

Abstract
Hypothalamic proopiomelanocortin (POMC) neurons release the endogenous opioid beta-endorphin and POMC neuron activity is inhibited by opioids, leading to the proposal that beta-endorphin acts to provide feedback inhibition. However, both intrinsic properties and synaptic inputs contribute to the regulation of POMC neurons such that attributing an autoregulatory role to opioids must include consideration of opioid receptor localization and sensitivity at both presynaptic and postsynaptic sites. In the present study, whole-cell recordings were made in POMC cells in mouse brain slices and the presynaptic and postsynaptic regulation of POMC neurons was examined using selective agonists for mu, kappa, and delta opioid receptors. Activation of mu, but not kappa or delta, receptors induced a direct postsynaptic outward current. Agonists for each of the receptors inhibited the frequency of spontaneous IPSCs. Mu and kappa, but not delta, agonists reduced the amplitude of evoked IPSCs and appeared to colocalize in a significant portion of GABAergic terminals onto POMC neurons. The presynaptic inhibition caused by the mu agonist DAMGO had an EC50 of 80 nm, whereas the EC50 was 350 nm when measuring the postsynaptic outward current. This differential sensitivity adds an unexpected component of opioid-dependent feedback regulation, where low levels of opioid receptor activation would likely disinhibit POMC neuron activity and higher concentrations would result in an overall inhibition. The results may help explain why it has been difficult to clearly discern the role that opioids play in the regulation of food intake and other processes involving POMC neurons.
Introduction
Proopiomelanocortin (POMC) neurons release multiple peptide transmitters, including α-melanocyte-stimulating hormone and the endogenous opioid beta-endorphin, that modulate aspects of mood, motivation, food intake, and analgesia (Kieffer and Gavériaux-Ruff, 2002; Bertolini et al., 2009; Hegadoren et al., 2009). In addition to releasing an endogenous opioid, POMC neuron activity can be regulated by opioids. POMC neurons express postsynaptic mu opioid receptors (MOR) that are coupled to inhibitory G-proteins and activate a G-protein-coupled inwardly rectifying potassium (GIRK) conductance (Kelly et al., 1990); thus, MOR agonists hyperpolarize POMC neurons and inhibit action-potential firing (Slugg et al., 2000; Cowley et al., 2001; Ibrahim et al., 2003; Roseberry et al., 2004; Hentges et al., 2009). This direct postsynaptic inhibition of POMC neurons by opioids, together with the fact that POMC neurons release beta-endorphin, led to the proposal that POMC neurons may be regulated by feedback inhibition (Kelly et al., 1990; Garcia de Yebenes and Pelletier, 1993).
Opioids can exert their effects through multiple classes of opioid receptors, including mu, delta, and kappa receptors (MOR, DOR, and KOR, respectively). All three of these receptors are coupled to inhibitory G-proteins (Stevens, 2009) and can be found in the arcuate nucleus (Mansour et al., 1994), but only MOR activation has been specifically shown to directly hyperpolarize POMC neurons (Slugg et al., 2000; Ibrahim et al., 2003). Opioid receptors are also expressed presynaptically to POMC neurons and their activation inhibits transmitter-mediated postsynaptic currents in POMC neurons (Hentges et al., 2004, 2009). Studies examining presynaptic regulation of POMC neurons by opioids have used the nonspecific opioid receptor agonist [Met5]-enkephalin, leaving it unclear which opioid receptors are present on terminals presynaptic to POMC neurons.
The expression of various opioid receptors could have functional consequences in terms of presynaptic and postsynaptic regulation, sensitivity to endogenous ligands and administered agonists, and receptor desensitization with chronic exposure to agonist. Thus, the goal of the current study was to determine the distribution and relative contribution of presynaptic and postsynaptic opioid receptors in the regulation of POMC neurons. The data demonstrate that all three opioid receptors examined can act presynaptically to inhibit transmitter release, but only MORs directly inhibit POMC neurons via a postsynaptic mechanism. In addition to differential expression of opioid receptors at presynaptic and postsynaptic sites, the data indicate that there is a significant difference in the sensitivity of presynaptic and postsynaptic MORs.
Materials and Methods
Animals.
Transgenic mice expressing the fluorescent protein Discosoma red (DsRed) in POMC neurons were produced by standard techniques as previously described (Hentges et al., 2009) and backcrossed onto the C57BL/6J background. All experiments were performed in brain slices prepared from 6- to 12-week-old mice. Both male and female mice were used and the data were pooled since there was no significant difference in opioid responses between cells from each sex by any measure compared [inhibition of the amplitude of evoked IPSCs (eIPSCs) by 10 μm DAMGO, p = 0.19; inhibition of the amplitude of eIPSCs by 50 nm DAMGO, p = 0.31; inhibition of the amplitude of eIPSCs by 500 nm U69593, p = 0.32; amplitude of GIRK-mediated outward current induced by 10 μm DAMGO, p = 0.22). All mice were housed under controlled temperatures (22–24°C) with a constant 12 h light/dark cycle. Mice were given standard chow and tap water ad libitum. All animal procedures met United States Public Health Service guidelines and were approved by the Institutional Animal Care and Use Committee
Drugs.
Stock solutions of 6,7-dinitroquinoxaline-2,3(1H,4H) (DNQX; Sigma), (+)-MK-801 (Sigma), and (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide (U69593; Biomol International) were prepared with DMSO at no lower than 10,000× the final concentration. Stock solutions of [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin (DAMGO; Sigma), d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP; Tocris Bioscience), nor-binaltorphimine (nor-BNI; Sigma), [D-Pen2,5]enkephalin (DPDPE; Bachem), naltrindole (Sigma), and bicuculline methiodide (Tocris Bioscience) were prepared with distilled water.
Electrophysiology.
Sagittal brain slices (240 μm) containing the arcuate nucleus were prepared using a Leica VT1200S vibratome. Slices were prepared in ice-cold artificial CSF (aCSF) solution containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 glucose (saturated with 95% O2 and 5% CO2). Slices were allowed to rest in warm (37°C) aCSF solution containing 15 μm MK-801 for at least 45 min before being transferred to the recording chamber, which was continuously perfused with aCSF solution at 37°C saturated with 95% O2 and 5% CO2.
Whole-cell voltage-clamp recordings were made with an internal recording solution containing the following (in mm): 57.5 KCl, 57.5 K-methyl sulfate, 20 NaCl, 1.5 MgCl2, 5 HEPES, 0.1 EGTA, 2 ATP, 0.5 GTP, 10 phosphocreatine, pH 7.3. The cesium-based internal solution had KCl and K-methyl sulfate replaced with 57.5 CsCl and 57.5 Cs-methane sulfonate. For some experiments, GDPβS (1 mm) was used in place of GTP in the internal solution. Recording pipettes had a tip resistance of 1.5–2.3 MΩ when filled with internal solution. Cells were held at −60 mV for recording spontaneous or evoked postsynaptic currents (sPSCs and ePSCs, respectively). Postsynaptic currents (PSCs) were evoked using a 0.5 ms stimulus delivered every 20 s to the slice from a bipolar stimulating electrode. The intensity of the stimulus varied cell-to-cell (∼10–700 μA) and was set at a level that evoked ∼50% of the maximal current for the cell. The stimulating electrode was generally placed in the dorsal arcuate nucleus in the center of the rostral–caudal extent of the POMC cells that were identified based on the presence of the DsRed fluorophore. Recordings were made from cells either rostral or caudal to the stimulating electrode. PSCs were evoked for at least 5 min under baseline conditions to ensure a stable recording. The three ePSCs recorded each minute were averaged together to give a single point to plot for that minute. For analysis, 2 or 3 min of baseline were averaged and compared with the last 1 or 2 min in drug after a steady-state response was reached. sPSCs were collected at 10 kHz and digitally filtered at 1 kHz. sPSCs were detected using Axograph X software based on rise-time kinetics. Recordings were accepted for analysis if access resistance did not exceed 20 MΩ and did not change significantly during the recording. All recordings that tested the effects of the DOR agonist DPDPE were performed in the presence of 500 nm CTAP to block MORs.
Nor-BNI treatment.
POMC–DsRed transgenic mice received a single intraperitoneal injection of the KOR antagonist nor-BNI (10 mg/kg, ∼0.1 ml) or saline 5–7 d before slice preparation.
Statistics.
All comparisons between two datasets were analyzed using t tests. Paired t tests were used when a single cell received two treatments or served as its own control. Multiple groups were compared with one-way ANOVA with Newman–Keuls post hoc paired analysis. Dose–response curves (best fit) were constructed and analyzed using GraphPad Prism software. All data are presented as mean ± SEM. Differences between groups were considered significant if p < 0.05.
Results
Postsynaptic opioid receptors
Postsynaptic opioid receptors are functionally coupled to GIRK channels such that opioids induce a potassium-mediated outward current (Williams et al., 2001). To determine whether specific opioid receptors are functionally expressed in POMC neurons, the ability of selective opioid receptor agonists to induce a GIRK channel-mediated outward current was examined. Whole-cell voltage-clamp recordings (holding potential of −60 mV) were made in POMC cells in the presence of the GABAA receptor antagonist bicuculline (10 μm) and the AMPA receptor antagonist DNQX (10 μm) to block spontaneous IPSCs (sIPSCs) and spontaneous EPSCs (sEPSCs), respectively. The MOR agonist DAMGO (10 μm) induced an outward current of 26 ± 3 pA (n = 28, p < 0.001) (Fig. 1A, top) that was reversed by the MOR selective antagonist CTAP (500 nm).
Figure 1.

Postsynaptic regulation of POMC neurons by selective opioid receptor agonists. A, Example traces showing the direct postsynaptic current in POMC neurons induced by DAMGO (10 μm; top). DAMGO does not induce an outward current when potassium in the recording pipette is replaced with cesium (middle) or when GTP in the pipette is replaced with GDPβS (bottom). B, Example traces showing the lack of any direct postsynaptic current after the application of U69593 (500 nm) or DPDPE (1 μm). Gaps in the traces represent times when voltage steps were applied. C, Agonist-induced currents at voltages ranging from −50 to −130 mV. All data have baseline currents subtracted so that only the agonist-induced current is plotted. All recordings were done in the presence of MK-801 (15 μm), DNQX (10 μm), and bicuculline (10 μm). C, Inset, Sample traces from one cell before and after DAMGO. n = 4, 4, and 7 cells for DPDPE, U69593, and DAMGO, respectively. Error bars represent SEM.
Replacing GTP with GDPβS (1 mm) in the recording pipette prevented DAMGO from inducing a significant outward current (n = 3, p = 0.1, baseline vs DAMGO) (Fig. 1A, bottom). Use of a cesium-based internal recording solution rather than potassium-based internal also prevented DAMGO from inducing a detectable outward current (n = 4, p = 0.1, baseline vs DAMGO) (Fig. 1A, middle). Together, the data indicate that both potassium efflux and functional G-protein signaling are required for the mu agonist to cause an outward current in POMC–DsRed neurons, consistent with the literature for opioid-mediated currents in many other cell types. The outward current caused by DAMGO was not different when TTX (500 nm) was added to the bath solution (31 ± 8 pA compared with control 26 ± 3 pA, n = 6, p = 0.27) and was recorded in the presence of DNQX and bicuculline. Thus, presynaptic release does not appear to affect the postsynaptic response to DAMGO. Altogether, the data indicate that the outward current is a postsynaptic GIRK-mediated current. Neither the KOR agonist U69593 (500 nm) nor the DOR agonist DPDPE (1 μm) induced a postsynaptic current in POMC cells held at −60 mV (n = 4 per group) (Fig. 1B).
The ability of opioid receptor agonists to activate a GIRK conductance was also examined by constructing current/voltage (IV) curves. Current was recorded at voltages ranging from −130 to −50 mV in 10 mV increments. Baseline currents were subtracted from the currents recorded in agonist and plotted (Fig. 1C). The subtracted IV curve for DAMGO (10 μm, n = 7) shows inward rectification and a reversal potential near that of K+. Neither U69593 (500 nm) nor DPDPE (1 μm) produced postsynaptic currents at any of the holding potentials tested (n = 4) (Fig. 1C). Thus, POMC cells express functional postsynaptic mu, but not kappa or delta, opioid receptors that activate a hyperpolarizing current in POMC neurons.
Presynaptic opioid receptors
Opioid receptor activation can cause a robust decrease in transmitter release (Williams et al., 2001). Thus, to determine whether specific opioid receptor subtypes were present on presynaptic inputs to POMC neurons, the ability of selective opioid receptor agonists to inhibit eIPSC and sIPSC was examined. DAMGO (10 μm) reduced the amplitude of eIPSCs to 31 ± 2% of baseline (n = 59, p < 0.001) (Fig. 2A) and inhibited the frequency of sIPSCs to 23 ± 4% of baseline (n = 5, p < 0.001) (Fig. 2B). The selective KOR agonist U69593 (500 nm) also inhibited both the amplitude of eIPSCs (48 ± 5% of baseline, n = 10, p < 0.001) (Fig. 2A) and the frequency of sIPSCs (38 ± 8% of baseline, n = 8, p = 0.001) (Fig. 2B). The DOR selective agonist DPDPE (100 nm) did not inhibit the amplitude of eIPSCs (102 ± 9% of baseline, n = 3, p = 0.38) (Fig. 2A) but did inhibit the frequency of sIPSCs (60 ± 7% of baseline, n = 5, p < 0.01) (Fig. 2B). This difference in the regulation of evoked and spontaneous events may suggest that the terminals containing DORs presynaptic to POMC neurons may be anatomically restricted to a region beyond that affected by the stimulating electrode. However, moving the electrode to the rostral or caudal portion of the nucleus (rather than the central portion) did not change the lack of a response to DPDPE (95 ± 4% of baseline, n = 3; p = 0.18). However, the DOR-containing terminals could terminate on the POMC cells very distant from the soma or approach POMC cells from the ventral aspect where it is difficult to place the stimulating electrode. The delta receptor selective antagonist naltrindole (30 nm) alone had no effect on eIPSC amplitude (n = 7, p = 0.07; data not shown).
Figure 2.
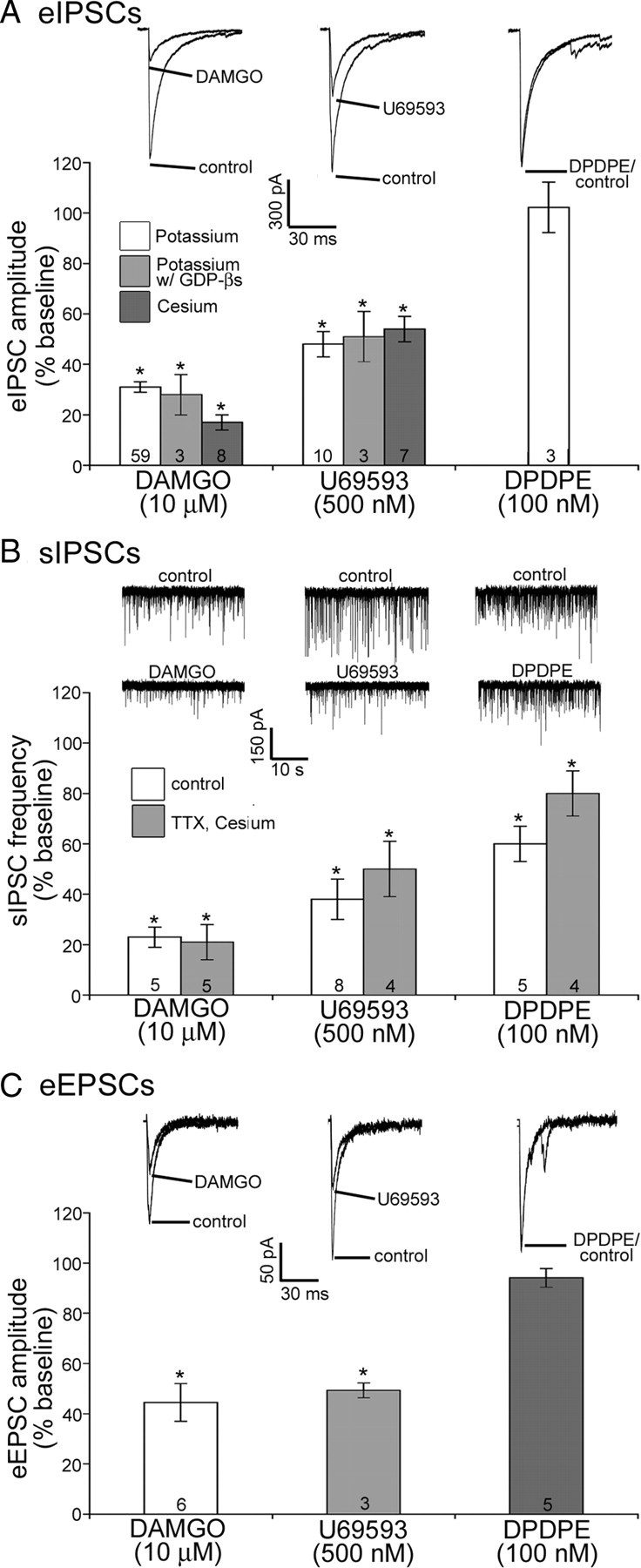
Presynaptic effects of selective opioid receptor agonists. A–C, Example traces and compiled data representing the effects of the MOR agonist DAMGO (10 μm), the KOR agonist U69593 (500 nm), or the DOR agonist DPDPE (100 nm) on the amplitude of evoked IPSCs (A), the frequency of spontaneous IPSCs (B), and the amplitude of eEPSCs (C). Numbers shown in the bar graph indicate number of cells tested for that group. Example traces for evoked data are averaged from three to six consecutive sweeps under control conditions and with drug applied. Error bars represent SEM. *p < 0.05.
DAMGO (10 μm) reduced the amplitude of eIPSCs equally well when GTP was replaced with GDPβS in the internal recording solution (Fig. 2A). U69593 also inhibited eIPSCs to the same extent when GDPβS was substituted for GTP (Fig. 2A) as expected since U69593 did not induce a detectable postsynaptic effect. When cesium was used rather than potassium in the recording pipette DAMGO (10 μm) and U69593 (500 nm) inhibited eIPSCs to an extent similar to that observed with potassium (Fig. 2A).
The majority of spontaneous PSCs in POMC neurons are TTX-insensitive. TTX (500 nm) did not alter the frequency of sIPSCs (93 ± 10% of baseline, n = 13, p = 0.5), consistent with previous reports (Pinto et al., 2004; Hentges et al., 2005). When TTX (500 nm) was added to the bath solution and a cesium-based internal recording solution was used rather than a potassium-based internal solution, all three opioid receptor agonists inhibited the frequency of spontaneous IPSCs comparably to that observed when a potassium-based internal was used in the absence of TTX (Fig. 2B). Thus, the inhibition in sIPSC frequency by opioid agonists appears to be presynaptic and not affected by postsynaptic factors.
There is relatively little glutamatergic input to POMC neurons compared with GABAergic (Pinto et al., 2004, Hentges et al., 2009). Nonetheless, when POMC neurons with substantial EPSCs were found, the ability of selective opioid receptor agonists to inhibit the EPSC was examined. Both DAMGO (10 μm) and U69593 (500 nm) reduced the amplitude of eEPSCs (45 ± 8% of baseline, n = 6, p < 0.01 compared with baseline and 49 ± 3% of baseline, n = 3, p < 0.01, respectively) (Fig. 2C), whereas the DOR selective agonist DPDPE (100 nm) did not inhibit eEPSC amplitude (94 ± 3% of baseline, n = 6, p = 0.25) (Fig. 2C). Altogether, the data indicate that both GABAergic and glutamatergic inputs to POMC neurons express mu and kappa opioid receptors, and suggest that a select population of GABAergic inputs may also express DORs.
Sensitivity of presynaptic and postsynaptic mu opioid receptors
High concentrations of MOR agonist affect both presynaptic and postsynaptic opioid receptors to inhibit transmitter release and activate an outward current, respectively (Figs. 1, 2). To examine presynaptic and postsynaptic receptor sensitivity, dose–response curves were constructed for responses to the MOR selective agonist DAMGO (10 nm-30 μm) (Fig. 3A). In each experiment, the cell was treated with a submaximal concentration of DAMGO followed by a maximal concentration (10 or 30 μm DAMGO). The results are presented as the percentage of maximal response to account for variability between cells. Using this assay, there is no difference in the percentage maximal inhibition of eIPSC amplitude to a low concentration of DAMGO (50 nm) using either a cesium- (63 ± 5% of baseline, n = 8) or potassium-based internal solution (70 ± 3% of baseline, n = 23, p = 0.2). Dose–response curves were constructed using a potassium-based internal so that both presynaptic and postsynaptic responses could be assayed simultaneously. DAMGO (50 nm) inhibited the eIPSC to 70 ± 3% of baseline (n = 23, p ≤ 0.001), but did not induce a notable postsynaptic outward current (3.8 ± 1.3 pA, n = 9) whereas a high concentration of DAMGO (10 μm) inhibited eIPSCs (to 30 ± 2% of baseline, p ≤ 0.001, n = 64) and activated a GIRK conductance (26.2 ± 2.9 pA, p < 0.001, n = 28) (Fig. 3B,C). The dose–response curve is significantly left-shifted for presynaptic inhibition (EC50 = 80 nm) versus outward postsynaptic current (EC50 = 353 nm, p < 0.0001, compared with presynaptic curve) (Fig. 3A), indicating that, although high concentrations of agonist elicit both presynaptic and postsynaptic responses, low concentrations of agonist will likely inhibit presynaptic transmitter release without causing a direct postsynaptic hyperpolarization.
Figure 3.

Dose–responses for presynaptic and postsynaptic MORs. A, Dose–response curves for presynaptic (circles) and postsynaptic (squares) responses induced by DAMGO. Data are plotted as a percentage of the inhibition of the amplitude of eIPSCs or outward current induced by a maximal concentration of DAMGO (10 or 30 μm). Numbers shown in parenthesis indicate number of cells for that group. Error bars represent SEM. B, An example trace showing the attenuation of eIPSC amplitude induced by 50 nm and 10 μm DAMGO and the reversal by naloxone (NLX). C, An example trace showing the ability of 10 μm, but not 50 nm, DAMGO to induce an outward postsynaptic current. Evoked IPSCs were recorded while monitoring whole-cell current such that the downward deflections that occur at even (20 s) intervals and have the same amplitudes as one another represent the transient current induced by the stimulus pulse. Example traces for evoked data are averaged from three to six consecutive sweeps under control condition and with the given dose of drug applied.
Presynaptic colocalization of MORs and KORs
In POMC neurons, both mu and kappa opioid receptor agonists caused a robust (>50%) decrease in eIPSC amplitude (Fig. 2A), raising the possibility that the two receptors could be expressed on the same presynaptic terminals. To determine whether KORs are localized to terminals that also express MORs, a maximal concentration of DAMGO (10 μm) was applied until the amplitude of the eIPSCs reached peak inhibition (three consecutive sweeps with no further reduction in amplitude). Once peak inhibition (28 ± 5% of baseline, p < 0.001) (Fig. 4A) was achieved with DAMGO, a maximal concentration of U69593 (500 nm) was applied in the presence of DAMGO (10 μm). U69593 was applied for at least 5 min without further reducing eIPSC amplitude compared with DAMGO alone (24.1 ± 6% of baseline vs 27.8 ± 4.8% of baseline, p > 0.05). After at least 5 min of coapplication of DAMGO and U69593, DAMGO was replaced by the MOR selective antagonist CTAP (500 nm), which only partially reversed the inhibition observed in the presence of both DAMGO and U69593 (to 55 ± 7% of baseline, p < 0.01). Although DAMGO was not coapplied with CTAP in this experiment, the reversal observed is attributed to CTAP since CTAP causes a maximal reversal within 3.8 ± 0.1 min and the DAMGO-induced inhibition of eIPSC amplitude is stable for >5 min after application is stopped (27 ± 8% of baseline at end of DAMGO treatment vs 27 ± 5% of baseline 5 min into wash, n = 3, p = 0.5). CTAP was perfused over the slice for at least 5 min and the concentration of CTAP used is sufficient to reverse the DAMGO-induced inhibition of eIPSC amplitude in the absence of U69593 (104 ± 7% of baseline, n = 6, p = 0.35 compared with baseline) (Fig. 4B, inset). Thus, the remaining inhibition of eIPSC amplitude after MORs are antagonized must be due to the inhibition induced by the KOR agonist U69593. Indeed, the addition of the KOR antagonist nor-BNI (100 nm), along with the MOR antagonist CTAP, caused a complete reversal of the inhibition of eIPSC amplitude caused by the agonists (105 ± 5% of baseline, p < 0.001) (Fig. 4A,B). The peak reversal induced by nor-BNI occurred within 3.8 ± 0.5 min whereas it took 9 ± 3 min to return to baseline after termination of nor-BNI in the absence of antagonist. Therefore, the washing out of U69593 does not contribute to the reversal observed with nor-BNI. Altogether, the data suggest that KOR and MOR agonists inhibit the same inhibitory presynaptic terminals.
Figure 4.

Colocalization of presynaptic MORs and KORs. A, A plot of eIPSC amplitudes over time from a representative experiment showing the effects of DAMGO, DAMGO plus U69593, U69593 plus CTAP, and CTAP plus nor-BNI on eIPSC amplitude in a POMC neuron. Each point represents three averaged sweeps. B, Compiled data from eight cells treated as in A. B, Inset, DAMGO (10 μm) alone can be reversed by CTAP (500 nm, n = 6). C, Spontaneous IPSC frequency under the same conditions as in A and B (n = 5). a, Significantly different from baseline; b, significantly different from DAMGO plus U69593; c, significantly different from U69593 plus CTAP. Error bars represent SEM.
To verify that mu and kappa opioid receptor colocalization may occur on the majority of inputs to POMC neurons, not just the specific inputs that were near the stimulating electrode, the above experiments were repeated examining spontaneous rather than evoked IPSCs (Fig. 4C). DAMGO (10 μm) reduced sIPSC frequency to 23 ± 4% of baseline (p < 0.001). The addition of U69593 (500 nm) caused a further inhibition of sIPSC frequency (to 6 ± 1% of baseline, p < 0.01). Replacing DAMGO with CTAP (500 nm) partially reversed the inhibition caused by DAMGO and U69593 together (to 33 ± 8% of baseline, p < 0.05). When U69593 was replaced by nor-BNI (100 nm) while still in the presence of CTAP, there was a complete reversal of the inhibition of sIPSC frequency (98 ± 3% of baseline, p = 0.001). Thus, spontaneous IPSCs had the same pattern of responses as found for eIPSCs. All cells in both the eIPSC and sIPSC experiments received each of the treatments so that each cell served as its own control (n = 8 for eIPSCs; n = 5 for sIPSCs). Together, the data indicate that a significant portion of inhibitory inputs to POMC neurons express both mu and kappa opioid receptors.
Possible interaction between mu and kappa opioid receptors
Opioid receptors can form functional dimers in various combinations (Alfaras-Melainis et al., 2009); thus, the possibility that the presynaptic colocalization of MORs and KORs could be functionally significant was explored. A single intraperitoneal injection of nor-BNI at a dose of 10 mg/kg has been shown to be sufficient to completely eliminate KOR function for over a week after the injection (Bruchas et al., 2007). POMC–DsRed transgenic mice received a single intraperitoneal injection of nor-BNI (10 mg/kg) and brain slices were prepared 5–7 d later. U69593 (500 nm) did not reduce eIPSC amplitude in cells from nor-BNI-treated mice (Fig. 5B) as it did in untreated mice (103 ± 3% of baseline, n = 4, p = 0.32 vs 48 ± 5% of baseline in controls) (Fig. 5B, inset), confirming that the nor-BNI treatment left the KORs nonfunctional. Evoked IPSCs were recorded and the effects of low and high doses of DAMGO on eIPSC amplitude were examined to determine whether functional removal of KOR signaling altered either sensitivity or efficacy of DAMGO at MORs. There was no significant difference in the inhibition of the amplitude of eIPSCs by 50 nm DAMGO between control and nor-BNI-treated mice (29 ± 3% in controls, n = 23 vs 28 ± 4% in nor-BNI-treated, n = 6, p = 0.93) (Fig. 5), or between the maximal inhibition of eIPSC amplitude by 10 μm DAMGO (70 ± 2% in controls, n = 64 vs 66 ± 6% in nor-BNI-treated, n = 6, p = 0.68). Thus, effectively inactivating KORs did not alter the sensitivity or efficacy of MOR agonist, suggesting that although these two receptors can serve similar functions, their activities are independent from one another.
Figure 5.

Loss of KOR signaling does not affect MOR function. A, B, DAMGO (50 nm and 10 μm) induced inhibition of eIPSC amplitude in a POMC neuron from an untreated (A) or nor-BNI-treated (B) mouse. Each point represents three averaged sweeps. B, Inset, Example trace showing the effect of U69593 in a cell from an untreated mouse. C, Mean responses to 50 nm and 10 μm DAMGO in POMC neurons from untreated (open bars) and nor-BNI-treated (filled bars) mice. Number of cells in each group is noted in the bar. Error bars represent SEM.
Postsynaptic MORs display acute desensitization, as demonstrated by a reduction in the GIRK-mediated current elicited by a high concentration of agonist over a few minutes (10 or 30 μm DAMGO caused a peak outward current of 19 ± 8 pA at minute 2 that desensitized to 4 ± 6 pA at minute 10, n = 5, p < 0.01). However, presynaptic responses do not display acute desensitization to a maximal concentration of DAMGO using ePSC amplitude as the assay (Fig. 5A). To determine whether KOR signaling might play a role in preventing presynaptic MOR desensitization, the high concentration of MOR selective agonist (DAMGO, 10 μm) was perfused onto slices from untreated and nor-BNI-treated mice for 10 min. The peak inhibition of eIPSC amplitude was reached by 2 min and there was no difference in amplitude between minutes 2 and 10 of treatment in slices from either untreated mice (35 ± 5% vs 34 ± 3% of baseline for minutes 2 and 10, respectively; n = 5, p = 0.77) (Fig. 5A) or treated mice (32 ± 8% vs 36 ± 5% of baseline for minutes 2 and 10, respectively; n = 5, p = 0.55) (Fig. 5B). Therefore, it appears that KOR signaling alone is not responsible for the lack of presynaptic desensitization observed with this high dose protocol. Altogether, the data from nor-BNI-treated mice suggest that elimination of KOR function has little or no effect on the MORs even though they are colocalized at inhibitory terminals presynaptic to POMC neurons.
Discussion
The present data demonstrate that MORs alone mediate the opioid-induced hyperpolarization of POMC neurons, whereas mu, kappa, and delta opioid receptors mediate the presynaptic inhibition by opioids. The EC50 for presynaptic actions at MORs is ∼4.4-fold lower than for postsynaptic actions. Altogether, the results suggest that opioid receptor agonists with selectivity for specific subtypes of opioid receptors may differentially regulate POMC neurons. Additionally, the concentration and localization of MOR agonist present may be an important determinant of whether presynaptic or postsynaptic actions will predominate. Although the present study focused on regulation of POMC neurons by exogenously applied opioids, the data suggest that the various endogenous opioids may have distinct actions depending on concentration and localization of release (i.e., to act primarily presynaptically or postsynaptically).
Opioid receptor localization
Previous studies using nonspecific opioid receptor agonists have indicated the presence of functional postsynaptic opioid receptors in mouse POMC neurons (Cowley et al., 2001; Hentges et al., 2004, 2009; Roseberry et al., 2004). The direct postsynaptic actions of MOR-selective agonists found here are consistent with prior reports in mouse POMC neurons (Slugg et al., 2000; Ibrahim et al., 2003). The present data show that, unlike the MOR selective agonist, neither KOR nor DOR selective agonists elicit a postsynaptic outward current, indicating that the predominant opioid receptor coupled to GIRK channels in mouse POMC neurons is the MOR.
Evidence for presynaptic regulation of POMC neurons by opioids is sparse and limited by the use of a nonselective opioid receptor agonist (Roseberry et al., 2004; Hentges et al., 2009). The present study took a systematic approach to examine the involvement of mu, kappa, and delta opioid receptors in the presynaptic regulation of POMC neurons. The data not only demonstrate that all three receptors are present on presynaptic GABAergic terminals, but also indicate two particularly interesting aspects of presynaptic opioid regulation. First is the finding that presynaptic terminals can coexpress both mu and kappa receptors. The presence or absence of functional kappa receptors does not appear to overtly alter the function of MORs and both MORs and KORs serve to inhibit presynaptic transmitter release with similar maximal effects. However, these receptors have varying affinities for endogenous ligands found in the arcuate, with beta-endorphin being selective for MORs and dynorphin for KORs, for example. Thus, presynaptic regulation of POMC neurons by opioids may be determined by the distribution of endogenous opioids in addition to the localization of presynaptic and postsynaptic opioid receptors.
The second interesting finding regarding presynaptic opioid receptors is that the DOR-selective agonist inhibited spontaneous but not evoked IPSCs. The simplest explanation for this would be that the DOR-containing fibers terminate on POMC neurons in a region that was not recruited by the stimulating electrode. Along with the other data here, this would suggest that there are three populations of opioid-sensitive terminals: those expressing only MORs, those expressing both MORs and KORs, and a third population expressing only DORs. However, the present data cannot rule out the possibility that DORs could function to inhibit spontaneous transmitter release, but not evoked in the same terminal. Additionally, it may be that functional DORs would be more heavily expressed after MORs are desensitized (Chieng and Christie, 2009) and that increasing the number of functional DORs could enable inhibition of evoked transmitter release. Future studies will need to further explore the nature of the DOR-mediated inhibition.
Activation of presynaptic and postsynaptic MOR receptors
MORs mediated both presynaptic and postsynaptic effects. However, the EC50 at presynaptic and postsynaptic MORs differed by ∼4.4-fold (∼80 and 350 nm, respectively). The EC50 for GIRK conductance activation found in the present study is higher than reported in many cell types, but is consistent with other reports in POMC neurons (Slugg et al., 2000; Ibrahim et al., 2003). Interestingly, under the same recording conditions, the presynaptic inhibition mediated by DAMGO occurred at much lower concentrations. The explanation for the relatively high sensitivity of presynaptic MORs compared with postsynaptic MORs remains to be determined, but differences in receptor number, reserve, or coupling to effectors could be involved.
The finding that POMC neurons express MORs in addition to producing the endogenous MOR ligand beta-endorphin suggests that MORs may act as autoreceptors on POMC neurons (Kelly et al., 1990; Garcia de Yebenes and Pelletier, 1993). The present data do not exclude that possibility, but indicate that an autoregulatory mechanism may involve both a presynaptic and postsynaptic component with presynaptic inhibition predominating when opioid levels are insufficient to activate postsynaptic MORs. In the presence of high concentrations of agonist, the postsynaptic activation of a potassium conductance and the resulting hyperpolarization would likely minimize the significance of inhibited presynaptic transmitter release. However, high concentrations of agonist cause postsynaptic MORs to desensitize in many cells, including POMC neurons [current data and Roseberry et al. (2004)], whereas the presynaptic receptor-mediated inhibition of PSCs does not seem to desensitize under the same conditions (Fig. 5A and data in text). Therefore, presynaptic MOR agonists may play an important role in regulating POMC neuron activity when high levels of agonist persist long enough to cause postsynaptic receptor desensitization as well as when agonist concentrations are too low to affect postsynaptic MORs. The differential sensitivity of presynaptic and postsynaptic MORs may partly explain why opioids acutely stimulate food intake and yet antagonist therapy is not effective for weight loss (Levine et al., 1985). The current results suggest that high concentrations of opioids acutely inhibit anorexigenic POMC neurons via postsynaptic MORs. However, opioid receptor antagonists would only block endogenous opioid actions, which would be predominately presynaptic due to low levels of endogenous opioids or because postsynaptic responses would be desensitized if high local concentrations of endogenous opioids existed. Further experiments to determine whether other neurons in the food intake and reward pathways also display differential expression and sensitivity at presynaptic and postsynaptic opioid receptors will help further clarify the specific contribution of opioids in the regulation of food intake.
Conclusions
POMC neurons play an important role in essential processes, such as the regulation of food intake, reward, and antinociception. POMC neurons not only produce an endogenous opioid, but are also heavily regulated by opioids. The results here demonstrate that opioid regulation of POMC neurons occurs through postsynaptic MORs and presynaptic mu, kappa, and delta opioid receptors. The restricted expression of KORs and DORs on presynaptic terminals and the increased sensitivity of presynaptic MORs compared with postsynaptic MORs indicate that the regulation of POMC neurons by opioids involves both presynaptic and postsynaptic actions. The data suggest that the effects that endogenous or exogenous opioids will have on POMC neuron regulation will depend on receptor specificity and the concentration of agonist.
Footnotes
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK0798749 to S.T.H. We thank Connie King and Alyssa Strieby for providing excellent technical assistance, Drs. Malcolm J. Low and Veronica Otero-Corchon for providing the POMC-DsRed transgenic mouse line, and Dr. John T. Williams for helpful discussions and suggestions.
References
- Alfaras-Melainis K, Gomes I, Rozenfeld R, Zachariou V, Devi L. Modulation of opioid receptor function by protein-protein interactions. Front Biosci. 2009;14:3594–3607. doi: 10.2741/3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini A, Tacchi R, Vergoni AV. Brain effects of melanocortins. Pharmacol Res. 2009;59:13–47. doi: 10.1016/j.phrs.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ. Chronic morphine treatment induces functional delta-opioid receptors in amygdala neurons that project to periaqueductal grey. Neuropharmacology. 2009;57:430–437. doi: 10.1016/j.neuropharm.2009.06.034. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Garcia de Yebenes E, Pelletier G. Opioid regulation of proopiomelanocortin (POMC) gene expression in the rat brain as studied by in situ hybridization. Neuropeptides. 1993;25:91–94. doi: 10.1016/0143-4179(93)90087-q. [DOI] [PubMed] [Google Scholar]
- Hegadoren KM, O'Donnell T, Lanius R, Coupland NJ, Lacaze-Masmonteil N. The role of beta-endorphin in the pathophysiology of major depression. Neuropeptides. 2009;43:341–353. doi: 10.1016/j.npep.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ. GABA release from proopiomelanocortin neurons. J Neurosci. 2004;24:1578–1583. doi: 10.1523/JNEUROSCI.3952-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Low MJ, Williams JT. Differential regulation of synaptic inputs by constitutively released endocannabinoids and exogenous cannabinoids. J Neurosci. 2005;25:9746–9751. doi: 10.1523/JNEUROSCI.2769-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci. 2009;29:13684–13690. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Ronnekleiv OK. Opioids hyperpolarize beta-endorphin neurons via mu-receptor activation of a potassium conductance. Neuroendocrinology. 1990;52:268–275. doi: 10.1159/000125597. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gavériaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Levine AS, Morley JE, Gosnell BA, Billington CJ, Bartness TJ. Opioids and consummatory behavior. Brain Res Bull. 1985;14:663–672. doi: 10.1016/0361-9230(85)90116-9. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, Watson SJ. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350:412–438. doi: 10.1002/cne.903500307. [DOI] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Roseberry AG, Liu H, Jackson AC, Cai X, Friedman JM. Neuropeptide Y-mediated inhibition of proopiomelanocortin neurons in the arcuate nucleus shows enhanced desensitization in ob/ob mice. Neuron. 2004;41:711–722. doi: 10.1016/s0896-6273(04)00074-1. [DOI] [PubMed] [Google Scholar]
- Slugg RM, Hayward MD, Ronnekleiv OK, Low MJ, Kelly MJ. Effect of the mu-opioid agonist DAMGO on medial basal hypothalamic neurons in beta-endorphin knockout mice. Neuroendocrinology. 2000;72:208–217. doi: 10.1159/000054589. [DOI] [PubMed] [Google Scholar]
- Stevens CW. The evolution of vertebrate opioid receptors. Front Biosci. 2009;14:1247–1269. doi: 10.2741/3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]