. [DOI] [PubMed] [Google Scholar]
- 10.Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas. 2003;27:235–238. doi: 10.1097/00006676-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Song MH, Kim MH, Lee SK, et al. Regression of pancreatic fibrosis after steroid therapy in patients with autoimmune chronic pancreatitis. Pancreas. 2005;30:83–86. [PubMed] [Google Scholar]
- 12.Okazaki K, Kawa S, Kamisawa T, et al. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol. 2006;41:626–631. doi: 10.1007/s00535-006-1868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Notohara K, Burgart LJ, Yadav D, et al. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol. 2003;27:1119–1127. doi: 10.1097/00000478-200308000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno N, Bhatia V, Hosoda W, et al. Histological diagnosis of autoimmune pancreatitis using EUS-guided trucut biopsy: a comparison study with EUS-FNA. J Gastroenterol. 2009;44:742–750. doi: 10.1007/s00535-009-0062-6. [DOI] [PubMed] [Google Scholar]
- 15.Kitagawa M, Naruse S, Ishiguro H, et al. Evaluating exocrine function tests for diagnosing chronic pancreatitis. Pancreas. 1997;15:402–408. doi: 10.1097/00006676-199711000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Yatabe Y, Koga T, Mitsudomi T, et al. CK20 expression, CDX2 expression, K-ras mutation, and goblet cell morphology in a subset of lung adenocarcinomas. J Pathol. 2004;203:645–652. doi: 10.1002/path.1566. [DOI] [PubMed] [Google Scholar]
- 17.Homma T, Harada H, Koizumi M. Diagnostic criteria for chronic pancreatitis by the Japan Pancreas Society. Pancreas. 1997;15:14–15. doi: 10.1097/00006676-199707000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Ito T, Nakano I, Koyanagi S, et al. Autoimmune pancreatitis as a new clinical entity. Three cases of autoimmune pancreatitis with effective steroid therapy. Dig Dis Sci. 1997;42:1458–1468. doi: 10.1023/a:1018862626221. [DOI] [PubMed] [Google Scholar]
- 19.Ko SB, Naruse S, Kitagawa M, et al. Aquaporins in rat pancreatic interlobular ducts. Am J Physiol. 2002;282:G324–G331. doi: 10.1152/ajpgi.00198.2001. [DOI] [PubMed] [Google Scholar]
- 20.Burghardt B, Elkaer ML, Kwon TH, et al. Distribution of aquaporin water channels AQP1 and AQP5 in the ductal system of the human pancreas. Gut. 2003;52:1008–1016. doi: 10.1136/gut.52.7.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hori Y, Fukumoto M, Kuroda Y. Enrichment of putative pancreatic progenitor cells from mice by sorting for prominin1 (CD133) and platelet-derived growth factor receptor beta. Stem Cells. 2008;26:2912–2920. doi: 10.1634/stemcells.2008-0192. [DOI] [PubMed] [Google Scholar]
- 22.Koblas T, Pektorova L, Zacharovova K, et al. Differentiation of CD133-positive pancreatic cells into insulin-producing islet-like cell clusters. Transplant Proc. 2008;40:415–418. doi: 10.1016/j.transproceed.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Immervoll H, Hoem D, Sakariassen PØ, et al. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer. 2008;8:48. doi: 10.1186/1471-2407-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Soyombo AA, Shcheynikov N, et al. Slc26a6 regulates CFTR activity in vivo to determine pancreatic duct HCO3− secretion: relevance to cystic fibrosis. EMBO J. 2006;25:5049–5057. doi: 10.1038/sj.emboj.7601387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt M, Klonowski-Stumpe H, Eckert M, et al. Disruption of paracellular sealing is an early event in acute caerulein-pancreatitis. Pancreas. 2004;28:181–190. doi: 10.1097/00006676-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- 27.Cohn JA, Friedman KJ, Noone PG, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658. doi: 10.1056/NEJM199809033391002. [DOI] [PubMed] [Google Scholar]
- 28.Cohn JA, Neoptolemos JP, Feng J, et al. Increased risk of idiopathic chronic pancreatitis in cystic fibrosis carriers. Hum Mutat. 2005;26:303–307. doi: 10.1002/humu.20232. [DOI] [PubMed] [Google Scholar]
- 29.Ko SB, Zeng W, Dorwart MR, et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. 2004;6:343–350. doi: 10.1038/ncb1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang D, Shcheynikov N, Zeng W, et al. IRBIT coordinates epithelial fluid and HCO3− secretion by stimulating the transporters pNBC1 and CFTR in the murine pancreatic duct. J Clin Invest. 2009;119:193–202. doi: 10.1172/JCI36983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li C, Naren AP. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol Ther. 2005;108:208–223. doi: 10.1016/j.pharmthera.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Forsmark CE. Chronic pancreatitis. In: Feldman M, Friedman LS, Sleisenger MH, editors. Gastrointestinal and liver disease. 7th. Philadelphia: Saunders; 2002. pp. 943–969. [Google Scholar]
- 33.Vitali C, Moutsopoulos HM, Bombardieri S, The European Community Study Group on diagnostic criteria for Sjogren's syndrome Sensitivity and specificity of tests for ocular and oral involvement in Sjogren's syndrome. Ann Rheum Dis. 1994;53:637–647. doi: 10.1136/ard.53.10.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidler U, Lenzen H, Cinar A, et al. Molecular mechanisms of disturbed electrolyte transport in intestinal inflammation. Ann N Y Acad Sci. 2006;1072:262–275. doi: 10.1196/annals.1326.024. [DOI] [PubMed] [Google Scholar]
- 35.Chmiel JF, Konstan MW. Anti-inflammatory medications for cystic fibrosis lung disease: selecting the most appropriate agent. Treat Respir Med. 2005;4:255–273. doi: 10.2165/00151829-200504040-00004. [DOI] [PubMed] [Google Scholar]
- 36.Auerbach HS, Williams M, Kirkpatrick JA, et al. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet. 1985;28:686–688. doi: 10.1016/s0140-6736(85)92929-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Primers Used in this Study to Amplify CFTR Gene Fragments
Supplementary Table 2. CFTR Genotypes in Japanese Patients With Autoimmune Pancreatitis