

Abstract
This perspective surveys the history of- and recent advances in- metallacycle-mediated coupling chemistry of substituted alkenes. While the reaction of preformed metal–π complexes with ethylene was reported nearly 30 years ago, the generalization of this mode of bimolecular C–C bond formation to the regio- and stereoselective union of complex substrates has only recently begun to emerge. This perspective discusses early observations in this area, the challenges associated with controlling such processes, the evolution of a general strategy to overcome these challenges, and a summary of highly regio- and stereoselective convergent coupling reactions that are currently available by metallacycle-mediated cross-coupling with substituted alkenes.
1. Introduction
The chemical sciences are advancing at an impressive pace, with frequent reports describing novel patterns of reactivity and selectivity in organic chemistry. While contributions that impact synthesis at the tactical level are abundant, those that offer advances at the strategic level (via the establishment of novel bond constructions) are rare, yet have a unique and perhaps more significant impact on the evolution of chemical synthesis.1 Modern advances in this vein include asymmetric olefin oxidation,2 metal-catalyzed cross-coupling (Pd-, Ni-, and Cu- catalysis),3 and olefin metathesis chemistry.4 While much recent attention has been given to the manner in which these processes proceed (i.e. reagent stoichiometry), their broad impact in chemical synthesis is arguably derived from the novel synthetic transformations that they define. It is from this perspective that the current review is written, presenting a brief history of- and recent advances in- metallacycle mediated cross-coupling of substituted alkenes.
Bimolecular, or convergent, C–C bond formation defines the backbone of organic synthesis, and chemical advances that provide new avenues for such bond construction can broadly impact strategic planning in organic synthesis. It is widely accepted that points of convergency (enabled by such processes) in synthesis pathways greatly enhance efficiency and throughput, in part by decreasing the longest linear sequence of steps required to prepare a target. Despite their rather central role in chemistry, one can argue that only a few general modes of reactivity are typically exploited for such bond construction:
Nucleophilic substitution,
Nucleophilic addition to a polarized π-bond,
Cycloaddition,
Metal-catalyzed cross-coupling, and more recently,
Crossed olefin metathesis.
While carbometalation chemistry of alkenes and alkynes (Figure 1A) could be considered as another mode of reactivity suitable to be included in this list, such processes are often significantly limited in substrate scope.5 This rather general statement is supported by the accepted difficulty associated with carbometalation of highly substituted and electronically unactivated π-systems (i.e. those that are not highly polarized), and the challenges associated with the control of site- and stereoselectivity in the C–C bond-forming event. Despite these barriers to utility, significant advances have been made in harnessing this mode of reactivity for the functionalization of alkynes.6 Unfortunately, similar success has not been realized in the related transformations of substituted and electronically unactivated alkenes. The focus of this Perspective is to present the potential utility of metallacycle-mediated bond construction for such transformations (Figure 1B), outline a selected historical account of advances in the field, and conclude by highlighting a collection of synthetic methods that have emerged from our studies in this area (Figure 2; eqs 1-6):
Figure 1. Functionalization of electronically unactivated π-systems by carbometalation.

Figure 2. Examples of metallacycle-mediated coupling by way of olefin functionalization.

Stereodefined alkenes from homoallylic alcohol–alkyne coupling,
1,4-dienes from allylic alcohol–alkyne coupling,
(Z)-trisubstituted alkenes by allylic alcohol–vinylsilane coupling,
Stereodefined 1,5-amino alcohols through homoallylic alcohol–imine coupling,
Homoallylic amines via allylic alcohol–imine coupling,
Skipped dienes or trienes from the coupling of vinylcyclopropanes with vinylsilanes or alkynes.
2. Background and Challenges
2.1 Initial observations in metal-centered [2+2+1] for alkene functionalization
An early example of metallacycle-mediated coupling of alkenes is the Pauson-Khand reaction, where treatment of a preformed Co–alkyne complex and a reactive alkene result in the formation of a cyclopentenone (Figure 3).7 While this process was initially reported in the context of intermolecular annulation, the utility of the intramolecular Pauson Khand reaction has far exceeded the bimolecular process in part due to the challenges with controlling regioselection.8 That said, strategies are emerging to control a subset of intermolecular variations of this cyclopentenone annulation.9
Figure 3. Cyclopentene annulation via Co-centered [2+2+1].

The development of related metal-centered [2+2+1] chemistry for alkene functionalization based on the reactivity of Group (IV) metals soon followed. In 1974, Whitesides hypothesized that the reaction of ethylene with Cp2TiN=NTiCp2 (2) leads to an equilibrium mixture of a saturated titanacyclopentane (3) and the corresponding bis-ethylene complex (4) (Figure 4).10 Two years later, he demonstrated the ability to dimerize alkenes through the in situ reduction of Cp2TiCl2 with Li-naphthalenide, formation of intermediate metallacyclopentanes (6), and carbonylation en route to substituted cyclopentanones (7).11 Soon thereafter, Grubbs described a dimerization process that proceeded by exposure of a preformed metallacyclopentane (3) to an excess of the terminal olefin of interest (8).12 Overall, hydrocarbon products of homodimerization (10) resulted via olefin exchange with the preformed titanacyclopentane.
Figure 4. Homodimerization via saturated metallacyclopentanes.

While these initial reports documented the interesting behavior of metallacyclopentanes based on Cp2Ti, their apparent limitation to homodimerization of terminal- or strained alkenes greatly constrained their potential impact in synthetic organic chemistry. A significant advance was described by Erker in 1979 that documented the ability to control this mode of reactivity for effective cross-coupling reactions of benzyne.13 As depicted in Figure 5, mild heating of Cp2ZrPh2 (11) resulted in the formation of benzyne-Zr complex (12) that, when exposed to a terminal alkene, underwent cross-coupling to generate a substituted metallaindane (13). Interestingly, exposure of this complex to strained or unhindered terminal alkenes resulted in an olefin exchange reaction, presumably via regeneration of a reactive Zr-benzyne complex (12) and subsequent carbometalation (→14).
Figure 5. Cross-coupling with alkenes via metallacyclopentenes.

The conclusion of this discussion, regarding the early development of metal-centered [2+2+1] chemistry as a means for cross-coupling of alkenes, is suitably focused on contributions from the Bercaw laboratory. After confirming the structure of a (Cp*)2Ti-ethylene complex by X-ray diffraction, Bercaw and coworkers demonstrated that this organometallic species was a viable intermediate for the generation of saturated metallacyclopentanes (17).14 Unfortunately, while insertion of ethylene was effective, the reaction was not suitable for higher olefins. That said, carbometalation of a range of other π -systems, including internal alkynes (i.e. →18), nitriles, carbon dioxide and acetaldehyde were successful.
By 1985, a series of early observations regarding the basic reactivity of Cp2Ti- and Cp2Zr- in metallacycle-mediated bimolecular C–C bond formation had been reported. However, the use of this chemistry in organic synthesis was restricted to: 1) cross-coupling of benzene with terminal or strained alkenes, and 2) reductive cross-coupling reactions of ethylene.
2.2 The emergence of more general cross-coupling procedures: Cp2Zr- and Cp2Ti-mediated alkyne–alkene and imine–alkene coupling
Over the next fifteen years, progress from Erker's Zr–benzyne and metallaindane chemistry (illustrated in Figure 5) fueled the development of some general classes of cross-coupling based on the chemistry of zirconacyclopropenes and azazirconacyclopropanes – prepared from the decomposition of Cp2Zr(Me)-R complexes (Figure 6A-D).15 Here, Buchwald demonstrated that intermediate zirconacyclopropanes undergo bimolecular coupling with ethylene and a small subset of unhindered terminal alkenes. As previously established by Erker, the regiochemistry of insertion with terminal alkenes was such that C–C bond formation occured at the internal carbon of the terminal alkene (i.e. 27 and 30).
Figure 6. Early Cp2Zr-based methods for metallacycle-mediated coupling of ethylene and some unhindered terminal alkenes.

Overall, the contributions depicted in Figure 6 defined a significant step forward from the initial findings of Whitesides, Grubbs, Erker, and Bercaw. These advances provided a means to overcome the problems associated with homodimerization of alkenes or alkynes in Group (IV) metal-mediated coupling, yet substantial barriers associated with substrate scope and selectivity remained unanswered for years to come:
Can the control of regiochemistry in metallacycle-mediated cross-coupling between two unsymmetrical systems be general, where regioselection is observed in the functionalization of each unsymmetrical π -system? Very few examples of such complex coupling chemistry had been described (for one example, see Figure 6D).
Can metal-centered [2+2+1] chemistry be utilized in coupling reactions that engage complex (substituted and branched) alkenes? To date, nearly all reported olefin functionalization reactions based on metal-centered [2+2+1] chemistry employ ethylene as a substrate, with only a handful exploring the reactivity of terminal unbranched olefins.
If internal alkenes can be rendered reactive to such metallacyclopropanes, how can regioselection be controlled?
Related to the question above, if internal alkenes could be employed, how could relative and/or absolute stereochemistry be controlled in the insertion process?
From a practical perspective, is there a means to avoid the use of sensitive solid reagents required to generate the metallacyclopropane intermediates (i.e. without the requirement of glovebox techniques)?
Could one get away from the requirement of using metallated substrates (vinyl- and aryl- Grignard and organolithiums) as starting materials? Substrate scope could be markedly enhanced if coupling reactions did not require that one of the substrates be a highly reactive organometallic.
2.3 Metallacycle-mediated functionalization of allylic- and homoallylic alcohols and ethers
In the 1990's, a number of reports surfaced describing metallacycle-mediated functionalization of allylic- and homoallylic alcohols/ethers. Among the first of these, described by Hoveyda and coworkers, was Zr-catalyzed and Zr-promoted addition reactions of EtMgCl to stereodefined allylic and homoallylic systems.16 As illustrated in Figure 7, Zr-catalyzed addition of EtMgCl to 31a produces the syn-product 32 with up to 95:5 levels of diastereoselection, while the same addition reaction to the allylic ether 31b delivers the anti-product 33 with selectivities up to ≥ 99:1. Interestingly, these authors found that a related stoichiometric process proceeds with an opposite sense of regioselection (36 → 37 + 38). Here, the major products derive from formal Sn2′ alkylation. While no comment was made concerning the stereochemistry of the product mixture from this reaction, the related Cp2Zr-promoted coupling of an allylic benzyl ether (40) with a TMS-alkyne (41) delivered a nearly 1:1 mixture of stereoisomeric 1,4-dienes (42 + 43).17 The lack of stereoselection associated with these Zr-promoted processes has been uniformly observed with related metal-promoted transformations: 1) Ta-promoted coupling of alkynes with allylic alcohols,18 and 2) Ti-promoted coupling of Grignard reagents with allylic alcohols.19
Figure 7. Metallacycle-mediated coupling of allylic alcohols and ethers.

As illustrated in Figure 8, similar addition reactions of EtMgCl have been realized with homoallylic alcohols and ethers.16c Zr-catalyzed ethylmagnesiation of homoallylic alcohol 45a or 45b provided the anti product 46, albeit with varying levels of stereoselection (from 92:8 to ≥ 95:5).
Figure 8. Metallacycle-mediated coupoling of homoallylic alcohols and ethers.

In studies focused on the use of TaCl5 for metallacycle-mediated bond construction, Utimoto has reported the cross-coupling of internal alkynes with homoallylic alcohols (Figure 8).20 Formation of a Ta–alkyne complex (51) was followed by the addition of a homoallylic alkoxide to generate an intermediate fused bicyclyic metallacycle 52. Interestingly, the regiochemical course of this coupling reaction (with respect to the alkene) is opposite to that seen in related Cp2Zr- and Cp2Ti-mediated chemistry (see Figure 6C and D). Unfortunately, this alkoxide-directed coupling reaction proved to be significantly limited in substrate scope. As in all other bimolecular carbometalation reactions of electronically unactivated alkenes, this Ta-promoted coupling reaction was limited to the functionalization of terminal alkenes.
2.4 Metal-catalyzed Alder-Ene chemistry for alkyne–alkene cross-coupling
The Alder-Ene reaction represents an alternative metallacycle-mediated process for the functionalization of alkenes, where bimolecular C–C bond formation is accompanied by the formation of a 1,4-diene.21 Ru-catalysis has rendered a collection of Alder-Ene chemistry between monosubstituted alkenes and terminal alkynes possible under relatively mild conditions (Figure 9; 53 + 54 → 55). The application of this type of bimolecular coupling reaction is often challenging in complex settings, in some cases requiring an extensive screen of substrates, solvent and additive to produce high yields and levels of regioselection (56 + 57 → 58).22 Recent advances in this area of cross-coupling include the development of reaction conditions suitable for the regioselective coupling of internal alkynes with terminal alkenes (i.e. 60 and 63).23 Finally, novel catalysts based on Co have emerged that define similarly effective regioselective Alder-Ene reactions between conjugated alkynes and terminal alkenes (65 + 66 → 67).24
Figure 9. Alkene–alkyne coupling via Alder-Ene chemistry.

2.5 Ni-catalyzed cross-coupling reactions
Recently, progress in the chemistry of Ni has led to the elucidation of reaction pathways for the coupling of terminal (monosubstituted) alkenes 68 with aldehydes (Figure 9). In coupling reactions promoted by 20 mol% of Ni(cod)2 along with 40 mol% of dicyclohexylphenylphosphine, a mixture of regio and stereoisomeric products 70 and 71 (rs = 2:1 to 5:1) result in yields ranging from 45-95%.25 While advances have been described in an effort to address regioselection, a few notable characteristics of this method warrant consideration: 1) yields are optimal when the alkene is used as co-solvent, 2) Ni(cod)2 is a sensitive starting material (glovebox techniques were reported for experiment set up), and 3) while the reactions are substoichiometric in Ni(cod)2, turnover numbers typically range from 2 to 5, thereby requiring a significant quantity of a toxic and sensitive metal reagent.
In related studies, it was discovered that replacing Cy2PhP with EtOPPh2 in the coupling of terminal alkenes with aldehydes altered the regiochemical course of C–C bond formation.25b,c This variation of the coupling protocol delivers homoallylic silyl ethers with regioselection up to 95:5; unfortunately, the alkene is typically generated with low levels of stereoselection (E:Z from 2:1 to ca. 5:1 when R1 = alkyl).26
Most recently, a modified Ni(cod)2-catalyzed coupling reaction was reported for allylic substitution of simple alkenes.27 While most examples to date describe addition reactions of ethylene to allylic ethers and carbonates, a few coupling processes were reported with terminal (monosubstituted) alkenes (Figure 11). As seen in other Ni-catalyzed cross-coupling reactions with alkenes, a large excess of the terminal alkene is required.
Figure 11. Terminal alkene–allylic carbonate coupling by Ni-catalysis.

2.6 Ta-catalyzed cross-coupling reactions
Tantalum-catalyzed cross-coupling of secondary amines to alkenes represents a unique collection of metallacycle-mediated bimolecular C–C bond-forming processes.28a,b Early accomplishments in this area by Maspero and Nugent led to bond formation, but reactions were plagued with generally low yields, extended reaction times, and low catalyst turnover. Recently, Herzon and Hartwig described significant advances in this area. First, 4-8 mol% Ta[N(CH3)2]5 was found to promote coupling of N-methyl aniline with a range of alkenes at 160-165 °C.28c While requiring only 1.25 equiv of alkene, coupled products were formed in 66-96% yields in 27-67 h (eq 1). Subsequently, these authors reported that this type of coupling reaction was possible with a range of secondary amines using 2 mol% of [TaCl3(NEt2)2]2 (eq 2).28d
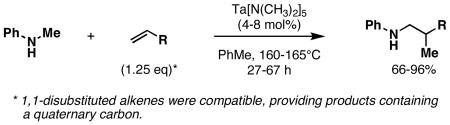 |
(1) |
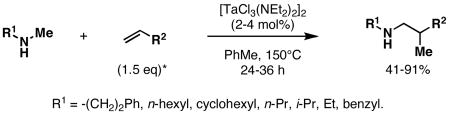 |
(2) |
2.7 Summary of background
While effort has been taken to highlight a collection of metallacycle-mediated alkene functionalization reactions, we recognize that this discussion is not comprehensive. Rather, the selected examples serve to document some of the most relevant history and shed light on the relatively small subset of metallacycle-mediated coupling reactions that are suitable for complex stereoselective fragment coupling. What should be immediately evident is that this entire class of intermolecular C–C bond forming processes is significantly limited in scope – a limitation that is independent of the nature of the metal employed, or whether that metal is used in a substoichiometric fashion.
2.7 Summary of Challenges
Metallacycle-mediated bond construction for the functionalization of alkenes has great potential to define powerful fragment union reactions for convergent synthesis. To realize this potential, a variety of problems must be overcome that currently limit this class of chemical reactivity to a boutique collection of coupling processes. While much attention has been given to rendering metallacycle-mediated coupling chemistry substoichiometric with respect to the central metal component, major limitations in this broad area of chemical reactivity are independent of such considerations. To date, the development of metallacycle-mediated coupling chemistry of alkenes is truly in its infancy, with most reactions defining effective solutions to the functionalization of ethylene. In fact, only a handful of reactions are currently available for complex fragment union, and these reside firmly in the coupling chemistry of terminal alkenes with alkynes via Alder-Ene chemistry.
The major barriers that complicate all reaction development in this area are:
Low levels of reactivity of substituted olefins in metallacycle-mediated chemistry. Not only are substituted olefins poor substrates for the formation of metal–π complexes, they are also poor substrates for reactions with preformed metal–π complexes.
Control of regioselection. While reactivity issues stand first and foremost as a barrier to achieving novel bond construction processes with alkenes, if one could access sufficient reactivity to employ substituted alkenes in metallacycle-mediated coupling, regioselectivity arises as a major unadressed challenge.
Control of diastereoselection. Similarly, if one could use substituted alkenes in metallacycle-mediated bond construction, the issue of diastereoselection emerges as an additional unsolved problem.
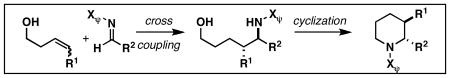
3. Design of Alkoxide-Directed Reductive Cross-Coupling Reactions of Alkenes
3.1. Our Goal
In designing a general process to overcome the limitations of pre-existing methods in reductive cross-coupling technology, we targeted a metal-based system that could be used for the stoichiometric pre-activation of one of the coupling partners – a strategy that would provide a general solution to the control of cross-coupling over homodimerization. Given this initial design criteria, the metal-based system needed to be: 1) inexpensive, 2) non-toxic, and 3) result in byproducts that are both non-toxic and easy to remove from the products of interest. In addition, we favored the selection of a metallic system compatible with a range of Lewis-basic functionality, as we aimed to develop coupling reactions of utility in complex molecule synthesis (i.e. natural product synthesis). Furthermore, we desired a system that would be capable of forging C-C bonds in the presence of unprotected heteroatom-based functionality. Finally, we set out to devise a collection of heteroatom-directed reductive cross-coupling reactions where the control of reactivity and selectivity would be possible based on the strategic placement of a suitable directing group.29
3.2. The selection of a titanium alkoxide-based system
Given our goal of providing chemical methods useful for the convergent synthesis of complex natural products, we desired a system capable of being directed by functionality commonly found embedded in the backbone of such systems. Based on the prevalence of oxygen and nitrogen functionality in natural products of biomedical relevance, we focused our efforts on defining a collection of directed reductive cross-coupling methods where such functionality could serve to direct C-C bond formation - in preference to functionality based on phosphorous, sulfur, aromatic heterocycles or simple π-unsaturation (i.e. a remote alkene).30
To identify a suitable system based on these requirements, one needs to consider only a handful of relevant precedent. First, the ability of Ti alkoxides to undergo rapid and reversible ligand exchange has been well-showcased in the Sharpless epoxidation.2a,b Second, Cp2Ti–π complexes are known to participate in reductive coupling chemistry.10-12, 14 In fact, this type of system was first demonstrated in the reductive dimerization of diphenylacetylene, later studied by Bercaw,14 and subsequently employed in a variety of intramolecular reductive coupling reactions.31 Finally, the pioneering studies of Kulinkovich and Rothwell demonstrated that titanium aryloxides and titanium alkoxides could be employed to access similar reactivity seen with Cp2Ti–π complexes.32 Subsequent investigation of the chemistry of Ti-alkoxides in metallacycle-mediated C–C bond-forming processes, primarily in the laboratories of Professors Sato and Cha, has led to the discovery and development of a wide range of novel reactions.33 While the contributions to date have established a solid foundation of Ti-mediated metallacycle-based bond constructions in organic chemistry, the previously described barriers related to the control of reactivity and selectivity have remained firmly in place.
As we further considered the use of Ti(Oi-Pr)4 as a stoichiometric component of our reaction design, issues of cost and toxicity were at the forefront of our thoughts. First, the byproducts from aqueous work-up of stoichiometric Ti(Oi-Pr)4-mediated coupling reactions (i.e. the Kulinkovich reaction) are i-PrOH and TiO2. Fortunately i-PrOH is a relatively benign solvent and TiO2 is a species encountered daily in most of our lives, as it is a component of products like toothpaste, chewing gum, sunscreen and paint. The accompanying reducing metal typically employed alongside the titanium(IV) alkoxide is a Grignard reagent, the byproducts of which are simple magnesium salts and hydrocarbons.
Although we will describe Ti(Oi-Pr)4-based reductive cross-coupling methods that provide a means to couple π -systems not currently possible with catalytic methods, a cost analysis between projected use of stoichiometric Ti(Oi-Pr)4 versus known catalysts (based on Zr, Ni, Rh and Ru) for metallacycle-mediated functionalization of alkenes is informative.34 If one were to conduct a hypothetical metallacycle-mediated coupling reaction on a 1 mole scale, stoichiometric use of Ti(Oi-Pr)4 would cost $18 (Strem 2008) while the catalytic use of 20 mol% Ni(cod)2 would cost over $1,000 (Strem 2008). In the case of Zr-catalyzed transformations, 5 mol% Cp2ZrCl2 would be competitive with stoichiometric use of Ti(Oi-Pr)4 ($27 – Strem 2008). Alternatively, if a CpRu(cod)Cl-promoted reaction was performed at the typically published 5 mol%, this catalyst would have to cost ca. $1/g in order to compete with the stoichiometric use of Ti(Oi-Pr)4.35
The challenges associated with the development of metal-catalyzed C–C bond formation is a significant and popular concern in current organic chemistry. While much scientific inquiry has embraced this pursuit, our interests were focused on defining a broad class of reductive cross-coupling reactions that extend beyond the basic bond constructions afforded by current methods. Given the considerations described above that are based on reaction design, cost, toxicity, and the ease with which Ti(Oi-Pr)4 can be handled, we embraced a program aimed at defining alkoxide-directed reductive cross-coupling reactions mediated by stoichiometric Ti(Oi-Pr)4 as a strategy to overcome the barriers that have broadly limited all intermolecular metallacycle-mediated coupling reactions.
3.3. The design of alkoxide-directed coupling reactions of alkenes
Two strategies were conceived for alkoxide-directed metallacycle-mediated coupling of alkenes. First, we imagined preformation of a Ti–olefin complex followed by alkoxide-directed coupling with a second π -system (Figure 12A). This reaction design was thought to define a very limited subset of synthetically useful fragment coupling reactions for the following reasons: 1) internal alkenes are typically not effective for the generation of metallacyclopropanes,36 2) even if producing titanacyclopropanes from disubstituted alkenes was efficient, a method for the control of stereochemistry at each Ti–C bond would have to be established, and 3) while directed carbometalation with unsaturated alkoxides (77) was anticipated to deliver metallacyclopentane products, regiochemical control with respect to the alkene (75) would likely be a function of the differential steric environment around the preformed metallacyclopropane 76.
Figure 12. Basic considerations in the design of alkoxide-directed metallacycle-mediated coupling of alkenes.

Alternatively, we imagined that introduction of the alkene as the second π -system in a metallacycle-mediated cross-coupling could have significant advantages (Figure 12B). First, the formation of the initial Ti–π complex could be achieved with establishd protocols (i.e. internal alkyne, terminal alkene, imine). Second, the intramoleculare nature of the carbometalation process, achieved by transient formation of a mixed titanate ester between 81 and 82, should provide a means to achieve bond constructions typically not accessible in bimolecular processes.37 Third, the regiochemical control of alkene functionalization would be based on the position of a pendant hydroxyl. Finally, stereochemical control in the carbometalation of the alkene was thought feasible by translation of stereochemical information from the allylic and/or homoallylic positions of 82.
4. New Regio- and Stereoselective Metallacycle-Mediated Coupling Reactions of Alkenes
4.1. Ti-mediated coupling of homoallylic alcohols with internal alkynes
We initiated our investigations with the coupling of homoallylic alcohols and internal alkynes. As depicted in Figure 13, in-situ formation of a Ti–alkyne complex 85 (alkyne, Ti(Oi-Pr)4, c-C5H9MgCl) would be followed by addition of a suitably functionalized alkene (84). Metal-centered [2+2+1] would then furnish functionalized metallacyclopentenes (86 or 87), protonation of which would deliver cross-coupled product 88 or 89.
Figure 13. Basic reaction pathways for the coupling of alkenyl alcohols with internal alkynes.

As illustrated in Figure 14, our initial studies into this reaction process confirmed that the order of bond forming processes targeted was sufficient to overcome the poor levels of reactivity associated with substituted alkenes.38 Here, the structures of all products isolated (i.e. 90-95) were consistent with the formation of a fused bicyclic titanacyclopentene (i.e. 86; Figure 13), delivering products where C–C bond formation occurred distal to the pendant hydroxyl. This coupling reaction was demonstrated to be compatible with a range of alkene substitution, however, with alkenes bearing terminal substituents that were α-branched (i.e. i-Pr), the reaction was not effective. The coupling of a hydroxymethyl-substituted cyclohexene and a symmetrical alkyne provided additional evidence that this C–C bond-forming reaction was directed by the neighboring hydroxyl group. Here, 93 was formed with very high levels of regio- and stereoselection (C–C bond formation occurred syn- to the neighboring hydroxymethyl substituent).
Figure 14. Examples of cross-coupling reactions between homoallylic alcohols and alkynes.

As evidenced by the highly selective formation of 94 and 95, this alkene–alkyne coupling reaction can be used for highly complex fragment coupling reactions. Chiral homoallylic alcohols were coupled with a chiral and unsymmetrically substituted internal alkyne. In each case, no evidence for the production of minor stereoisomeric products was observed, indicating that the cross-coupling process proceeded with very high levels of regioselection with respect to both unsymmetrically substituted π-systems, while also occuring with very high levels of diastereoselection (presumably derived from the minimization of A-1,3 strain in the (Z)-homoallylic alcohol starting materials – not shown).
Recently, this regioselective alkene–alkyne cross-coupling reaction was a central feature of a novel convergent pathway to substituted spiroketals (Figure 15).39 Here, TMS-substituted alkynes, derived from a formal hydroalkynylation of a homoallylic ether, were coupled to stereodefined homoallylic alcohols to deliver intermediate vinylsilanes. Subsequent oxidative cleavage and acid-promoted spiroacetylization then delivered complex stereodefined spiroketals (i.e. 98, 101, 103 and 106) that contained a substitution pattern that would be quite challenging to prepare with available aldol- and dithiane-based methods. In all cases, the central Ti-mediated alkene–alkyne coupling reaction proceeded with high levels of regio- and stereocontrol (no evidence was found for the production of isomeric products).
Figure 15. A convergent synthesis of spiroketals via alkene–alkyne coupling.

4.2. Ti-mediated coupling of homoallylic alcohols with imines
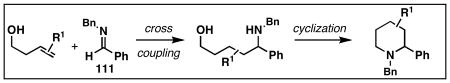
While azatitanacyclopropanes have been prepared from imines,40 and demonstrated to participate in metallacycle-mediated coupling chemistry with terminal alkynes, their potential utility in alkene functionalization had not been demonstrated. This absence was thought to be related to the particularly low levels of reactivity associated with these complexes, as bimolecular carbometalation reactions with internal alkynes are known to be challenging.40 Building on our success with homoallylic alcohol–alkyne coupling, we speculated that a similar reaction strategy may be effective for the functionalization of imines (Figure 16). Here, selective formation of a fused bicyclic azametallacyclopentane would be coupled to a complex stereochemical issue that ultimately results in the formation of the substituted 1,5-aminoalcohol 110.
Figure 16. Homoallylic alcohol–imine coupling.

Our initial study of this alkoxide-directed cross-coupling reaction began with evaluation of reactivity and relative stereoselection for the coupling of a series of simple achiral homoallylic alcohols with aromatic imines.41 As illustrated in Table 1, this coupling reaction proved effective for the union of terminal-, (Z)-, (E)-, and 1,1-disubstituted alkenes. Interestingly, the coupling reactions of the (Z)-, and (E)-disubstituted alkenes proceeded with exquisite levels of stereoselection and delivered anti- products with ≥ 20:1 selectivity (in neither instance was any evidence found for the production of regio- or diastereomeric products). In the case of 1,1-disubstituted alkenes, similarly high levels of regioselection were observed, albeit with dimished levels of stereoselection. In this case the syn- isomer 122 was produced as the major product alongside minor quantities of the anti-product (dr = 4:1). As demonstrated, these coupling reactions define a convergent stereoselective path to the assembly of substituted piperidines (114, 117, 120, and 123) by a simple two-step sequence: 1) reductive cross-coupling, and 2) cyclization (PPh3, imidazole, CCl4).
Table 1. 1,5-amino alcohols and piperidines via homoallylic alcohol– imine coupling.
 | ||||||
|---|---|---|---|---|---|---|
| entry | homoallylic alcohol | 1,5-aminoalcohola | yield (%) | rr | dr | piperidineb(yield) |
| 1 |  |
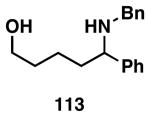 |
76 | ≥95:5 | n/a | 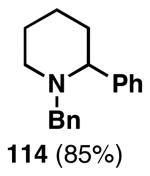 |
| 2 |  |
 |
73 | ≥95:5 | ≥95:5 | 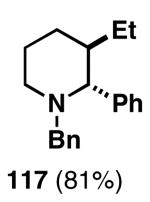 |
| 3 | 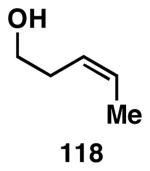 |
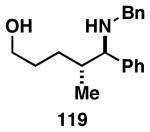 |
63 | ≥95:5 | ≥95:5 | 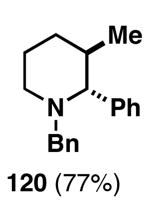 |
| 4 | 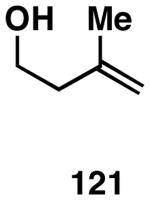 |
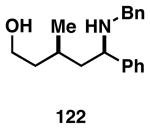 |
67 | ≥95:5 | 4:1 | 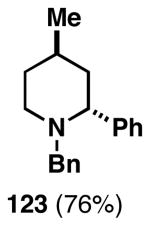 |
Reaction conditions for cross coupling: imine (1 eq), Ti(Oi-Pr)4 (1.5 eq), c-C5H9MgCl (3.0 eq), Et2O (−78 to −40 °C), then add alkoxide (1.5 eq) (−40 to −20 °C, 0 °C, or rt).
Reaction conditions for cyclization: PPh3, imidazole, CCl4, reflux.
The origin of stereoselection in this coupling reaction remains under investigation, yet the observed selectivity is consistent with a proposal based on kinetically controlled C–C bond formation. As illustrated in Figure 17, anti-selectivity in the coupling of (E)- or (Z)- alkenes is consistent with an empirical model whereby non-bonded steric-interactions about the developing C–C bond are minimized. Here, avoiding eclipsing interactions between the alkene- and imine-substituent are reasoned to result in the observed stereoconvergence. In the coupling reaction of 1,1-disubstituted alkenes, stereoselection is thought to derive from the preferred orientation of the imine substituent in the cis-fused bicyclo[3.3.0] transition state geometry F.
Figure 17. Empirical model for simple diastereoselection in reductive cross-coupling between homoallylic alcohols and imines.

While it is well-established that saturated metallacyclopentanes can be in rapid equilibrium with expulsion of an alkene and generation of a metallacyclopropane,42 recent investigations have provided support for the proposition that the coupling reactions depicted in Table 1 proceed under reaction conditions that do not promote such fragmentation.43 These observations are consistent with the assumed underlying principles that govern the empirical model depicted in Figure 17.
The imine–allylic alcohol coupling reaction can also be conducted in an asymmetric fashion. As highlighted in Table 2 entries 1-3, single asymmetric coupling reactions proceeded with very high levels of stereoselection, defining a unique stereoselective convergent coupling process for the synthesis of optically active 1,5-aminoalcohols and substituted piperidines. Interestingly, this coupling process is also highly effective in a double asymmetric mode. Coupling of the chiral homoallylic alcohols 129 and 132 with a chiral imine, delivers either the 1,5-syn-1,4-anti-1,5-aminoalcohol product 130 with ≥50:1 selectivity, or the 1,5 -anti-1,4-syn-1,5-aminoalcohol product 133 with high, but somewhat lower, selectivity (dr = 34:4:1). Conversion to chiral tetrasubstituted piperidines 131 and 134 was possible by a sequence consisting of: 1) selective reductive removal of the phenylglycine motif, 2) formation of a sulfonamide, and 3) cyclization.
Table 2. Single and double asymmetric homoallylic alcohol–imine coupling reactions en route to chiral piperidines.
 | ||||||
|---|---|---|---|---|---|---|
| entry | homoallylic alcohol | imine | 1,5-aminoalcohola | yield (%) | dr | piperidineb(yield) |
| 1 | 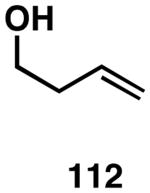 |
 |
 |
83 | 24:1 | 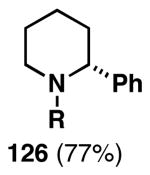 |
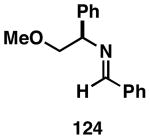
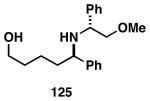
| 2 |  |
124 |  |
75 | 20:1 |  |
| 3 |  |
124 | 127 | 61 | 25:4:1 | |
| 4 | 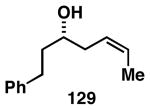 |
ent-124 | 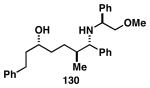 |
76 | ≥50:1 | 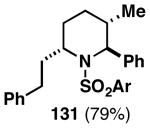 |
| 5 | 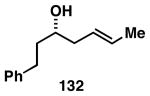 |
124 | 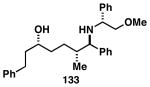 |
88 | 35:4:1 | 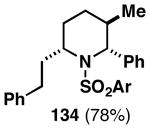 |
Reaction conditions for cross coupling: imine (1 eq), Ti(Oi-Pr)4 (1.5 eq), c-C5H9MgCl (3.0 eq), Et2O (−60 to −30 °C), then add alkoxide (1.5 eq) (−40 to 0 °C or rt).
Reaction conditions for cyclization: For 126 - 2-NsCl, Et3N, DMAP, CH2Cl2, rt. For 128 -MsCl, Et3N, CH2Cl2, rt. R = CH(Ph)CH2OMe. For 131 and 134 - 1) H2 (1 atm), Pd(OH)2, AcOH/MeOH (1:10 v/v), rt, 2) 2-NsCl, Et3N, DMAP, CH2Cl2, 0 °C to rt, 3) PPh3, DIAD, THF, rt.
4.3. Ti-mediated coupling of allylic alcohols with alkynes
While allylic alcohols and ethers have been defined as substrates for metallacycle-mediated coupling, transformations that proceed with allylic transposition have been uniformly unselective, delivering products as mixtures of olefin isomers.17-19 In an effort to define a series of stereoselective and synthetically useful cross-coupling reactions based on this mode of reactivity, we aimed to design a means to control the regio- and stereochemical course of such processes. As illustrated in Figure 18, we speculated that alkoxide-directed carbometalation may provide a means to access a rigid boat-like transition state geometry en route to a fused bicyclo-[3.2.0] system (135). Here, the boat-like geometry would be enforced by the mechanistic requirements for bond formation that include: 1) ligand exchange at Ti, and 2) alignment between σTi–C with π C=C. Once achieved, the resulting fused metallacycle was anticipated to undergo a stereospecific syn-elimination akin to that proposed in the Tebbe olefination.44 In this manner, the relative stereochemistry of the oxatitanacyclobutane intermediate should correlate to the alkene geometry in the product.
Figure 18. Reaction design for the stereoselective functionalization of allylic alcohols.

Our initial studies in this area resulted in the elucidation of highly stereoselective coupling reactions for the synthesis of 1,4-dienes.45 As depicted in Figure 19, (Z)-allylic alcohol starting materials (137) can be selectively coupled with internal alkynes (138) to deliver 1,4-dienes possessing one (E)-disubstituted alkene, one (E)-trisubstituted alkene, and a central C3-substituent (R2; 139).
Figure 19. (E)-selective allylic alcohol–alkyne coupling.

Alternatively, the stereochemical course of this cross-coupling reaction can be controlled by minimization of A-1,2 strain. Here, allylic alcohols bearing 1,1-disubstituted alkenes (140), are coupled to internal alkynes (138) en route to 1,4-dienes that contain one (Z)-trisubstituted alkene (141; Figure 20).
Figure 20. (Z)-selective allylic alcohol–alkyne coupling.

While these initial studies defined the first highly stereoselective coupling reactions of its kind, recent studies have shed light on chemoselectivity associated with this process.46 As depicted in Table 3, 1,5-dienes that possess one alkene that is part of an allylic alcohol functionality, and one alkene that is part of a homoallylic alcohol motif, can be employed in highly selective coupling reactions. When the diene substrates have related levels of substitution, highly chemo- and stereoselective cross-coupling is observed at the allylic alcohol, resulting in the formation of stereodefined skipped polyenes (144, 147, 149, 151, and 153).
Table 3. Chemoselective reductive cross-coupling reactions of 1,5-dienes as a route to stereodefined skipped trienes.
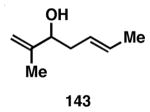
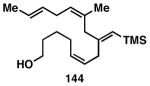
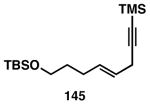
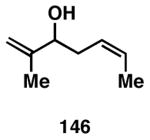
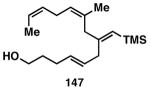
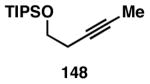
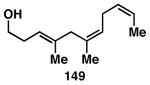
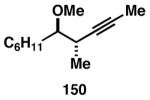
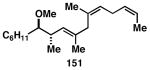

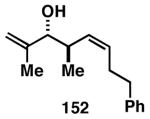
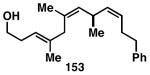
Reaction conditions: alkyne (2-3 equiv.), Ti(Oi-Pr)4 or ClTi(Oi-Pr), c-C5H9MgCl, PhMe (−78 to −35 °C), then cool to −78 °C and add Li alkoxide of the allylic alcohol as a solution in THF (warm to 0 °C).
Yield reported is over two steps: chemoselective reductive cross-coupling and silyl deprotection with TBAF in THF.
This coupling process has been demonstrated to be of great utility in natural product synthesis. As depicted in Figure 21, the first total synthesis and structure elucidation of a phorbasin was accomplished by a chemical pathway that employed an allylic alcohol–alkyne coupling reaction to establish the oxygenated cyclohexene core.47 In this process, a highly stereoselective addition reaction, controlled by the stereochemistry of the allylic alcohol 154, was coupled to a bond construction where high levels of site-selectivity for C–C bond formation with respect to each reaction component was observed. In an unrelated campaign in total synthesis, a late stage allylic alcohol–alkyne coupling reaction was used to establish the stereodefined skipped polyene backbone of lehualide B.48 Interestingly, the potentially sensitive γ-pyrone moiety of 157 was compatible with this metallacycle mediated coupling process.
Figure 21. Application of allylic alcohol–alkyne coupling in natural product total synthesis.

4.4. Ti-mediated coupling of allylic alcohols with vinylsilanes: A stereochemically orthogonal reaction to the Claisen rearrangement
The unique stereochemical course of the alkyne–allylic alcohol coupling reaction, where 1,1-disubstituted olefins delivered (Z)-trisubstituted alkene-containing 1,4-diene products, prompted us to consider the potential of a related coupling reaction for the stereoselective synthesis of isolated alkenes. While Claisen chemistry has been established as a strategy for the synthesis of stereodefined trisubstituted olefins, this process typically delivers (E)-trisubstituted alkenes with high levels of stereochemical control. We wondered whether a Ti-promoted reductive coupling process could define a stereochemically orthogonal pathway to related products.
As depicted in Figure 22, use of chlorodimethylvinylsilane in titanium alkoxide-mediated reductive cross-coupling with allylic alcohols bearing 1,1-disubstituted alkenes was anticipated to deliver products containing a (Z)-trisubstituted alkene (160). This expectation was in accord with the empirical model for the allylic alcohol–alkyne coupling reaction previously discussed. As depicted in Figure 23, this coupling reaction proceeds with uniformly high levels of stereoselection, and delivers products not accessible with modern methods based on the Claisen-rearrangement (161-166).49
Figure 22. Allylic alcohol–vinylsilane coupling as a stereochemically orthogonal reaction to the Claisen rearrangement.

Figure 23. (Z)-selective allylic alcohol–vinylsilane coupling.

4.5. Ti-mediated coupling of allylic alcohols with imines, a unique pathway to stereodefined homoallylic amines
Based on the high degree of stereochemical control observed in the reductive cross-coupling reactions of substituted allylic alcohols with alkynes and vinylsilanes, we wondered whether the aspects of stereochemical control observed in homoallylic alcohol–imine coupling chemistry would translate to a new coupling process. In short, we anticipated that preformation of a Ti–imine complex, followed by addition of an allylic alkoxide would result in a highly controlled allyl transfer process proceeding by way of directed carbometalation and syn-elimination. We recognized that this subset of cross-coupling would be significantly more complex than previously discussed allylic alcohol-based transformations, as stereochemical control would have to be addressed in the functionalization of each reaction component.
The following discussion breaks down our observations in this interesting coupling reaction by basic considerations associated with product structure and control of the bimolecular process:
Imine prenylation
While our initial investigations demonstrated that Ti-mediated reductive cross-coupling of allylic alcohols with imines was successful in delivering homoallylic amines (111 + 167 → 168; Figure 24),50 this basic coupling reaction is possible with a host of methods based on the reactivity of allylic organometallic reagents. A significantly more difficult bond construction is the prenylation of imines. It is well known that allylic organometallic reagents are typically not useful for imine prenylation, as these reagents typically react to deliver products of inverse prenylation.51 Based on the unique mechanistic course of reductive cross-coupling reactions, we speculated that imine prenylation could derive from reaction with a simple commercially available tertiary allylic alcohol (169). In fact, these processes are equally effective with aromatic and aliphatic imines, delivering products of imine prenylation (170, 172, and 174) in 80-92% yield (rs ≥ 20:1).
Figure 24. Imine–allylic alcohol coupling reactions – a path to imine prenylation.

Imine allylation with control of relative stereochemistry
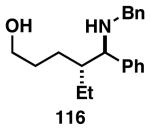
Reductive cross-coupling of a range of substituted allylic alcohols proceeds in a highly stereoselective manner (Figure 25).50 As seen in previous coupling reactions of allylic alcohols bearing 1,1-disubstituted alkenes, union of imine 111 with allylic alcohol 175 delivers a homoallylic amine product 176 possessing a (Z)-trisubstituted alkene in 87% yield. Interestingly, when the allylic alcohol contains an internal alkene (177 or 179), the reductive cross-coupling reaction proceeds with both high stereoselectivity for the formation of a substituted alkene product (E-disubstituted or Z-trisubstituted), and high anti-stereochemistry between the allylic and homoallylic stereocenters. This sense of stereoselection is consistent with the empirical models depicted in Figure 26.
Figure 25. Diastereoselective allylic alcohol–imine coupling reaction.

Figure 26. Empirical model in support of the relative stereochemistry observed in imine–allylic alcohol reductive cross-coupling.

Enantioselective imine allylation by reductive cross-coupling
Based on the emerging empirical model for stereochemical control of these imine–allylic alcohol reductive cross-coupling reactions, we speculated that asymmetric allyltransfer chemistry would be possible with chiral allylic alcohols. As depicted in Figure 27, complex asymmetric imine allylation proceeds with outstanding transfer of stereochemical information, in this case delivering the chiral homoallylic amine 178 with ≥ 95% ee.52
Figure 27. Asymmetric reductive cross-coupling.

While a more thorough discussion of asymmetric reductive cross-coupling reactions of imines with allylic alcohols is not within the scope of this review, recent studies in our laboratory have found that a range of double asymmetric coupling processes are useful for perturbing the stereochemical course of these allyltransfer processes.
Application of allylic alcohol–imine coupling for the synthesis of stereodefined heterocycles
The significant functional group tolerance of Ti-mediated reductive cross-coupling chemistry, in combination with the stereochemical control possible in alkoxide-directed processes, defines a fertile area for the development of new synthetic pathways to complex heterocycles. As illustrated in Figure 28A, a two-step process consisting of chemoselective coupling and intramolecular aza-Sakurai allylation delivers complex indolizines and quinolizidines from simple starting materials.53 In an unrelated study, chemoselective coupling of allylic alcohols containing a pendent vinylhalide serves as the first step of a one-pot process for the synthesis of substituted lactams (Figure 28B).54 Here, formation of the intermediate azametallacyclopentane, from the union of 111 with 185, is followed by addition of water, then 2 mol% of a PdCl2, 6 mol% of t-Bu3P, Et3N, and CO. Overall, an one-pot annulation results, delivering the fused γ-lactam 186 in 73% yield (dr ≥ 20:1).
Figure 28. Routes to stereodefined heterocycles enabled by a stereoselective reductive cross-coupling between allylic alcohols and imines.

4.6. Ti-mediated coupling of vinylcyclopropanes with alkynes as a route to stereodefined skipped trienes
In a recent study aimed at defining novel stereoselective routes to the assembly of skipped trienes, we elucidated a stereospecific reductive cross-coupling reaction of vinylcyclopropanes with alkynes.55 As depicted in Figure 29A, preformation of a Ti–alkyne complex, followed by alkoxide-directed carbometalation of a vinylcyclopropane was expected to deliver an unstable tricyclic metallacyclopentane. Decomposition of this intermediate (189) by a six electron process would then deliver, after protonation of the terminal organometallic product, a stereodefined skipped triene.
Figure 29. Skipped trienes from the stereoselective reductive cross-coupling of vinylcyclopropanes with alkynes.

One representation of the power of this bond construction is shown in Figure 29B. Here, union of the stereodefined vinylcyclopropane 191 (itself derived from a four step-functionalization of a simple allylic alcohol) with a TMS-alkyne delivers the complex stereodefined skipped triene 193 in 82% yield based on recovered starting material. While the conversion for this transformation remained relatively low (36% isolated yield), the triene product was formed as a single isomer. Note that this bimolecular C–C bond-forming reaction delivers a skipped triene product that contains three stereodefined alkenes (one (Z)-disubstituted, one (E)-disubstituted, and one (E)-trisubstituted), while also harboring stereodefined substitution at each central sp3 carbon of the isolated 1,4-dienes.
In a brief study to explore the utility of this new reductive cross-coupling process, we pursued the synthesis of a collection of complex and diverse polyunsaturated fatty acids (194-196; Figure 29C). Interestingly, the stereochemical course of this coupling reaction is unique compared to the chemoselective reactions of 1,5-diene-3-ols described in Table 3, as all examples explored provide a central (E)- alkene (dior trisubstituted). While these polyunsaturated acyclic molecules may appear as relatively simple architectures, one should not overlook the complexity associated with these structures, or the difficulties that one would incur in attempted synthesis using available chemical methods.
Conclusions
Chemo-, regio- and stereoselective intermolecular C–C bond formation represents the backbone of organic synthesis. Despite the central role of these bond-forming processes in organic chemistry, relatively few general areas of chemical reactivity are typically employed for such bond construction. Carbometalation chemistry is one area of chemical reactivity with great potential to impact the manner in which complex molecules are forged. Unfortunately, major hurdles have stood in the way of advancing broadly useful methods in this area based on the often low reactivity of electronically unactivated alkenes and internal alkynes, as well as the often harsh reaction conditions required to enable addition of preformed organometallic reagents to these systems.
While the reactivity of metallacyclopropanes in addition reactions to ethylene, and simple unfunctionalized terminal alkenes have been known for over 20 years, these reactions have not proven to have a significant impact on complex organic synthesis due to challenges asociated with the control of such processes. Recently, a program aimed at defining complex fragment union reactions based on the chemistry of metallacyclopropanes has resulted in the discovery of a collection of novel synthetic methods that, for the first time, harnesses the potential of reactive metallacyclopropanes for defining highly controlled bimolecular C–C bond forming reactions. In short, a series of alkoxide-directed Ti-mediated coupling reactions have been established as a means to achieve the synthesis of a range of complex functionality from fragment coupling reactions of homoallylic alcohols and allylic alcohols with alkynes, vinylsilanes and imines. We look forward to the growth of new reaction methods and applications in target-oriented synthesis that follow from these initial discoveries.
Figure 10. Alkene–aldehyde coupling by Ni-catalysis.

Notes and references
‡ Footnotes should appear here. These might include comments relevant to but not central to the matter under discussion, limited experimental and spectral data, and crystallographic data.
- 1.a) Corey EJ, Cheng X. The Logic of Chemical Synthesis. John Wiley & Sons, Inc.; New York: 1989. p. 436. [Google Scholar]; b) Nicoloau KC, Sorensen EJ. Classics in Total Synthesis. Wiley-VCH; Weinheim: 1996. p. 798. [Google Scholar]
- 2.a) Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974. [Google Scholar]; b) Katsuki T, Martin VS. Org React. 1996;48:1. [Google Scholar]; c) Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]; d) Wong OA, Shi Y. Chem Rev. 2009;108:3958. doi: 10.1021/cr068367v. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews, see: de-Meijere A, Diederich F. Metal-catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 2004. p. 938.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368.Negishi E. Bull Chem Soc Jpn. 2007;80:233.Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505.Beletskaya IP, Cheprakov AV. Coordin Chem Rev. 2004;248:2337.
- 4.For recent reviews, see: Hoveyda AH, Zhugralin AR. Nature. 2007;450 doi: 10.1038/nature06351.Grubbs RH. Tetrahedron. 2004;60:7117.. For a review of metathesis reactions in total synthesis, see: Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4490. doi: 10.1002/anie.200500369.. For a review of enyne metathesis, see: Diver ST, Giessert AJ. Chem Rev. 2004;104:1317. doi: 10.1021/cr020009e.. For a review of olefin cross-metathesis, see: Connon SJ, Blechert S. Angew Chem Int Ed. 2003;42:1900. doi: 10.1002/anie.200200556.
- 5.For reviews see: Dénés F, Pérez-Luna A, Chemla F. Chem Rev. 2010;110:2366. doi: 10.1021/cr800420x.Fallis AG, Forgione P. Tetrahedron. 2001;57:5899.Marek I. J Chem Soc, Perkin Trans. 1999;1:535.Nakamura E. Pure & Appl Chem. 1996;68:123.Negishi E. Acc Chem Res. 1987;20:65.Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048.
- 6.For reviews, see: Normant JF, Alexakis A. Synthesis. 1981:841.Lipshutz BH, Sengupta S. Org React. 1992;41:135.Marek I, Normant JF. In: Metal-catalyzed Cross-coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 1998. pp. 271–337.Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Eur J Org Chem. 2010. p. 391.
- 7.a) Khand IU, Knox GR, Pauson PL, Watts WE. J Chem Soc, Chem Commun. 1971:36. [Google Scholar]; b) Khand IU, Knox GR, Pauson PL, Watts WE, Foreman MI. J Chem Soc Perkin Trans. 1973;1:977. [Google Scholar]
- 8.For recent reviews on metal-mediated [2+2+1] annulations, see: Croatt MP, Wender PA. Eur J Org Chem. 2010:19.Gibson SE, Stevenazzi A. Angew Chem Int Ed. 2003;42:1800. doi: 10.1002/anie.200200547.Pauson PL. Tetrahedron. 1985;41:5855.
- 9.For a recent review of the intermolecular Pauson-Khand annulation, see: Gibson SE, Mainolfi N. Angew Chem Int Ed. 2005;44:3022. doi: 10.1002/anie.200462235.. For recent examples, see: Wender PA, Deschamps NM, Williams TJ. Angew Chem Int Ed. 2004;43:3076. doi: 10.1002/anie.200454117.. For an intermolecular example of [2+2+1] between a 2,3-disubstituted diene and norbornene, see: Wender PA, Croatt MP, Deschamps NM. J Am Chem Soc. 2004;126:5948. doi: 10.1021/ja0489487.
- 10.McDermott JX, Whitesides GM. J Am Chem Soc. 1974;96:947. [Google Scholar]
- 11.McDermott JX, Wilson ME, Whitesides GM. J Am Chem Soc. 1976;98:6529. [Google Scholar]
- 12.Grubbs RH, Miyashita A. Chem Commun. 1977:864. [Google Scholar]
- 13.Erker G, Kropp K. J Am Chem Soc. 1979;101:3659. [Google Scholar]
- 14.a) Cohen SA, Auburn PR, Bercaw JE. J Am Chem Soc. 1983;105:1136. [Google Scholar]; b) Cohen SA, Bercaw JE. Organometallics. 1985;4:1006. [Google Scholar]
- 15.a) Buchwald SL, Lum RT, Dewan JC. J Am Chem Soc. 1986;108:7441. [Google Scholar]; b) Buchwald SL, Watson BT, Huffman JC. J Am Chem Soc. 1987;109:2544. [Google Scholar]; c) Buchwald SL, Watson BT, Wannamaker MW, Dewan JC. J Am Chem Soc. 1989;111:4486. [Google Scholar]; d) Cámpora J, Buchwald SL. Organometallics. 1993;12:4182. [Google Scholar]; e) Aoki K, Peat AJ, Buchwald SL. J Am Chem Soc. 1998;120:3068. [Google Scholar]
- 16.a) Hoveyda AH, Xu Z. J Am Chem Soc. 1991;113:5079. [Google Scholar]; b) Hoveyda AH, Xu Z, Morken JP, Houri AF. J Am Chem Soc. 1991;113:8950. [Google Scholar]; c) Houri AF, Didiuk MT, Xu Z, Horan NR, Hoveyda AH. J Am Chem Soc. 1993;115:6614. [Google Scholar]
- 17.Suzuki N, Kondakov DY, Kageyama M, Kotora M, Hara R, Takahashi T. Tetrahedron. 1995;51:4519.; stereoselection was enhanced if an allylic chloride was used in place of an allylic ether.
- 18.Takai K, Yamada M, Odaka H, Utimoto K, Fujii T, Furukawa I. Chem Lett. 1995:315. [Google Scholar]
- 19.Kulinkovich OG, Epstein OL, Isakov VE, Khmel’nitskaya EA. Synlett. 2001:49. [Google Scholar]
- 20.Takai K, Yamada M, Odaka H, Utimoto K. J Org Chem. 1994;59:5852. [Google Scholar]
- 21.Trost BM, Indolese AF, Müller TJJ, Treptow B. J Am Chem Soc. 1995;117:615. [Google Scholar]
- 22.Trost BM, Probst GD, Schoop A. J Am Chem Soc. 1998;120:9228.. After a thorough investigation of this Alder-Ene reaction, optimal levels of regioselectivity were obtained with the diol of 56 (removal of acetonide), 11 mol% of the catalyst, 22 mol% of NH4PF6, and 1.1 eq of the alkyne (rs = 12.5:1)
- 23.Trost BM, Gunzner JL. J Am Chem Soc. 2001;123:9449. doi: 10.1021/ja011424b.Trost BM, Machacek MR, Ball ZT. Org Lett. 2003;5:1895. doi: 10.1021/ol034463w.. For an interesting example with alkynylborates, see: Hansen EC, Lee D. J Am Chem Soc. 2005;127:3252. doi: 10.1021/ja0424629.
- 24.Hilt G, Treutwein J. Angew Chem Int Ed. 2007;46:8500. doi: 10.1002/anie.200703180. [DOI] [PubMed] [Google Scholar]
- 25.Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194. doi: 10.1021/ja055363j.Ho CY, Ng SS, Jamison TF. J Am Chem Soc. 2006;128:5362. doi: 10.1021/ja061471+.Ng SS, Jamison TF. J Am Chem Soc. 2006;128:11513. doi: 10.1021/ja062866w.Ho CY, Jamison TF. Angew Chem Int Ed. 2007;46:782. doi: 10.1002/anie.200603907.. For a recent review of Ni-catalyzed coupling reactions of alkenes, see: Ng SS, Ho CY, Schleicher KD, Jamison TF. Pure Appl Chem. 2008;80:929. doi: 10.1351/pac200880050929.
- 26.High levels of stereoselection were observed when R1 = aromatic or t-Bu.
- 27.Matsubara R, Jamison TF. J Am Chem Soc. 2010;132:6880. doi: 10.1021/ja101186p.. Allylic alcohols, ethers, carbonates and chlorides were also examined in this coupling reaction.
- 28.a) Clerici MG, Maspero F. Synthesis. 1980:305. [Google Scholar]; b) Nugent WA, Ovenall DW, Holmes SJ. Organometallics. 1983;2:161. [Google Scholar]; c) Herzon SB, Hartwig JF. J Am Chem Soc. 2007;129:6690. doi: 10.1021/ja0718366. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Herzon SB, Hartwig JF. J Am Chem Soc. 2008;130:14940. doi: 10.1021/ja806367e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.For reviews, see: Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307.Catellani M, Chiusoli GP, Costa M. J Organomet Chem. 1995;500:69.Breit B. Chem Eur J. 2000;6:1519. doi: 10.1002/(sici)1521-3765(20000502)6:9<1519::aid-chem1519>3.3.co;2-y.Oestreich M. Eur J Org Chem. 2005:783.
- 30.For an example of a P-directed hydroformylation for the synthesis of stereodefined polyketides, see: Krauss IJ, Wang CCY, Leighton JL. J Am Chem Soc. 2001;123:11514. doi: 10.1021/ja016978t.. For an example of a pyridyl-directed Pauson Khand annulation, see: Itami K, Mitsudo K, Fujita K, Ohashi Y, Yoshida J. J Am Chem Soc. 2004;126:11058. doi: 10.1021/ja047484+.. For an example of an alkene-directed reductive coupling, see: Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342. doi: 10.1021/ja0446799.
- 31.For examples of Cp2Ti-promoted cyclization reactions, see: Nugent WA, Calabrese JC. J Am Chem Soc. 1984;106:6422.Nugent WA, Thorn DL, Harlow RL. J Am Chem Soc. 1987;109:2788.RajanBabu TV, Nugent WA, Taber DF, Fagan PJ. J Am Chem Soc. 1988;110:7128.Berk SC, Grossman RB, Buchwald SL. J Am Chem Soc. 1993;115:4912.Berk SC, Grossman RB, Buchwald SL. J Am Chem Soc. 1994;116:8593.Hicks FA, Kablaoui NM, Buchwald SL. J Am Chem Soc. 1996;118:9450.Sturla SJ, Kablaoui NM, Buchwald SL. J Am Chem Soc. 1999;121:1976.Hicks FA, Kablaoui NM, Buchwald SL. J Am Chem Soc. 1999;121:5881.. See also, refs 11 and 12.
- 32.For chemistry of Ti-aryloxides, see: Chamberlain LR, Durfee LD, Fanwick PE, Kobriger L, Latersky SL, McMullen AK, Rothwell IP, Folting K, Huffman JC, Streib WE, Wang R. J Am Chem Soc. 1987;109:390. doi: 10.1021/ja00236a017.Chamberlain LR, Durfee LD, Fanwick PE, Kobriger LM, Latesky SL, McMullen AK, Steffey BD, Rothwell IP, Folting K, Huffman JC. J Am Chem Soc. 1987;109:6068. doi: 10.1021/ja00236a017.Thorn MG, Hill JE, Waratuke SA, Johnson ES, Fanwick PE, Rothwell IP. J Am Chem Soc. 1997;119:8630.. For early examples documenting the reactivity of Ti-alkoxides, see: Kulinkovich OG, Sviridov SV, Vasilevski DA, Pritytskaya TS. J Org Chem USSR (Eng Transl) 1989;25:2027.
- 33.For recent reviews, see: Wolan A, Six Y. Tetrahedron. 2010;66:15.Wolan A, Six Y. Tetrahedron. 2010;66:3097.Kulinkovich O. Eur J Org Chem. 2004:4517.Sato F, Urabe H, Okamoto S. Chem Rev. 2000;100:2835. doi: 10.1021/cr990277l.Kulinkovich OG, de Meijere A. Chem Rev. 2000;100:2789. doi: 10.1021/cr980046z.
- 34.For a related discussion in the context of regioselective reductive cross-coupling reactions of internal alkynes, see Ref 6d.
- 35.While we were unable to locate a commercial vendor for CpRu(cod)Cl, the cost of [Cl2Ru(cod)]n (a potential precursor to CpRu(cod)Cl) is nearly $70/g.
- 36.For a discussion regarding the limitations associated with the Kulinkovich and related reactions, see ref 32d and 32f.
- 37.The ease with which intramolecular metallacycle-mediated bond formation occurs is supported by the large collection of cyclization reactions reported. For examples based on Ti, see ref 30.
- 38.Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440. doi: 10.1002/anie.200603515. [DOI] [PubMed] [Google Scholar]
- 39.Canterbury DP, Micalizio GC. J Am Chem Soc. 2010;132:7602. doi: 10.1021/ja102888f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Y, Yoshida Y, Sato F. Synlett. 1997:1353. [Google Scholar]
- 41.Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514. doi: 10.1021/ja071974v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill JE, Fanwick PE, Rothwell IP. Organometallics. 1992;11:1775. [Google Scholar]
- 43.Takahashi M, Micalizio GC. Chem Commun. 2010;46:3336. doi: 10.1039/b927430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.a) Tebbe FN, Parshall GW, Reddy GS. J Am Chem Soc. 1978;100:3611. [Google Scholar]; b) Petasis NA, Bzowej EI. J Am Chem Soc. 1990;112:6392. [Google Scholar]
- 45.Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112. doi: 10.1021/ja075678u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diez PS, Micalizio GC. J Am Chem Soc. 2010:132. doi: 10.1021/ja103836h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macklin TK, Micalizio GC. J Am Chem Soc. 2009;131:1392. doi: 10.1021/ja809491b. [DOI] [PubMed] [Google Scholar]
- 48.Jeso V, Micalizio GC. J Am Chem Soc. 2010:132. doi: 10.1021/ja104782u. 2010. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belardi JK, Micalizio GC. J Am Chem Soc. 2008;130:16870. doi: 10.1021/ja8074242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648. doi: 10.1002/anie.200900236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prenylation of imines has represented a significant challenge in organic chemistry. For a route to products like 170, 172 and 174, based on the reactivity of allylic barium reagents, see: Yanagisawa A, Ogasawara K, Yasue K, Yamamoto H. J Chem Soc Chem Commun. 1996:367.
- 52.Chen MZ, McLaughlin M, Takahashi M, Tarselli MA, Yang D, Umemura S, Micalizio GC. 2010 doi: 10.1021/jo101535d. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang D, Micalizio GC. J Am Chem Soc. 2009;131:17548. doi: 10.1021/ja908504z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Umemura S, McLaughlin M, Micalizio GC. Org Lett. 2009;11:5402. doi: 10.1021/ol9022134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Macklin TK, Micalizio GC. Nature Chem. 2010:2. doi: 10.1038/nchem.665. [DOI] [PMC free article] [PubMed] [Google Scholar]