

Abstract
Background. High mortality in the 2001 US and recent European anthrax outbreaks suggests that better understanding of the effects of the toxins produced by this bacterium is needed to improve treatment.
Methods and results. Here, 24-h edema (ETx) and lethal (LeTx) toxin infusions were investigated for 96 h in sedated canines receiving mechanical ventilation. The initial study compared similarly lethal doses of ETx (n=8) or LeTx (n=15) alone. ETx was 24 times less lethal than LeTx, and the median time to death in nonsurvivors (n=6 and n=9, respectively) was shorter with ETx (42 vs 67 h; P=.04). Compared with controls (n=9), both toxins decreased arterial and central venous pressures and systemic vascular resistance and increased heart rate, cardiac index, blood urea nitrogen (BUN) level, creatinine (Cr) concentration, BUN:Cr ratio, and hepatic transaminase levels (P ⩽ .05 for toxin effect or time interaction). However, ETx stimulated early diuresis, reduced serum sodium levels, and had more pronounced vasodilatory effects, compared with LeTx, as reflected by greater or earlier central venous pressures, systemic vascular resistance, and changes in the BUN:Cr ratio (P ⩽ .01). LeTx progressively decreased the left ventricular ejection fraction (P ⩽ .002). In a subsequent study, a lethal dose of LeTx with an equimolar nonlethal ETx dose (n=8) increased mortality, compared with LeTx alone (n=8;P=.05).
Conclusion. Shock with ETx or LeTx may require differing supportive therapies, whereas toxin antagonists should likely target both toxins.
Mortality among patients with shock during the 2001 US anthrax outbreak was high [1, 2]. Although the mechanisms and optimal management of this shock remain unclear, anthrax risk persists, as demonstrated by recent cases of highly fatal soft-tissue infection among intravenous drug users in the United Kingdom and Europe [3, 4].
Lethal (LeTx) and edema (ETx) toxins are important in the pathogenesis of anthrax [5, 6]. Both are binary-type toxins that, in addition to lethal factor and edema factor, include protective antigen (PA), the component necessary for binding and uptake by host cells. Lethal factor is a zinc endopeptidase that cleaves mitogenactivated protein kinase kinases, and edema factor is a potent calmodulin-dependent adenyl cyclase.
Despite the importance of LeTx and ETx during anthrax infection, there has been limited characterization of their cardiovascular effects [6–9]. Most studies are in small animals, in which measurement of cardiac preload, afterload, and output are difficult and monitoring is of limited duration. The effect of cardiovascular changes with toxin on noncardiac organ dysfunction has also not been assessed. Incomplete understanding of the pathophysiologic effects of LeTx and ETx hampers the development of beneficial supportive strategies. Therefore, in the present investigation recombinant LeTx, ETx, or control challenge was infused over 24 h in sedated and instrumented canines receiving mechanical ventilation; canines are an anthrax-susceptible species [10]. Cardiopulmonary, renal, and hepatic measurements were performed up to 72 h after completion of toxin infusions. To define the influence of fluid administration on the hemodynamic effects of the toxins, measurements were repeated daily after volume loading. An initial study (study 1) investigated and compared the individual effects of similarly lethal dose ranges of each toxin, and 50% lethal doses (LD50) were determined. A second study (study 2) investigated the effects of an LD50 of LeTx and an equimolar dose of ETx administered alone and together.
Methods
Study design. The National Institutes of Health Clinical Center Animal Care and Use Committee approved all experiments. Study 1 (experiments 1–11) used a modified up-down method to determine the LD50 of LeTx or ETx infusions given over 24h and provided data to investigate similarly lethal dose ranges of each toxin (Table 1) [11]. Study 2 (experiments 12–20) compared an LD50 LeTx dose, a similar molar ETx dose, and the combination.
Table 1.
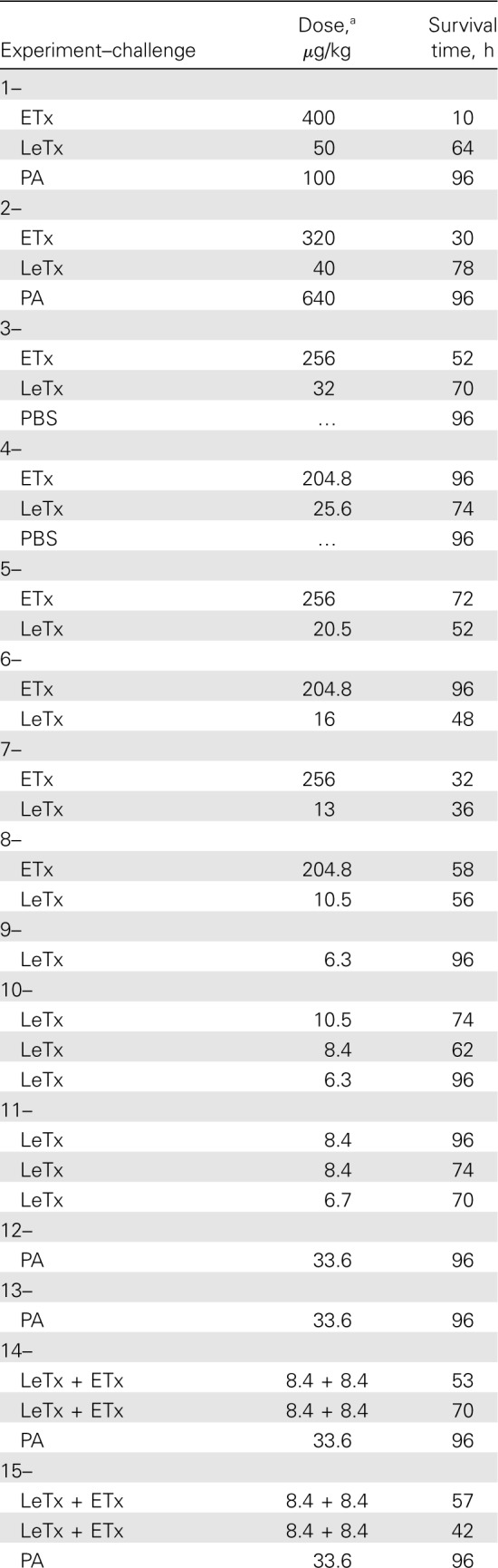
Summary of the Challenge Type, Dose, and Associated Survival in Individual Animals in Sequential Experiments
Weekly, 1–3 anesthetized purpose-bred beagles had venous, pulmonary and systemic arterial, and urinary catheters and tracheostomy placed as described elsewhere [12]. Anesthesia was weaned, and sedation, maintenance fluid, and mechanical ventilation were initiated and continued using standardized intensive care unit protocols. Maintenance fluid and ventilatory support were maintained at constant levels and were similar in all groups. Cardiopulmonary, renal, hepatic, and complete blood count measurements were obtained immediately before and at 8, 24, 48, 72, and 96 h after initiation of a 24-h toxin or control (diluent [phosphate-buffered saline] or PA alone) infusion. Some measurements were also obtained serially at 8- h intervals. Once daily after initial measurements, animals received normal saline volume loading (40 mL/kg intravenously over 40 min), and cardiopulmonary measures were repeated. Ejection fraction was determined using ultrasonography. Surviving animals were euthanized after final measures.
Toxin. Recombinant toxin components were prepared as described elsewhere [13–15]. Toxin doses are reported as the amount of edema factor or lethal factor used. All animals received similar volumes of toxin or control agent. Control PA doses were equal to doses administered with a toxin factor in the same experiment (Table 1). In some animals from study 1, serum PA was measured (by enzyme-linked immunosorbent assay), in addition to other parameters.
Figure 1.

A and B, Survival times associated with decreasing doses of edema (ETx) or lethal (LeTx) toxin in individual animals from study 1 (reported as the dose of edema or lethal factor used). Gray circles indicate doses that were associated with both survivors (survival time, 96 h) and nonsurvivors (survival time, <96 h), and black circles indicate doses for which no animal survived (termed low- and high-dose groups, respectively). C and D, Proportion of animals surviving over time with the administration of either low or high doses of ETx or LeTx or diluent only (controls) in study 1. E, Proportion of animals surviving over time in study 2, which compared LeTx and ETx alone or together with controls. Control animals (C, D, and E) represent animals investigated in both studies 1 and 2. The horizontal arrow (†) indicates the 24-h period over which toxin or control challenge was infused.
Statistical methods. In study 1, initial toxin doses used for the up-down design were based on those used in prior rat studies [11, 16–18]. Doses in subsequent experiments were either increased or decreased by 20% on the basis of the survival results from the preceding experiment. The final LD50 was determined after survival was reversed in 2 successive experiments. Survival times (study 1) and survival curves (study 2) were analyzed with rank and Wilcoxon tests, respectively.
Serial changes from baseline were analyzed for all parameters using repeated-measures analysis of variance with PROC MIXED in SAS software (version 9.1; SAS Institute). However, for clarity in figures serial toxin effects (ie, toxin minus control) at each time point are shown. Fluid effects represent changes from before to after fluid. It was determined a priori that if changes in controls across the 2 studies were similar they would be combined for analysis, and this was the case. Three animals receiving an estimated LD50 of LeTx in study 1 were combined with animals receiving LeTx alone (n=5) in analysis in study 2. Differences with P ⩽ .05 were considered to be statistically significant, and P values were unadjusted for multiple comparisons. All animals from each group that were alive at a particular time point were included in analysis (Figure 1C–1E).
Results
Study 1: Effects of Similarly Lethal Dose Ranges of ETx and LeTx
Survival. Fewer dose adjustments were required to establish LD50 values with ETx (205 µg/kg) than with LeTx (8.4 µg/kg) (Table 1 and Figure 1A and 1B). For subsequent analyses of study 1, doses of each toxin associated with no survivors were combined in groups termed “high dose,” and doses associated with both survivors and nonsurvivors were combined in groups termed “low dose.” Overall, the median time to death in nonsurvivors receiving ETx was shorter than that in nonsurvivors receiving LeTx (42 h [interquartile range, 30–58 h] vs 67 h [interquartile range, 54–74 h]; P=.04) (Figure 1C and 1D).
Cardiovascular measures. Compared with controls (data not shown), both low- and high-dose ETx decreased mean arterial pressure (MAP), central venous pressure (CVP), and systemic vascular resistance index (SVRI) and increased heart rate (HR), and low-dose ETx increased cardiac index (CI; P ⩽ .02 for each) (Figure 2). Decreases in MAP with low-dose ETx and increases in HR with high-dose ETx were resolving later (P ⩽ .02 for the time interactions). Neither ETx dose altered pulmonary artery occlusion pressure (PAOP), stroke volume index (SVI), or left ventricular ejection fraction (LVEF) significantly, although high-dose ETx had variable effects on LVEF (P=.06 for a time interaction).
Figure 2.

Mean serial effects (± standard error of the mean) in a comparison to controls (see Methods) with low (LD) and high (HD) doses of edema (ETx) and lethal (LeTx) toxin on changes from baseline in study 1, for mean arterial blood pressure (MAP), heart rate (HR), central venous pressure (CVP), cardiac index (CI), systemic vascular resistance index, and left ventricular ejection fraction (LVEF). The shaded area indicates the 24-h toxin infusion period. P values are shown for the overall effect of toxin compared to control (pα) and for the interaction between time and this effect (pβ). Increases or decreases with toxin, compared with controls, are indicated by symbols above or below the dashed horizontal no-effect line, respectively. Because measurements were performed in all animals available for study at each time point, the number of measures used for analysis are shown by the number of animals surviving from each group at a particular time point in the Kaplan-Meier curves in Figure 1C and 1D. For clarity of presentation, however, the toxin effects (ie, toxin minus control) are shown.
Compared with controls, both LeTx doses decreased MAP and CVP and increased HR (P ⩽ .05). With low-dose LeTx, MAP decreases lessened late, whereas the effect of both doses on HR progressively increased (P ⩽ .03 for the time interactions). After an initial small increase in LVEF, both LeTx doses caused decreases in LVEF (P ⩽ .01) that progressed (P ⩽ .02 for the time interactions). Low-dose LeTx increased CI and reduced SVRI (P ⩽ .02), and changes in the latter progressed (P=.03 for the time interaction). Neither dose of LeTx altered PAOP or SVI significantly.
In a comparison of either the low or high doses of the 2 toxins, during infusion (4–24 h) both ETx doses produced greater increases in HR and decreases in CVP than did LeTx (P ⩽ .02) (Figure 2). Both ETx doses also produced greater increases in CI and decreases in SVRI than did LeTx, but these differences were statistically significant only with the high doses (P ⩽ .02). After toxin infusion (32–96 h), low and high ETx doses produced greater reductions in SVRI than did low and high LeTx doses (P=.06 and Pp.03, respectively).
Renal and hepatic measures. Compared with controls, both low- and high-dose ETx caused increases in blood urea nitrogen (BUN) level (P < .0001) that resolved with the former but progressed with the latter (P < .0001 for the time interactions) (Figure 3). High-dose ETx caused increases in creatinine (Cr) level (P ⩽ .001) that progressed (P=.001 for a time interaction). Both doses increased the BUN:Cr ratio (P < .0003), although the effect of low-dose ETx decreased late (P= .0001 for a time interaction). Low-dose ETx increased urine output (P=.01) early (0–24 h) and late (72–96 h), whereas high-dose ETx only increased output early (P < .0003 for the time interactions). Low-dose ETx caused marked decreases in serum sodium level (P < .0001) early (P=.01 for a time interaction).
Figure 3.

Mean serial effects (± standard error of the mean) in a comparison of controls with low (LD) or high (HD) doses of edema (ETx) and lethal (LeTx) toxin on changes from baseline in study 1 for blood urea nitrogen (BUN) level, creatinine (Cr) level, BUN:Cr ratio, sodium (Na; 24-h urine output), and aspartate aminotransferase (AST) level. The format is similar to Figure 2.
Both LeTx doses produced progressive increases in BUN and Cr levels (P ⩽ .002 for the time interactions). Whereas low-dose LeTx increased BUN:Cr (P=.01), high-dose LeTx did not. Neither LeTx dose altered urine output or serum sodium level. Both doses of ETx and LeTx caused increases in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels (P ⩽ .01) that progressed (P < .0001 for the time interactions).
In a comparison of toxins, during infusion both ETx doses increased urine output (measured at 24 h) and AST level and decreased serum sodium level, compared with LeTx (P ⩽ .02 for each). After toxin infusion, both ETx doses produced greater increases in BUN level and BUN:Cr than did LeTx (P ⩽ .04, except for BUN:Cr with high-dose LeTx [P=.17]).
Arterial blood gas, other pulmonary measures, and circulating cells. Compared with controls, both doses of ETx and LeTx decreased pH and arterial base excess and high doses increased lactate levels (P ⩽ .01) in patterns that improved late with low doses but worsened with high doses (P < .0004 for the time interactions; data not shown). Low-dose LeTx increased lactate late (P=.02). Both ETx doses decreased plateau airway pressure (P ⩽ .01) but did not alter any other pulmonary parameter significantly. Although low-dose LeTx increased pulmonary artery mean pressure over time (P < .004 for a time interaction), neither LeTx dose altered any other pulmonary parameter significantly.
Compared with controls, both ETx doses reduced circulating neutrophil counts significantly (P=.003) and platelet counts (P=.06 and Pp.05 for low and high doses, respectively; data not shown). High-dose ETx also reduced lymphocyte counts late (P=.05 for the time interaction). Neither ETx dose altered hemoglobin levels significantly. LeTx did not alter these parameters significantly.
Protective antigen measures.Protective antigen measures. PA levels in phosphate-buffered saline controls (n=2) were undetectable. In toxin-challenged animals, PA levels increased, were at a maximum at 24 h, and subsequently decreased (data not shown). Mean levels (± standard error of the mean) at 24 h were 534±47 ng/mL (n=3) and1177±101 ng/mL (n=2) in the low- and highdose ETx groups, respectively, and 34±3 ng/mL (n=6) and 65±11 ng/mL (n=7) in the low- and high-dose LeTx groups, respectively.
Effects of fluid administration on cardiovascular measures. In controls, daily fluid challenges produced consistent increases in CVP, PAOP, MAP, and HR (P ⩽ .01 for the comparison of means of the prefluid measurements with means of the postfluid measurements, averaged over the daily infusions) but not in CI or other hemodynamic parameters (Figure 4). In patterns that did not differ significantly from those in controls, fluids with both doses of ETx and LeTx increased CVP, PAOP, and MAP. However, contrary to the findings in controls, fluids with both doses of ETx and LeTx increased CI throughout (P ⩽ .05 for the comparison of the effects of fluid with toxin with those of fluid with controls) or in patterns that varied with time (P ⩽ .02 for the time interactions). Similar to controls, fluids increased HR with low-dose ETx but decreased it with high-dose ETx (P ⩽ .03). Also different from controls, fluids increased SVI with high-dose ETx or LeTx (P ⩽ .02) and increased LVEF over time with high-dose LeTx (P=.01 for the time interaction). Fluids did not alter any pulmonary parameter significantly differently in a comparison of control and toxin animals (data not shown).
Figure 4.

Mean effects (± standard error of the mean) of a fluid bolus (40 mL/kg intravenously over 40 min) in study 1 for central venous pressure (CVP), pulmonary artery occlusion pressure (PAOP), mean arterial blood pressure (MAP), and cardiac index (CI) in animals receiving 24-h infusions with low (LD) or high (HD) doses of edema (ETx) or lethal (LeTx) toxin or in controls. P values are for the comparison between the effect of fluid in toxin animals versus control (pα) and for the interaction between time and these comparisons (pβ).
Study 2: Effects of an LD50 of LeTx and a Similar Molar Dose of ETx Alone or Together
Survival. Compared with controls (9 of 9 survived), the LD50 of LeTx caused significant mortality (3 of 8 survived; P= .01), whereas the eqimolar dose of ETx did not (4 of 4 survived) (Figure 1E). Infusion of both this ETx and LeTx LD50 together also reduced survival (0 of 8 survived), compared with controls (P < .0001) and LeTx alone (P=.05 for the comparison of LeTx with ETx and LeTx without ETx).
Cardiovascular, renal, hepatic, pulmonary, and circulating cell measures. Compared with controls, the nonlethal ETx dose administered alone decreased HR and CVP and increased hemoglobin (data not shown) and serum sodium levels (P ⩽ .03 for HR and HB; Pp.06 and Pp.07 for CVP and serum sodium level, respectively) but did not alter other measures significantly (Figures 5 and 6). LeTx alone reduced MAP late (P=.002 for the time interaction). Both LeTx alone or with ETx increased HR and reduced CVP, SVRI, and LVEF (P ⩽ .04); changes in HR and LVEF were greater late (P ⩽ .001 for the time interactions). Both LeTx alone or with ETx increased Cr levels late (P ⩽ .02 for the time interactions), whereas LeTx alone increased BUN levels late (P < .0001 for the time interactions) and the combination increased serum sodium levels (P=.01). Both LeTx alone or with ETx increased AST, ALT, and lactate levels and decreased pH and ABE (P ⩽ .05, except for ALT level with LeTx alone [P=.06]), and these changes increased over time (P ⩽ .5 for the time interactions, except for ALT level with LeTx and ETx [P=.06]). LeTx alone or with ETx did not significantly alter any change in pulmonary or cell measurements. Compared with LeTx alone, there were no changes in any parameter with LeTx combined with ETx that differed significantly except for small increases in BUN levels during infusion and increases in serum sodium levels late (P ⩽ .05).
Figure 5.

Mean serial effects (± standard error of the mean) in a comparison of controls with a median lethal dose (LD50) of lethal toxin (LeTx), an equal molar dose of edema toxin (ETx), or the combination (ETx + LeTx) on changes from baseline in study 2 for mean arterial blood pressure (MAP), heart rate (HR), central venous pressure (CVP), cardiac index (CI), systemic vascular resistance index, and left ventricular ejection fraction (LVEF). The format is similar to Figure 2.
Figure 6.

Mean serial effects (± standard error of the mean) in a comparison of controls with a median lethal dose (LD50) of lethal toxin (LeTx), an equal molar dose of edema toxin (ETx), or the combination (ETx + LeTx) on changes from baseline in study 2 for blood urea nitrogen (BUN) level, creatinine (Cr) level, BUN:Cr ratio, sodium (Na; 24-h urine output) level, and aspartate aminotransferase (AST) level. The format is similar to Figure 2.
Effects of fluid administration on cardiovascular measurements. Fluids increased CVP, PAOP, MAP, and HR but not other hemodynamic parameters in patterns that did not differ significantly in a comparison of controls with ETx-only animals (data not shown).With LeTx alone and LeTx with ETx, fluids also increased CVP, PAOP, MAP, and HR. Different from controls, fluids with LeTx alone or together with ETx increased CI for both, with LeTx alone increased LVEF, and with the toxins together increased SVI and decreased SVRI for both challenges (P ⩽ .06 for all). Compared with LeTx alone, fluids with LeTx and ETx together had effects that were not significantly different.
Postmortem Examination
At postmortem, each animal had thoracic and abdominal cavity fluid volumes quantified. Compared with controls, fluid volume from either cavity did not differ significantly with toxins.
Discussion
The present study is, to our knowledge, the first to use invasive hemodynamic monitoring and cardiac imaging in large animals to examine serial cardiopulmonary, renal, and hepatic effects of anthrax ETx and LeTx alone and together. Although ETx was less lethal than LeTx on a molar basis, similar to rat studies it produced deaths more rapidly [16, 19]. Notably, lethality with toxins occurred up to 54 h after completion of the infusions.
Consistent with its effect on mortality, ETx produced early decreases in MAP, CVP, and SVRI and increases in HR and CI. It also caused striking elevations in the BUN:Cr ratio, consistent with underperfusion of the renal vasculature. However, ETx did not increase hemoglobin or pleural or peritoneal fluid levels and did not worsen respiratory function. Thus, excessive extravasation of fluid did not appear to be a major contributor to the effects of ETx. Nonetheless, despite rapidly evolving hypotension ETx caused early increases in urine output and decreases in serum sodium levels. Therefore, shock with ETx in this canine model may relate to excessive arterial and venous dilation in addition to impaired renal handling of sodium or water.
Because both arteries and veins express anthrax toxin receptors and because edema factor has potent adenyl cyclase activity, ETx may have caused shock in this model via cAMP mediated vasodilation [20–22]. Intracellular cAMP lowers cytosolic calcium and myosin phosphorylation in vascular smooth muscle and causes dilation [23, 24]. Increases in cAMP in myocardial pacemaker cells may have also contributed to tachycardia and increased CI with ETx [25, 26]. Finally, as with cholera toxin increased urine output and reduced serum sodium levels with ETx may have been attributable to increases in renal tubular cell cAMP [27–29]. However, it is possible that ETx-induced adrenal injury may have also contributed to these changes [30]. Increases in AST and ALT levels might reflect either poor perfusion or direct hepatic effects of ETx [20, 30–32].
Although ETx and LeTx were studied over similarly lethal dose ranges in study 1, LeTx produced smaller or more gradual decreases in MAP, SVRI, and CVP and increases in HR and CI. Notably, both LeTx doses caused progressive reductions in LVEF. These LVEF changes are consistent with those in rodent studies, although their time course was delayed in canines [7–9]. Thus, although peripheral vascular effects may have contributed to LeTx-induced hemodynamic changes in the canine, myocardial dysfunction may have as well. Although inadequate preload (as reflected by PAOP) did not appear to underlie decreases in LVEF, CVP was reduced and fluid loading increased LVEF with high-dose LeTx. CI did not decrease with changes in LVEF, possibly because HR gradually increased.
Disruption of MAPKKs by lethal factor produces several maladaptive responses, including activation of apoptotic and pyroptotic pathways [33, 34]. The time course of such responses is consistent with changes in organ function noted in this model. LeTx-treated vascular endothelial cells underwent mitogen- activated protein kinase cleavage within 5 h, but subsequent apoptosis occurred over 3 days [35]. LeTx-treated endothelial monolayers demonstrated barrier dysfunction after 2–3 days [36]. Thus, LeTx could cause direct vascular endothelial injury manifesting over several days. Although extravasation of fluid did not appear to be pronounced with LeTx, endothelial injury could also disrupt regulation of vascular tone and myocardial function. Intact MAPKKs contribute to normal smooth muscle contraction [37]. With regard to reductions noted in LVEF, electron microscopy has shown early myocardial changes in mice receiving an LeTx bolus [7]. Also, inhibition of MAPK ERK1/2 produces stress-induced apoptosis and heart failure, whereas augmentation is protective [38–40]. Although hemodynamic dysfunction may have contributed to organ injury with LeTx, limited changes in the BUN:Cr ratio raises the possibility that renal and possibly liver dysfunction were related in part to direct cellular injury by toxin. In rats, although norepinephrine infusions decreased hypotension with LeTx challenge, a range of doses did not alter survival [41].
Consistent with findings in rats, lethal LeTx and ETx produced limited pulmonary changes in canines [16, 31]. Furthermore, postmortem exam did not demonstrate excessive pleural fluid. Thus, the pleural effusions and respiratory distress noted in patients with inhalational anthrax may relate to the site of toxin delivery or other factors. For example, purified anthrax cell wall produced substantial inflammatory lung injury in rats [42].
In this canine model, an LD50 of LeTx together with a nonlethal but equimolar ETx dose appeared to synergistically worsen survival, compared with LeTx alone. In rats, LeTx and ETx together also decreased survival more than was predicted on the basis of their individual effects, although the difference was not statistically significant [16]. Thus, even if smaller amounts of ETx are produced during infection than LeTx, it may still contribute to mortality [43]. Therefore, toxin-directed therapies for anthrax should likely target both toxins.
In rats, continuous 24-h fluid infusions with LeTx did not increase MBP and decreased survival and oxygenation. With LeTx in canines, however, daily 40-min infusions increased CVP, MBP, and CI and did not alter oxygenation. These differences in rat and canine models may relate to the regimen of fluid administration or to the absence or presence of ventilatory support. Understanding the effects of titrated hemodynamic support on shock during anthrax toxemia will be important.
Measurements of PA in patients with anthrax are not available. However, in cynomologous monkeys challenged with 200×LD anthrax spores, mean PA levels (± standard error 50 of the mean) increased from 1.3±2.3 ng/mL at 30 h to 178.3±37.4 ng/mL at 72 h (T.S.M. and G.M.S., unpublished data). Thus, although peak PA levels in canines challenged with ETx were almost a log higher than in those with spore challenge, levels with LeTx were more similar.
The present findings are relevant considering the recent softtissue anthrax infections in the United Kingdom [44]. Notably, after initial debridement, antibiotics, and 3–5 days of relative stability some nonsurvivors developed shock refractory to fluid resuscitation and vasopressors. These events may represent recurrent local infection with bacteremia and septic shock. However, results in this canine study raise 2 other possibilities. First, early recurrence of local infection with increasing intravascular release of toxin—most importantly ETx, with its potential to rapidly increase intracellular cAMP levels—could cause vasodilation and inhibition of vasopressors [37, 45, 46]. In this case, agents countering increased intracellular cAMP levels or their downstream effects might be considered. Second, LeTx produced early during infection may lead to progressive cellular dysfunction. Such changes might not be reversible with conventional hemodynamic support. As cited, although norepinephrine ameliorated shock with LeTx in rats, it did not improve survival. Therapy for this second possibility would require understanding the intracellular pathways disrupted by LeTx that have late, life-threatening manifestations.
In conclusion, the present study further delineates the differing effects that anthrax ETx and LeTx have on cardiovascular and organ dysfunction, as well as their potential synergistic effects on mortality. Developing optimal therapies to manage toxemia appears to be integral to improving the care of patients with anthrax.
Acknowledgments
We thank Deloris E. Koziol and Robert Wesley for their critical review of the statistical methods used in this study.
Footnotes
Potential conflicts of interest: Recombinant anthrax toxins were provided by Human Genome Sciences (Rockville, Maryland).
Financial support: National Institutes of Health (NIH) Clinical Center; Trans NIH-Food and Drug Administration Biodefense grant.
References
- 1.Barakat LA, Quentzel HL, Jernigan JA, et al. Fatal inhalational anthrax in a 94-year-old Connecticut woman. JAMA. 2002;287:863–868. doi: 10.1001/jama.287.7.863. [DOI] [PubMed] [Google Scholar]
- 2.Jernigan JA, Stephens DS, Ashford DA, et al. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Health Protection Scotland. Anthrax outbreak information. [Accessed 22 February 2010]. http://www.hps.scot.nhs.uk/anthrax/
- 4.BBC News. German death linked to Scots anthrax outbreak. [Accessed 22 February 2010]. http://news.bbc.co.uk/2/hi/uk_news/scotland/glasgow_and_west/8500848.stm.
- 5.Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 6.Sherer K, Li Y, Cui X, Eichacker PQ. Lethal and edema toxins in the pathogenesis of Bacillus anthracis septic shock: implications for therapy. Am J Respir Crit Care Med. 2007;175:211–221. doi: 10.1164/rccm.200608-1239CP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moayeri M, Crown D, Dorward DW, et al. The heart is an early target of anthrax lethal toxin in mice: a protective role for neuronal nitric oxide synthase (nNOS) PLoS Pathog. 2009;5:e1000456. doi: 10.1371/journal.ppat.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson LE, Kuo SR, Katki K, et al. Anthrax toxins induce shock in rats by depressed cardiac ventricular function. PLoS One. 2007;e2:e466. doi: 10.1371/journal.pone.0000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson LE, Mock J, Lal H, et al. Lethal and edema toxins of anthrax induce distinct hemodynamic dysfunction. Front Biosci. 2007;12:4670–4675. doi: 10.2741/2416. [DOI] [PubMed] [Google Scholar]
- 10.McGee ED, Fritz DL, Ezzell JW, Newcomb HL, Brown RJ, Jaax NK. Anthrax in a dog. Vet Pathol. 1994;31:471–473. doi: 10.1177/030098589403100411. [DOI] [PubMed] [Google Scholar]
- 11.Dixon WJ. The up-and-down method for small samples. J Am Stat Assoc. 1965;60:967–978. [Google Scholar]
- 12.Minneci PC, Deans KJ, Hansen B, et al. A canine model of septic shock: balancing animal welfare and scientific relevance. Am J Physiol Heart Circ Physiol. 2007;293:H2487–H2500. doi: 10.1152/ajpheart.00589.2007. [DOI] [PubMed] [Google Scholar]
- 13.Cooksey BA, Sampey GC, Pierre JL, et al. Production of biologically active Bacillus anthracis edema factor in Escherichia coli. Biotechnol Prog. 2004;20:1651–1659. doi: 10.1021/bp049725n. [DOI] [PubMed] [Google Scholar]
- 14.Gwinn W, Zhang M, Mon S, et al. Scalable purification of Bacillus anthracis protective antigen from Escherichia coli. Protein Expr Purif. 2006;45:30–36. doi: 10.1016/j.pep.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Laird MW, Zukauskas D, Johnson K, et al. Production and purification of Bacillus anthracis protective antigen from Escherichia coli. Protein Expr Purif. 2004;38:145–152. doi: 10.1016/j.pep.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Cui X, Li Y, Li X, et al. Bacillus anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J Infect Dis. 2007;195:572–580. doi: 10.1086/510856. [DOI] [PubMed] [Google Scholar]
- 17.Cui X, Moayeri M, Li Y, et al. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R699–R709. doi: 10.1152/ajpregu.00593.2003. [DOI] [PubMed] [Google Scholar]
- 18.Lichtman AH. The up-and-down method substantially reduces the number of animals required to determine antinociceptive ED50 values. J Pharmacol Toxicol Methods. 1998;40:81–85. doi: 10.1016/s1056-8719(98)00041-0. [DOI] [PubMed] [Google Scholar]
- 19.Stanley JL, Smith H. Purification of factor I and recognition of a third factor of the anthrax toxin. J Gen Microbiol. 1961;26:49–63. doi: 10.1099/00221287-26-1-49. [DOI] [PubMed] [Google Scholar]
- 20.Bonuccelli G, Sotgia F, Frank PG, et al. ATR/TEM8 is highly expressed in epithelial cells lining Bacillus anthracis' three sites of entry: implications for the pathogenesis of anthrax infection. Am J Physiol Cell Physiol. 2005;288:C1402–C1410. doi: 10.1152/ajpcell.00582.2004. [DOI] [PubMed] [Google Scholar]
- 21.Young JA, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 22.Stehlik J, Movsesian MA. Inhibitors of cyclic nucleotide phosphodiesterase 3 and 5 as therapeutic agents in heart failure. Expert Opin Investig Drugs. 2006;15:733–742. doi: 10.1517/13543784.15.7.733. [DOI] [PubMed] [Google Scholar]
- 23.Benoit JN, Taylor MS. Vascular reactivity following ischemia/reperfusion. Front Biosci. 1997;2:e28–e33. doi: 10.2741/a223. [DOI] [PubMed] [Google Scholar]
- 24.Griffith TM, Taylor HJ. Cyclic AMP mediates EDHF-type relaxations of rabbit jugular vein. Biochem Biophys Res Commun. 1999;263:52–57. doi: 10.1006/bbrc.1999.1313. [DOI] [PubMed] [Google Scholar]
- 25.Borer JS. Drug insight: if inhibitors as specific heart-rate-reducing agents. Nat Clin Pract Cardiovasc Med. 2004;1:103–109. doi: 10.1038/ncpcardio0052. [DOI] [PubMed] [Google Scholar]
- 26.Katz AM. Physiology of the heart. Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 255–286. [Google Scholar]
- 27.Friedler RM, Kurokawa K, Coburn JW, Massry SG. Renal action of cholera toxin. I. Effects on urinary excretion of electrolytes and cyclic AMP. Kidney Int. 1975;7:77–85. doi: 10.1038/ki.1975.12. [DOI] [PubMed] [Google Scholar]
- 28.Kurokawa K, Friedler RM, Massry SG. Renal action of cholera toxin. II. Effects on adenylate cyclase-cyclic AMP system. Kidney Int. 1975;7:137–144. doi: 10.1038/ki.1975.21. [DOI] [PubMed] [Google Scholar]
- 29.Pierce NF, Graybill JR, Kaplan MM, Bouwman DL. Systemic effects of parenteral cholera enterotoxin in dogs. J Lab Clin Med. 1972;79:145–156. [PubMed] [Google Scholar]
- 30.Firoved AM, Miller GF, Moayeri M, et al. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol. 2005;167:1309–1320. doi: 10.1016/S0002-9440(10)61218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dadachova E, Rivera J, Revskaya E, et al. In vitro evaluation, biodistribution and scintigraphic imaging in mice of radiolabeled anthrax toxins. Nucl Med Biol. 2008;35:755–761. doi: 10.1016/j.nucmedbio.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S, Bakre M, Yin H, Varner JA. Inhibition of endothelial cell survival and angiogenesis by protein kinase A. J Clin Invest. 2002;110:933–941. doi: 10.1172/JCI14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turk BE. Manipulation of host signalling pathways by anthrax toxins. Biochem J. 2007;402:405–417. doi: 10.1042/BJ20061891. [DOI] [PubMed] [Google Scholar]
- 34.Xu L, Frucht DM. Bacillus anthracis: a multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. Int J Biochem Cell Biol. 2007;39:20–24. doi: 10.1016/j.biocel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Kirby JE. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect Immun. 2004;72:430–439. doi: 10.1128/IAI.72.1.430-439.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warfel JM, Steele AD, D'Agnillo F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am J Pathol. 2005;166:1871–1881. doi: 10.1016/S0002-9440(10)62496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi O, Watanabe T, Nishida K, et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J Clin Invest. 2004;114:937–943. doi: 10.1172/JCI20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purcell NH, Wilkins BJ, York A, et al. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci U S A. 2007;104:14074–14079. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lips DJ, Bueno OF, Wilkins BJ, et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004;109:1938–1941. doi: 10.1161/01.CIR.0000127126.73759.23. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Cui X, Su J, et al. Norepinephrine increases blood pressure but not survival with anthrax lethal toxin in rats. Crit Care Med. 2009;37:1348–1354. doi: 10.1097/CCM.0b013e31819cee38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui X, Su J, Li Y, et al. Bacillus anthracis cell wall produces injurious inflammation but paradoxically decreases the lethality of anthrax lethal toxin in a rat model. Intensive Care Med. 2010;36:148–156. doi: 10.1007/s00134-009-1643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molin FD, Fasanella A, Simonato M, Garofolo G, Montecucco C, Tonello F. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon. 2008;52:824–828. doi: 10.1016/j.toxicon.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 44.Health Protection Scotland. Anthrax outbreak information: clinical guidelines for use of anthrax immune globulin intravenous (human) (AIGIV) in Scotland. [Accessed 27 October 2010]. http://www.hps.scot.nhs.uk/anthrax/documents/clinical-guidance-for-use-of-anthrax-immune-globulin-v12-1-2010-03-19.pdf.
- 45.McDaniel NL, Rembold CM, Richard HM, Murphy RA. Cyclic AMP relaxes swine arterial smooth muscle predominantly by decreasing cell Ca2+ concentration. J Physiol. 1991;439:147–160. doi: 10.1113/jphysiol.1991.sp018661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor MS, Benoit JN. Effect of milrinone on small mesenteric artery vasoconstriction: role of K(+) channels. Am J Physiol. 1999;277:G69–G78. doi: 10.1152/ajpgi.1999.277.1.G69. [DOI] [PubMed] [Google Scholar]