

Abstract
Genome-wide scans of nucleotide variation in human subjects are providing an increasing number of replicated associations with complex disease traits. Most of the variants detected have small effects and, collectively, they account for a small fraction of the total genetic variance. Very large sample sizes are required to identify and validate findings. In this situation, even small sources of systematic or random error can cause spurious results or obscure real effects. The need for careful attention to data quality has been appreciated for some time in this field, and a number of strategies for quality control and quality assurance (QC/QA) have been developed. Here we extend these methods and describe a system of QC/QA for genotypic data in genome-wide association studies. This system includes some new approaches that (1) combine analysis of allelic probe intensities and called genotypes to distinguish gender misidentification from sex chromosome aberrations, (2) detect autosomal chromosome aberrations that may affect genotype calling accuracy, (3) infer DNA sample quality from relatedness and allelic intensities, (4) use duplicate concordance to infer SNP quality, (5) detect genotyping artifacts from dependence of Hardy-Weinberg equilibrium (HWE) test p-values on allelic frequency, and (6) demonstrate sensitivity of principal components analysis (PCA) to SNP selection. The methods are illustrated with examples from the ‘Gene Environment Association Studies’ (GENEVA) program. The results suggest several recommendations for QC/QA in the design and execution of genome-wide association studies.
Keywords: GWAS, DNA sample quality, genotyping artifact, Hardy-Weinberg equilibrium, chromosome aberration
Introduction
The number of genome-wide association studies (GWAS) of complex human diseases has increased markedly in recent years with the availability of low-cost, high-density genotyping and large, well-characterized sample sets. Like other genomic applications, the production of GWAS data involves industrial-scale processes for which systems of quality control and quality assurance (QC/QA) are essential. This article describes a system of QC/QA for GWAS with a focus on the genotypic data. We define QC as steps taken to monitor and control the quality of a product as it is being produced, while QA is defined as a post-production review of product quality. In this case, the ‘product’ is a set of GWAS data produced for the purpose of detecting genotype-phenotype associations and gene-environment interactions. The QC/QA system is illustrated with examples from four projects in the ‘Gene Environment Association Studies’ (GENEVA) program [Cornelis, et al. 2010]. GENEVA projects consist of case-control, cohort, and family-based study designs. We focus here on QC/QA for case-control study designs, but many of the principles also apply to other designs.
The fundamental goal of a case-control association study is to test for an allelic frequency difference between cases and controls to find SNPs that affect disease susceptibility. Because GWAS typically involve large sample sizes to detect small effects and hundreds of thousands of polymorphisms are studied, even small artifactual differences in allelic frequency between cases and controls can generate false-positive results. Therefore, it is particularly important to avoid associations between case-control status and experimental factors that have potential effects on allelic frequency. Well-recognized artifacts occur when cases and controls differ in population structure [Cardon and Palmer 2003] or when case and control DNA samples are handled differently in ways that affect DNA quality [Clayton, et al. 2005]. Differences in DNA quality can result in differences in the frequency of missing genotype calls, which are often biased towards one genotype or another [Wellcome Trust Case Control Consortium 2007]. When studies contain related subjects, methods that implicitly assume independence of subjects may have inflated false-positive rates. False negative results may be increased by failure to control various experimental factors, leading to `noise’ in the system and thereby reducing power. Such factors include low quality DNA samples, poorly-performing SNP assays, and errors in sample identification. All of these problems are best handled by appropriate experimental design and quality control, but quality assurance also plays an important role in identifying biases that may be reduced or eliminated during the analysis phase of GWAS.
The need for careful QC/QA of genotypic data in GWAS is well recognized and several publications address various aspects [Broman 1999; Chanock, et al. 2007; Wellcome Trust Case Control Consortium 2007; Manolio, et al. 2007; McCarthy, et al. 2008; Miyagawa, et al. 2008; Ziegler, et al. 2008]. We have extended these methods to include some new approaches that (1) combine analysis of allelic probe intensities and called genotypes to distinguish gender misidentification from sex chromosome aberrations, (2) detect autosomal chromosome aberrations that may affect genotype calling accuracy, (3) infer DNA sample quality from relatedness and allelic intensities, (4) use duplicate concordance to infer SNP quality, (5) detect genotyping artifacts from dependence of Hardy-Weinberg equilibrium (HWE) test p-values on allelic frequency, and (6) demonstrate sensitivity of principal components analysis (PCA) to SNP selection. Here we describe the QC/QA process applied to GENEVA studies and provide some guidelines for the design of GWAS to avoid experimental artifacts.
Methods
Projects and subjects
The GENEVA projects covered in this article are (1) Addiction to alcohol and other drugs, (2) Lung Cancer, (3) Type 2 diabetes (T2D) with subjects from the Nurses’ Health Study (NHS) and (4) T2D with subjects from the Health Professionals Follow-up Study (HPFS). The Addiction and Lung Cancer projects were genotyped on the Illumina Human1Mv1_c and HumanHap550-2v3_B arrays, respectively, at the Center for Inherited Disease Research (CIDR). The T2D projects were genotyped on the Affymetrix Genome-Wide Human 6.0 array at the Broad Institute Center for Genotyping and Analysis (Broad). Details about GENEVA organization, study designs and subjects are described in Supporting Information, on the GENEVA website (see Web Resources), and in Cornelis et al [2010]. Data for these projects, along with QC/QA reports are posted in the NCBI database for Genotypes and Phenotypes [Mailman, et al. 2007] (dbGaP). The QC/QA reports are posted also on the GENEVA web site.
Definitions of Quality Measures
Missing call rate is either the fraction of missing calls per SNP over samples or the fraction per sample over SNPs. On a scatterplot of the normalized allelic probe intensities produced by SNP assays, θ is defined to be the polar coordinate angle of a point (i.e. a sample-SNP combination) and R is defined to be the sum of those intensities. BAlleleFreq (BAF) is a measure of allelic imbalance, defined as an estimate of the allelic frequency in a population of cells from a single individual. LogRRatio (LRR) is a measure of relative intensity, the logarithm (base 2) of the observed value of R divided by the expected value [Peiffer, et al. 2006]. The genotype cluster plot of a SNP displays either the intensities of the two alleles or R versus θ and the genotype calls of each sample. The confidence score is a measure related to the distance between a given data point and the centroid of the nearest genotype cluster in a cluster plot. The heterozygosity of a sample is the fraction of non-missing genotype calls that are heterozygous. The genotype discordance rate is the number of genotype calls that differ between a pair of samples divided by the total number of SNPs for which both calls are non-missing. A Manhattan signal plot is a genome-wide plot of −log10 of the p-value for SNP-phenotype association versus chromosomal position. A regional association plot is a similar to the Manhattan plot except that it zooms in on a small region showing trait associations and will usually include SNP-SNP correlation data. Contrast QC (CQC) measures the separation of raw allelic intensities from Affymetrix arrays into three clusters. Genomic inflation factor [Devlin and Roeder 1999] is the ratio of the median of the observed statistic for a set of genome-wide tests to the expected median of that statistic under the null hypothesis. More details for some of these terms are given in Supporting Information.
Genotyping batch design
A genoty ping batch consists of a set of samples that are processed together through the genotyping chemistry, hybridization and scanning stages. For the Affymetrix projects, a batch consisted of a 96-well plate and for the Illumina projects, a set of 24 samples (quarter-plate). Except for the Addiction project, samples were assigned to batches to balance case-control status and, in some projects, ethnicity. Each 96-well plate included one or more HapMap [International HapMap Consortium 2005] controls and some plates include study sample duplicates.
Genotyping Center QC
At both genotyping centers, DNA samples provided by study investigators are fingerprinted with a 24- or 79-SNP panel. The fingerprints are used to assess genotyping performance and to track sample identity. If gender discrepancies or unexpected duplicates are detected, study investigators may correct sample annotation or replace DNA samples prior to high density genotyping. Laboratory Information Management Systems (LIMS) are used to track and monitor samples, reagents and equipment at all stages of the genotyping process. Daily and monthly reports are reviewed to identify quality issues. Metrics based on raw intensities are used to eliminate low quality samples prior to genotype calling. Genotype calling was done using normalized intensities with the Birdseed algorithm [Korn, et al. 2008] for Affymetrix and with BeadStudio (see Web Resources) for Illumina projects. Genotypes were called by plate for Affymetrix and by project for Illumina. The software calculates a confidence score for each sample-SNP data point and those beyond a fixed threshold have their genotype set to missing. Samples with low quality scans are repeated. Post-production metrics are used to eliminate low quality samples and SNPs from the Genotyping Center data release, using project-specific thresholds. These metrics vary among platforms and projects, and may include call rate, allele frequency-plate associations, Hardy-Weinberg deviations, duplicate discordance of HapMap controls and sex differences in SNP metrics. More details of Genotyping Center QC are provided in Supporting Information.
QA Methods
The QA analyses were performed using two software packages, PLINK [Purcell, et al. 2007] and the R statistical package [R Development Core Team 2006]. For analysis in R, the genotypic and quantitative variables (probe intensities, etc.) are stored in netCDF files accessed with the ‘ncdf’ package (see Web Resources). netCDF is a compact storage format that allows rapid access to array-structured datasets. The results presented here were generated in R unless stated otherwise.
Most of the QA methods are described in the Results section below. More details on some methods are in the Supporting Information, which also includes an outline of the process (Table S1). Initially, the focus is on identifying samples of poor quality or questionable identity, which are removed for subsequent analyses and not posted on dbGaP. For the remaining samples and for SNPs, filters are provided to flag various features such as ethnicity, relatedness and data quality.
Results
Sample Quality
The missing call rate per sample is an informative indicator of sample quality. The Genotyping Centers generally fail samples with missing call rates >5%. During QA, we look for high outliers in the distribution of missing call rates and for low outliers in the distribution of the mean confidence score (over all non-missing genotypes) for each sample. None were found for the projects considered here.
Three additional quality measures are used in the QA process to detect mixed (contaminated) DNA samples. First, we identify outliers in autosomal heterozygosity within each ethnic group (for example, as points more than 1.5 inter-quartile ranges from the upper/lower quartile value). Second, we screen for samples with a high variance of BAF for non-homozygous SNPs, as described in Supporting Information. Third, we look for unusual patterns of relatedness, such as samples that appear to be related to many other samples. Samples with one or more of these characteristics may be mixtures of multiple DNA samples, which can be identified by examination of BAF/LRR plots. Figure S1 in Supporting Information shows examples of normal and low quality samples. The “Relatedness” section below describes an example of mixed sample detection.
Genotyping Batch Quality
All four studies have highly significant batch effects on the logarithm of missing call rate (ANOVA p-values < 10−100). In most cases, despite the high level of significance, the distribution of the mean missing call rate per batch is continuous with no obvious outliers. Therefore no batches were excluded by this criterion, except for one batch in the Addiction study in which only three of 24 samples passed QC at the genotyping center.
Another way to detect batch effects is to assess differences in allelic frequency between each batch and a pool of all other batches in a study, using a homogeneity test (see Supporting Information). This allelic frequency test can be affected not only by laboratory processing, but also by the biological characteristics of the samples in a batch, such as continental ancestry, other ethnic variation and relatedness. After taking continental ancestry into consideration, no batch outliers were found in any of the four projects, except for the batch with only three passing samples noted above (see Figure S2 and text in Supporting Information).
Gender Checks and Sex Chromosome Aneuploidy
Gender identity is usually inferred from X chromosome heterozygosity, but we find that this variable alone gives ambiguous results, because of sex chromosome aneuploidies and genotyping artifact. Therefore, we also use plots of the mean intensities of the X and Y chromosomes, as shown in Figure 1 for the Addiction and T2D NHS projects.
Figure 1. Gender and sex chromosome anomalies in the Addiction and T2D HPFS projects.
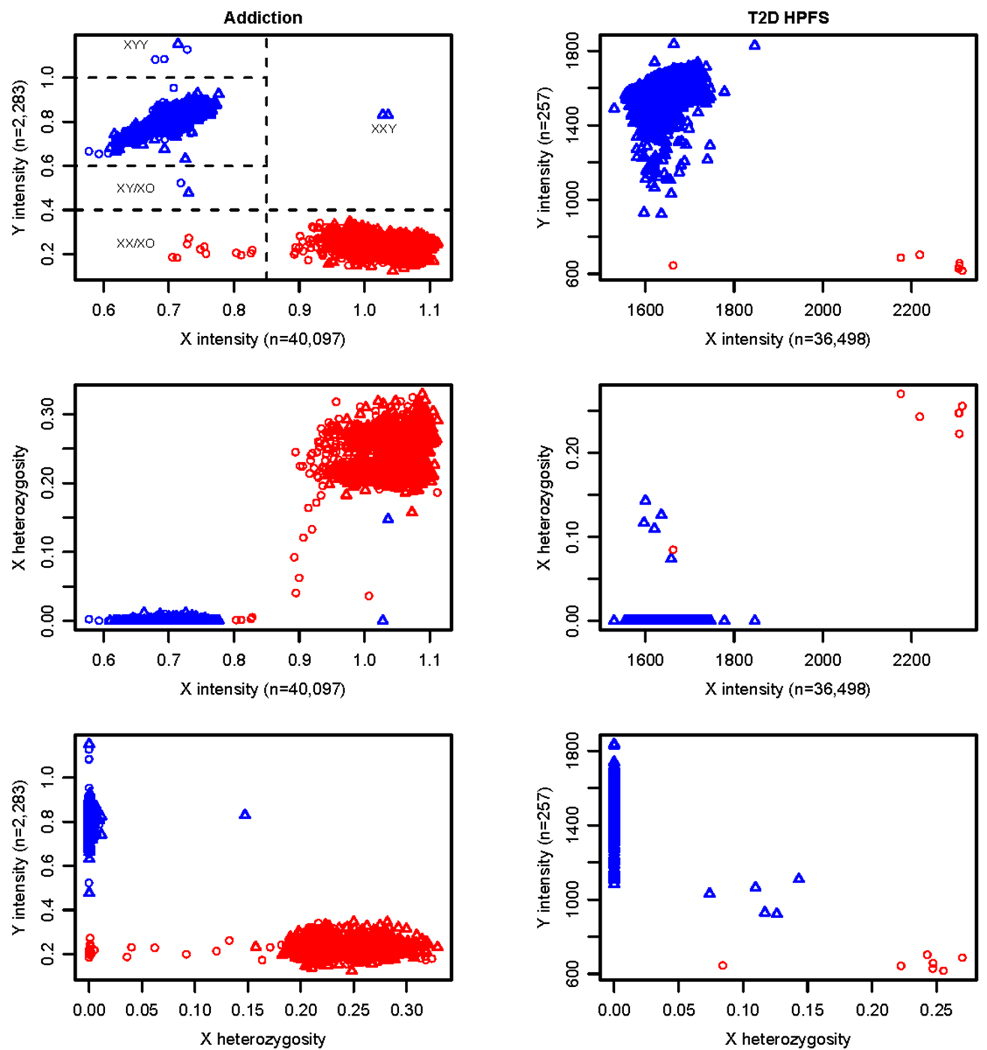
The X and Y probe intensities are calculated for each sample as the mean of the sum of the normalized intensities of the two alleles for each probe on those chromosomes. Probe pair samples sizes are given in the axis labels. In the Addiction project, the standard error of the mean intensity for each sample ranges from 0.002 to 0.004 for the X chromosome and 0.007 to 0.018 for the Y chromosome. In the T2D HPFS study, the standard error of the mean intensity for each sample ranges from 5 to 8 for the X chromosome and from 20 to 98 for the Y chromosome. X heterozygosity is the fraction of heterozygous calls out of all non-missing genotype calls on the X for each sample. Red/blue symbols are for subjects annotated as female/male. Symbols designate the tissue source of DNA samples, where triangle is for whole blood and circle is for lymphoblastic cell lines.
In the Addiction project, the majority of males and females fall into two very distinct clusters based on X and Y chromosome intensities. All samples annotated as males have a Y intensity greater than all samples annotated as females. Therefore, there is no evidence of gender misidentification. However, several samples (delineated by the dashed lines in Figure 1) are distinct from the majority of males and females. Two males with DNA samples from blood have an X chromosome intensity typical of females and a Y intensity typical of males. They appear to be XXY. One of these males has high X heterozygosity and might be mistaken for a female if X heterozygosity alone was used for checking gender. Four male samples (3 cell line and one blood) have unusually high intensities of the Y chromosome and may be either XYY or perhaps XY/XYY mosaics. Similarly, two males have a low Y intensity and may be XY/XO mosaics (one cell line, one blood). Several females have low X intensities and low X heterozygosity, indicating that they are XO or, perhaps more likely, XX/XO mosaics, since they are all cell line samples. As expected, many of the putative XX/XO mosaics show allelic imbalance in BAF plots of the X chromosome, which will be discussed in the next section. Data for samples with a sex (or other) chromosome abnormality are posted on dbGaP, but the affected chromosome is flagged.
In the T2D HPFS project, all subjects are male, but several HapMap females were genotyped as controls for the gender identity check. Figure 1 shows one HapMap female (a cell line sample) that appears to be XX/XO. The plot also shows five unusual males with low Y intensity and substantial levels of X heterozygosity, while all other males have zero heterozygosity. This situation is an artifact of the Birdseed method for calling X chromosome genotypes. In this algorithm, samples inferred to be males by annotation and/or Y chromosome intensity are analyzed with a prior assumption of two genotype clusters, while those inferred to be females have a prior assumption of three clusters. Based on their low Y intensities, these five males were mis-assigned as females during automated calling of the X chromosome genotypes. Consequently, the X chromosome SNPs of these five samples are flagged for omission from association analyses.
Chromosomal Aberrations
Previous studies have documented the use of measures of allelic imbalance (BAF) and relative intensity (LRR) for detecting chromosomal aberrations with SNP array data [Conlin, et al. 2010; Peiffer, et al. 2006]. Aneuploidy and large (multi-megabase) duplications and deletions have been detected in tumor cells and in lymphoblastoid cell lines [Simon-Sanchez, et al. 2007]. We also find such aberrations in blood and buccal cell samples. The frequencies and types of aberrations will be reported elsewhere, but some examples are given here from the Lung Cancer project. The upper panel of Figure 2 shows ‘sample 1’, a female with low X chromosome intensity and heterozygosity. Chromosome 8 in this sample has a normal pattern of three BAF bands, but on chromosome X the intermediate band (corresponding to heterozygous SNPs) is split into two, widely separated bands. These characteristics are expected for a mosaic population of disomic and monosomic cells in which monosomic cells predominate. The lower panel shows ‘sample 2’, which has a high intensity of chromosome 8 relative to its other chromosomes. In this case, the separation of the two intermediate bands is smaller. The positions of these bands (at about 0.4 and 0.6) and the high intensity indicate a mosaic population of trisomic and disomic cells. (A purely trisomic cell population would have the intermediate bands at 0.33 and 0.66.) Samples with a chromosome aberration are included in the posting on dbGaP, but we flag the affected chromosome to be filtered out during association analysis, since it is likely to have a high rate of genotyping errors. In addition, we suggest filtering out any sample-chromosome combination with a missing call rate greater than 5%, since such chromosomes may contain undetected aberrations.
Figure 2. Allelic imbalance reveals mosaic aneuploidy.
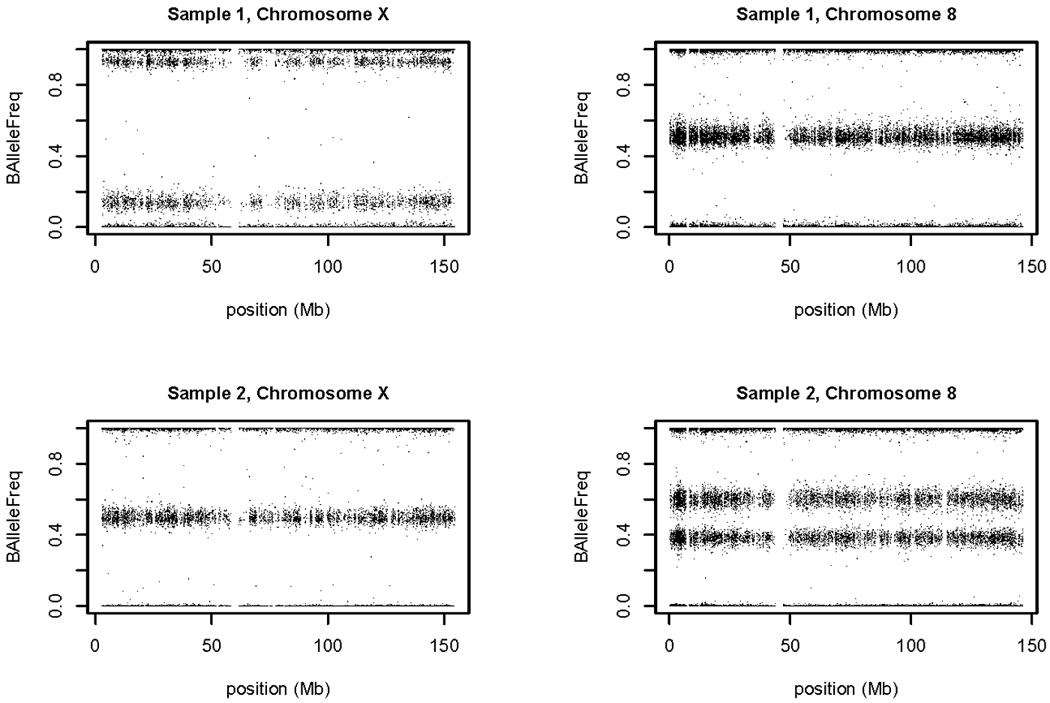
Scans of BAF for two blood samples in the Lung Cancer project indicate X chromosome aneuploidy in one and chromosome 8 aneuploidy in the other. In both cases, the evidence suggests cell populations that are mosaic for normal and aneuploid cells (see text).
Relatedness
We estimate the degree of relatedness between every pair of individuals in a study to identify unexpected relatedness. Three identity-by-descent (IBD) coefficients (Z0, Z1 and Z2), the probabilities of sharing 0, 1 or 2 alleles that are identical by descent, are estimated using a method of moments procedure implemented with PLINK software [Purcell, et al. 2007] and compared with their expectations and evolutionary variance (see Supporting Information). Some pedigree errors can be corrected by consulting original records, while others are corrected based on the inferred genetic relationships.
Figure 3 shows a plot of estimates of Z1 versus Z0 for all pairs of Lung Cancer study subjects with a kinship coefficient estimate greater than 0.025. All study subjects were expected to be unrelated, but this is clearly not the case. Based on expected values (+/− 2 SD), we inferred 14 pairs of full sibs and 5 pairs of half sibs. In addition, there is one parent-offspring pair and 36 pairs of duplicates, including a pair of identical twins. Two of the duplicate pairs could not be documented as coming from the same subject and were removed from the data set.
Figure 3. Relatedness inference from IBD estimates for the Lung Cancer project.
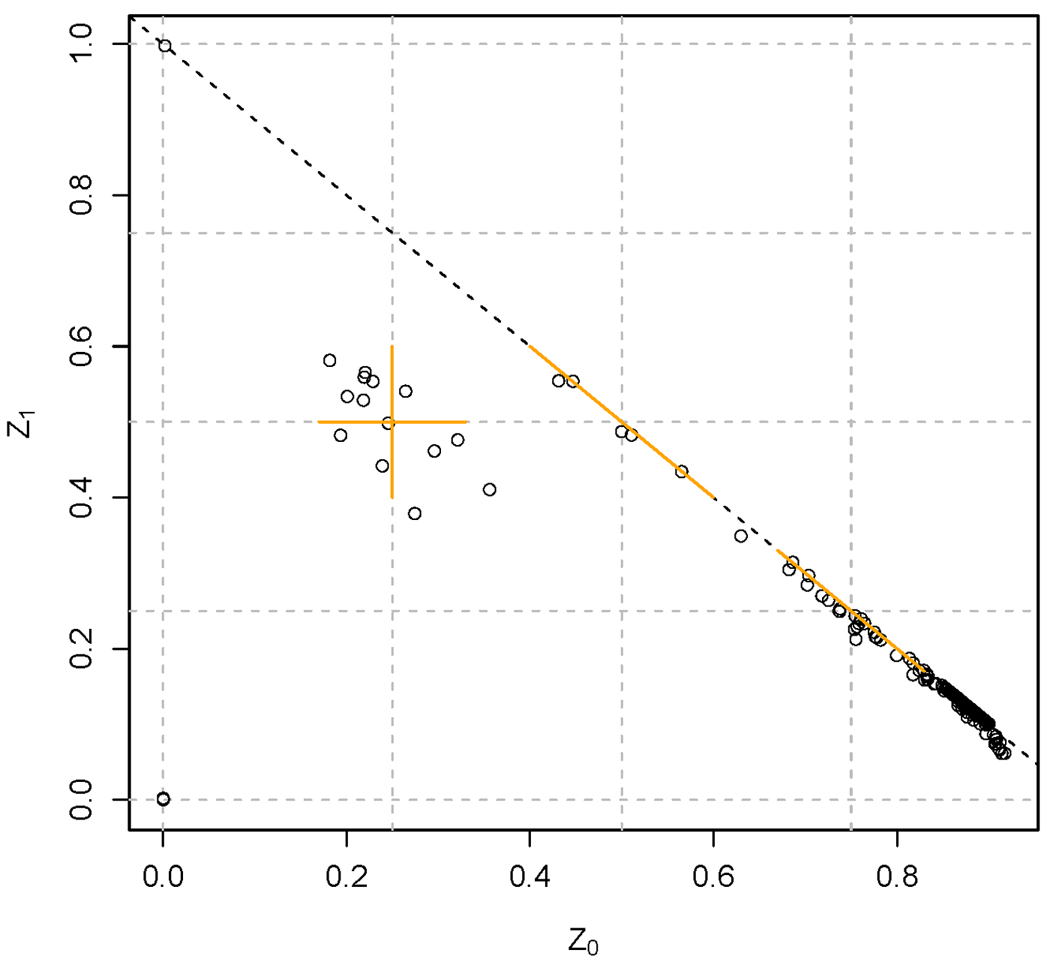
Estimates of the IBD coefficients, Z0 and Z1, are used to infer relatedness. Each point is for a pair of samples and the diagonal line is Z0 + Z1 = 1. The orange bars show the expected values +/− 2 standard deviations (SD) for full sibs (Z0 = 0.25 +/− 0.08, Z1 = 0.50 +/− 0.10), half sibs (Z1 = 0.5 +/− 0.10, Z0 = 1−Z1) and first cousins (Z1 = 0.25 +/− 0.08, Z0 = 1−Z1). Parent-offspring pairs are expected to occur at Z1=1 and duplicates (or identical twins) at Z0=Z1=0. Only pairs of samples with kinship coefficient estimates > 1/32 are plotted. (This truncation is responsible for the sharp downturn at the lower right end of the diagonal.)
In the T2D NHS project, the relatedness analysis revealed two samples that appeared to be related to nearly every other sample in the study with a kinship coefficient between the expected values of half sibs and first cousins (Figures S3 – S6). These samples have relatively high heterozygosity and their BAF plots indicate that they are mixed samples (Figure S1 d and e). Several other samples that appeared to have relatedness to a large number of other samples are not clearly mixed, but appear to be of low quality since they all have more than five chromosomes flagged for high variance of BAF (e.g. Figure S1 f). Similar samples were found in the T2D HPFS project and both projects are discussed further in Supporting Information.
Population Structure
To investigate population structure, we use principal components analysis (PCA), essentially as described by Patterson et al. [2006]. The choice of which SNPs to use for principal components analysis is not obvious. Using all SNPs on a whole-genome array is computationally demanding, but feasible, and would seem to be the best approach in terms of utilizing all available information about genetic relationships. However, whole-genome arrays contain clusters of highly correlated SNPs and a single cluster may have a very strong influence on certain PCs, as noted previously [Novembre, et al. 2008; Tian, et al. 2008]. For example, in the Lung Cancer project (which consists entirely of European-ancestry subjects), when using all autosomal SNPs with missing call rate less than 5% (~545k SNPs), the first two PCs separate U.S. and Italian subjects, while the third PC separates both U.S. and Italian subjects into three distinct groups. These three groups correspond to the genotypes of a cluster of highly correlated SNPs in 8p23, a region that contains a polymorphic inversion. The same result was found previously in PCA of other European-ancestry populations [Novembre, et al. 2008]. The highly localized features underlying some principal components may limit their usefulness in detecting and controlling for population structure. Moreover, they may even be counterproductive when used as covariates in association testing for traits affected by SNPs in those chromosomal regions. Therefore, when adjusting for potential population structure, we recommend against the use of PCs that are highly correlated with localized SNP clusters.
One approach to avoiding the strong influence of SNP clusters is to prune the full genome-wide SNP set before PCA to obtain a subset of SNPs in which all pairs have low correlations. In the T2D project, we compared two such SNP sets, one reported by Yu et al. [2008] selected to have pair-wise linkage disequilibrium (LD) of r2<0.004 and minor allele frequencies (MAF)>0.05 in a European-ancestry population. The other SNP set we selected, from among the 870,000 autosomal SNPs assayed on the NHS subjects, to have LD r2<0.04 and MAF>0.05. Both sets contained about 12,000 SNPs and the overlap is 445 SNPs. The first two eigenvectors obtained from the two SNP sets are very similar, whereas the third, fourth and fifth have much lower correlations (Figures S7 and S8). Similar sensitivity to SNP selection has been observed in other projects. These results suggest that, beyond the first one or two components, eigenvectors ordered by sample eigenvalues may not be robust indicators of population structure. However, we note that this does not rule out the eigensystem as a whole being similar across SNP sets. When subsets of SNPs are used for PCA, we recommend exploring the variability in PCA-based representations of the data under different SNP set selections.
Case-control associations with population structure and experimental factors
To check for association between case-control status and population structure, we test for an association between disease status and the first two eigenvectors from the PCA of each population subgroup of interest. No significant associations have been found so far, which may be a reflection of study designs that carefully match the geographic origins and other characteristics of cases and controls. We also test for a difference in missing call rate per sample between cases and controls, as a way of detecting association with experimental factors. No significant differences have been found so far, except for a special case in the Addiction project described below. In addition to case-control status, we test for correlation between missing call rate and quantitative traits of interest. For example, in the T2D NHS project, the correlation between body mass index and missing call rate is small and not significant (r=−0.01, p-value=0.28).
The Addiction project has three categories of case status: (a) alcohol and possibly other illicit drug dependence (‘case’), (b) controls exposed to alcohol, but never addicted to alcohol or illicit drugs (‘control’) and (c) addicted to illicit drug(s) but not to alcohol (‘other’). There is a significant association between case status and genotyping batch, which could lead to bias in case-control allelic frequencies, although the occurrence of 213 batches with median size of 21 samples reduces the magnitude of any potential problem. Using analysis of variance, there is a significant effect of case status on the logarithm of missing call rate (p<0.01), which is due to the ‘other’ category having a higher rate than the other two (0.12% versus 0.10%, Figure S9). This effect appears to be due to confounding with the DNA source, which was either blood or cell line. Among the DNA samples in the ‘other’ category, 77% are from cell lines, whereas the values for alcohol cases and controls are 34% and 25%, respectively. The missing call rate for cell lines is very significantly higher than for blood (p<9 × 10−14), as shown in Figure S9 (0.12% versus 0.10%). Therefore, allelic frequency differences between ‘other’ versus ‘case’ and ‘control’ categories are potentially biased by nonrandom missingness. In a situation like this, it may be useful to adjust for tissue type in the association analysis, by including it as a regression covariate.
We have found significant effects on the missing call rate of several experimental factors in multiple studies, including tissue type, tissue collection date, DNA extraction method and date, study site, plate, well and genotyping batch. Although confounding makes it difficult to distinguish causative factors, it is prudent to balance these factors with respect to phenotypic traits as much as possible in the design of GWAS experiments.
Genotyping Completeness and Accuracy
Current genotyping technology is very reliable and typically produces data with both high call rates and high accuracy. However, both types of measures should be evaluated for each project because genotyping processes, reagents and instrumentation may vary. The missing call rate is a measure of data completeness, but is also a measure of genotype quality because missingness is often nonrandom. Two methods can be used to assess genotyping accuracy, duplicate sample concordance and consistency with Mendelian transmission.
Genotyping error rates can be estimated from duplicate discordance rates. Each of the three genotypes may be miscalled as either of the other two genotypes, resulting in six potentially different error rates. For a given true genotype, we consider two error rates, α and β. The probability that duplicate genotyping instances of the same subject give a discordant genotype is 2[(1− α − β)(α + β) + αβ]. When α and β are very small, this is approximately 2(α + β) or twice the total error rate. In high-density genotyping, the number of SNPs per sample is so high that duplicating a single sample would give a good estimate of overall error rate, assuming that the rate was similar for every sample. However, DNA sample quality may vary considerably so that error rates can vary among samples. Therefore, we recommend using at least five study samples for estimating error rates.
For duplicate sample pairs, the median discordance rates (discordant calls per SNP) are 7 × 10−5 for Lung Cancer (33 pairs, Illumina HumanHap550 array) and 2 × 10−4 for Addiction (60 pairs, Illumina Human1M array), so the genotyping error rates are on the order of 10−4. The corresponding mean completion (call) rates are very high: 99.8% for Lung Cancer and 99.7% for Addiction samples. For the T2D projects run on the Affymetrix 6.0 array, study samples were not duplicated, but multiple replicates of a single HapMap control sample (NA12144) provide discordance rate estimates of 4 × 10−3 for NHS and 1 × 10−3 for HPFS. The corresponding mean completion rates are 99.6% for NHS and 99.7% for HPFS. It appears that the error rate is about an order of magnitude larger for the Affymetrix 6.0 than for the Illumina 1M arrays, although different sample sets and other factors could affect these results.
Duplicate discordance estimates for individual SNPs also can be used as a SNP quality filter. The problem here is to find a level of discordance that would eliminate a large fraction of SNPs with high error rates, while retaining a large fraction with low error rates. For example, if the mean error rate is 10−4, we may wish to retain greater than 99% of SNPs with error rates less than 10−3, while eliminating as many as possible of SNPs with error rates greater than 10−2. For the Addiction project, with 60 duplicates, a threshold of >1 discordant call seems appropriate, since it would eliminate 99.9% of SNPs with an error rate of 10−1, 33.5% with a rate of 10−2, 0.65% with a rate of 10−3 and <0.1% with an error rate of 10−4. Figure S10 shows the relationship between the probability of observing greater than 0, 1, 2, or 3 discordant calls and the number of duplicates for different genotyping error rates. These binomial calculations can be used to select the optimum threshold and number of duplicates to achieve various levels of distinction among different error rates. At least 30 pairs are indicated for most situations.
Mendelian Errors
Mendelian errors can be detected in parent-offspring pairs or trios. In principle, this method of error detection is less efficient than evaluating concordance of duplicate samples because some genotyping errors are consistent with Mendelian inheritance (e.g. offspring of AB and BB parents, with a true BB genotype called as AB). However, Mendelian errors can be used to detect clustering problems that are not detectable with duplicate concordance. For example, consider a SNP assay in which the AA and AB clusters merge together and are both called as AA, while the actual BB cluster is called as AB. In this case, two AB parents (both called as AA) with a BB offspring (called as AB) would generate a Mendelian error. Similarly, Mendelian errors can detect SNPs with null alleles (N) segregating. For example, one parent as AN, the other as BB and the offspring as BN would give apparently inconsistent genotypes of AA, BB and BB, respectively. In both cases, a duplicate sample would give concordant results
The GENEVA studies analyzed to date are not family-based, but the Addiction and Lung Cancer projects included a small number of HapMap trios as genotyping controls. For each SNP in the Addiction project, the Mendelian error rate was calculated as the number of errors detected divided by the number of families in which the offspring and at least one parent have non-missing genotypes. Among the 1,040,106 SNPs with a possibility for error detection, 99.1% have no errors and the mean error rate is 0.04%.
Hardy-Weinberg Equilibrium Testing
We use an exact test for Hardy-Weinberg equilibrium [Haldane 1954] (HWE) performed on unrelated control subjects with relatively homogenous ancestry. In quantile-quantile (QQ) plots, all four projects show deviations of observed from expected p-values at about 0.01 for autosomal SNPs (e.g. Figure S11). It is not clear how many of these deviations are due to genotyping artifacts and how many are due to true genotypic frequency deviations from HWE, but examination of cluster plots indicates that most of the extreme deviations are due to poorly performing SNP assays.
The HWE test appears to detect different types of genotyping artifact on the two genotyping platforms. Figure 4 shows the HWE p-value versus minor allele frequency for the Addiction and T2D NHS projects. The pattern of extreme HWE deviations is strikingly different. The Illumina data (Addiction) show a curve of low p-values that corresponds to SNPs for which one homozygous class is missing, or nearly so (as indicated by the theoretical plot in Figure S12 and the color-coding in Figure 4). This feature is not observed in the Affymetrix data (T2D NHS) and is likely due to the Illumina calling algorithm setting a limit on the distance between adjacent clusters (which may cause merging of adjacent clusters). The extreme HWE deviations in the Affymetrix data show a different pattern: SNPs with relatively low minor allele frequencies tend to have more very significant deviations than those with high frequency, and there are many SNPs in which the heterozygous class is deficient. This feature may be due to the Birdseed algorithm calling by 96-sample plate, which may make calling genotypes of SNPs with rare alleles more difficult. We should emphasize, however, that extreme deviations are rare with both platforms (<0.3% of SNPs having p-value< 10−10).
Figure 4. Exact HWE test statistic and minor allele frequency.
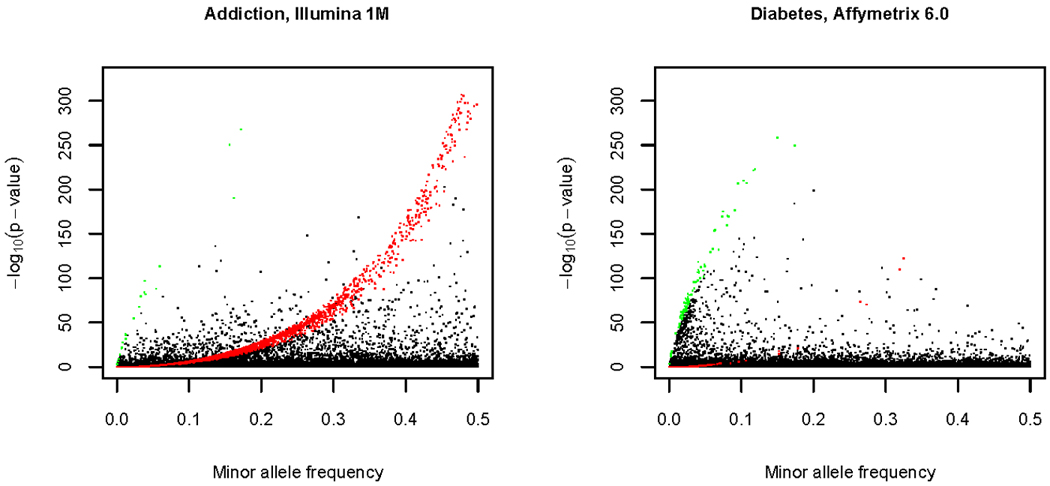
The data presented are for autosomal SNPs in European-ancestry subjects, either for the Addiction study (Illumina Human1M array) or for the NHS study of the Diabetes project (Affymetrix 6.0 array). The sample sizes are 1365 for Addiction and 1752 for the NHS study. The SNPs tested in the Addiction project (930,358) were filtered by excluding SNPs with a missing call rate greater than 15% (and some other criteria, Table I). The NHS SNP test results shown here (867,003) are filtered by exclude SNPs with a missing call rate greater than 15%. The plot for a completely unfiltered set is very similar. SNPs colored in red are those for which one of the two homozygotes occurs at less than 10% of the expected value, while those in green are those for which heterozygotes occur at less than 10% of the expected value. See Figure S13 for a version of this figure in which the Y- axis is focused on −log10(p-value) from 0 to 10. See Figure S12 for a theoretical explanation of curves highlighted in green and red.
One of the SNP filters that we recommend is based on HWE test p-value. Interpretation of these p-values is difficult because the choice of significance level depends on sample size [Wakefield 2009]. However, the purpose of the recommended filter is to flag poorly performing assays rather than detecting real deviations in the population, so we examine genotype cluster plots to set a threshold for filtering. In all four studies described here, these plots show that many assays with p-values between 10−3 and 10−4 have good clustering and genotype calling whereas many of those with p-values less than 10−4 are of poor quality. (For example, in the Lung Cancer project, among 48 plots in the range of p=10−6 to 10−4, 12 of 48 plots showed good clustering, whereas in the range of p=10−4 to 10−2, 42 of 48 showed good clustering.) Therefore, we recommend filtering at p=10−4 for these four studies. Other studies may require a different threshold to account for variations in sample size and genotyping technology.
Sample Exclusion and Filtering
Samples are designated for exclusion if they are of questionable identity (e.g. unresolved gender mismatch) or of unacceptable quality (e.g. appear to be contaminated). All remaining samples are posted on dbGaP, but we recommend that filters are applied prior to association testing. In some cases, the filters apply only to certain chromosomes of a sample (e.g. chromosome aberrations). We recommend filtering out samples with an overall missing call rate greater than 2% and those that are PCA outliers from all major ethnic groups in the study. The percentages of data lost by application of the sample filters is 1.6% for Addiction, 0.5% for Lung Cancer, 1.6% for T2D NHS and 0.5% for T2D HPFS.
The presence of low quality samples during genotype calling may affect the cluster definitions and, therefore, the accuracy of genotype calls for high quality samples. This effect was demonstrated by Pluzhnikov et al. [2008] in a project that used the Affymetrix 5.0 array with Birdseed v2 calling by plate. Eight low quality samples were detected with high heterozygosity and “unusual patterns of relatedness”. These samples were all on one plate, which had a disproportionately high number of cases. False positive associations were found and these remained after simply removing the eight low quality samples. Re-calling of the remaining samples on the affected plate was necessary to remove the false positives.
The GENEVA T2D projects were genotyped on the Affymetrix 6.0 array with Birdseed v1.33 calling by plate. A total of 28 (NHS) and 26 (HPFS) low quality samples were found with high heterozygosity, a high level of relatedness to other samples, and poor BAF plots (see Supporting Information for more details). However, important differences from the situation described by Pluzhnikov et.al [2008] are that the samples are distributed across many plates and we did not find evidence of spurious associations. The QQ plots show low genomic inflation (Figure S14) and genotypic cluster plots for the top hits (after SNP filtering described below) are generally of good quality. Nevertheless, the possible effect of low quality samples was investigated in two ways.
First, for the HPFS project, we estimated the concordance between a HapMap control (NA12144) run on each plate and the consensus genotype calls of 139 replicate genotyping instances of this subject from an independent study using the same array and calling algorithm (and using SNPs that pass the quality filters described below). The mean discordance is very low for plates with and without low quality samples and the difference is not significant (15.8 × 10−5 for 15 plates with low quality samples and 9.8 × 10−5 for 14 plates without such samples; p-value=0.72). Second, we re-called a sample of 8 plates from each study, four plates with and four plates without low quality samples. The maximum number of low quality samples per plate was five. The discordance between the original and recalled genotypes is significantly higher for plates from which low quality samples were removed (p-value=9 × 10−4) and it varies significantly with the number of samples removed (p-value=2 × 10−4). However, the discordance is very low for both types of plates. The highest median discordance between original and recalled genotypes is 6 × 10−5 for an HPFS plate with five low quality samples. For comparison, the median discordance between 15 HapMap control samples from HPFS (all from plates with no low quality samples) and the external consensus reference is 1.4 × 10−4. Therefore, the effect of recalling after low quality sample removal (measured as discordance between original and recalled genotypes) is less than independent genotyping of the same sample.
We concluded that the substantial effort required to recall and reanalyze all of the affected plates in the T2D studies (14/29 in HPFS and 15/41 in NHS) was very unlikely to make a significant improvement in the quality of the data, so these data sets were released to dbGaP without doing so. However, we advise GWAS analysts to consider re-calling a plate whenever one or more of the following occur: (a) a significant portion of the plate consists of low quality samples; (b) the plate is an outlier with respect to median missing call rate and/or the allelic frequency-plate association test; (c) the study has high genomic inflation and low quality cluster plots for association test hits; (d) case-control association with affected plates.
In the two Illumina projects, we did not detect unusual patterns of relatedness or evidence of mixed samples. No low quality samples were detected in the QA process for the Lung Cancer project. In the Addiction project, one problematic genotyping batch of 3 samples was detected and removed from the data set (of which only one sample was used in cluster definition). For Illumina genotype calling, all samples (except those with call rate < 98%) are used for cluster definition, so this system is much less susceptible to the influence of a few low quality samples than the by-plate Birdseed calling for Affymetrix projects. In another GENEVA Illumina project (performed by the Broad genotyping center and not described in this article), we evaluated the effect of recalling after removing 846 low quality scans (out of 2970) and replacing them with high quality scans from the same subjects. The discordance between genotype calls in the original and recalled genotypes was extremely low: 1594 of 2124 samples had no discordance and the highest discordance rate was 9 × 10−6.
SNP Filtering
Data for all SNPs released by the Genotyping Centers are posted on dbGaP, but we recommend filtering association test results based on missing call rate, duplicate discordance, Mendelian errors, sex differences in allelic frequency and heterozygosity, and MAF. The thresholds vary among studies according to quality metric distributions and genotyping platform. For both the Illumina Human1M and Affymetrix 6.0 arrays, the fraction of SNPs that were either failed by the Genotyping Center QC or flagged for filtering during QA is about 7% without the MAF > 0.01 filter (Table I), while the corresponding figure for the Illumina HumanHap550 array is 2%. The recommended MAF filter level is based on power to detect associations. However, for comparison among studies, the filters in Table I are all set to MAF>0.01 in subjects from the United States with primarily European ancestry. For this ethnic group and MAF criterion, the percent of SNPs lost from the Affymetrix 6.0 and Illumina Human1M arrays after both quality and MAF criteria are applied is about 20%, while that for the Illumina HumanHap550 array is 6%. The table also shows that genome coverage (estimated for HapMap II CEU subjects) is decreased by only 1–2% due to the recommended filters. Supporting information provides data about overlap of SNP filters within and among studies and the physical distribution of SNPs that fail to pass these filters.
Table I.
SNP failure and recommended filter criteria with results from 4 GENEVA projects.
| Remove SNPs with: | SNPs losta | |
|---|---|---|
| Projects using Illumina arrays | ||
| Addiction Illumina Human1M | Lung Cancer Illumina HumanHap550 | |
| Pre-release failures | ||
| Missing call rate > 15% | 4,434 | 417 |
| >1 discordance in replicate HapMap controls | 2,725 | 331 |
| Manual review and other criteriab | 1,743 | 213 |
| Total SNPs failed by Genotyping Center | 8,902 | 961 |
| Post-release recommended filters | ||
| MAF = 0 | 31,755 | 106 |
| Missing call rate ≥ 2% | 28,800 | 7,569 |
| Missing call rate ≥ 5% in one or both sexes | 0 | 0 |
| >1 family with Mendelian error(s) | 835 | 486 |
| >1 subject with discordant call(s) | 843 | 66 |
| Sex difference in allelic frequency ≥0.2 | 13 | 0 |
| Sex difference in heterozygosity > 0.3c | 0 | 0 |
| HWE p-value < 10−4 in study controls | 2,275 | 1,242 |
| MAF < 0.01 | 134,710 | 23,036 |
| Initial number of SNPsd | 1,049,008 | 561,466 |
| Percent of SNPs lost excluding MAF filter | 7.0% | 1.9% |
| Percent of SNPs lost including MAF filter | 19.8% | 6.0% |
| Genome coverage at r2>0.8 for all SNPs on the arraye | 91.2% | 87.4% |
| Genome coverage at r2>0.8 after filteringe | 90.0% | 86.8% |
| Projects with Affymetrix arrays | ||
| T2D - NHS Affymetrix 6.0 | T2D HPFS Affymetrix 6.0 | |
| Pre-release failures | ||
| Missing call rate > 5% | 23,859 | 26,872 |
| HWE p-value < 10−8 in all samples | 3,312 | 2,389 |
| Plate associations (single plate p<10−8, 2 or more p<10−4)f | 3,380 | 5,844 |
| Total SNPs failed by Genotyping Center | 30,551 | 35,105 |
| Post-release recommended filters | ||
| One member of each pair of duplicate probes (mostly AFFX)g | 2,839 | 2,903 |
| MAF=0 | 1,438 | 2,782 |
| Missing call rate ≥ 3% | 17,802 | 15,987 |
| > 1 discordance in replicate samples of NA12144 | 7,121 | 5,340 |
| HWE p-value < 10−4 in study controls | 540 | 513 |
| MAF < 0.01 | 126,331 | 121,469 |
| Initial number of SNPs | 909,622 | 909,623 |
| Percent of SNPs lost excluding MAF filter | 6.6% | 6.9% |
| Percent of SNPs lost including MAF filter | 20.5% | 20.2% |
| Genome coverage at r2>0.8 for all SNPs on the arraye | 80.0% | 80.0% |
| Genome coverage at r2>0.8 after filteringe | 78.1% | 77.9% |
The number of SNPs lost at each step is after losses at the previous step
Other criteria include gender difference in missing call rate and autosomal heterozygosity, male X heterozygosity, female Y heterozygosity
For autosomal and pseudo-autosomal SNPs only
The initial number of SNPs assayed is the total number of probes on the Illumina Human1M array (1,072,820) minus the number of intensity-only probes.
Calculated with HapMap II data for CEU subjects [Barrett and Cardon 2006] with software by Carl Anderson (see Web resources)
These plate association tests were conducted without adjustment for ethnicity differences among plates.
The Affymetrix 6.0 array has 3024 SNPs with the same ‘rs’ number and the same map position. Each of these SNPs is assayed with two different probes, one of which is ‘AFFX’, used for quality control.
Preliminary Association Tests
Our final data cleaning step is to perform preliminary association tests and then examine QQ, Manhattan signal, regional association and genotype cluster plots. We use logistic regression and likelihood ratio tests for case-control studies, using samples filtered by quality criteria and retaining unrelated subjects. Initially, we select which of the following covariates to include in the model: age, sex, recruitment center and the first several eigenvectors from the PCA. These potential covariates are analyzed in models that exclude genotype and those with significant effects are included in the final model. We then include the genotype (coded for an additive model) for each SNP in turn and test for SNP effects with a likelihood ratio test. We recommend examination of cluster plots for the ‘top hits’ (most significant SNPs) in an association study and flag results for any SNPs that show poor clustering. Examples of QQ and cluster plots are illustrated for the Addiction study in Figure S15. Another check on the quality of top hits is to examine Manhattan signal and regional association plots of association test p-value versus chromosomal position. As noted previously [Wellcome Trust Case Control Consortium 2007], valid associations are likely to appear as a small cluster of SNPs with low p-values, unless the sentinel polymorphism is in a SNP-poor region.
The benefits of attention to QC/QA of genotypic data are difficult to quantify, but some examples have been reported. Pluzhnikov et al. [2008] describe a genotyping plate effect (due to a small number of low quality samples) that resulted in spurious associations. The Wellcome Trust Case Control Consortium [2007] (supplement) reported a decrease in the genomic inflation factor with the application of a series of quality filters and we observed a similar effect in the GENEVA Addiction study, where the genomic inflation factor changes from 1.08 to 1.04 after filtering.
Discussion
The most effective QC/QA process starts with a good experimental design. Our experience with GENEVA and other projects has led to the following recommendations. Adherence to sound epidemiological principles in the recruitment of subjects for case-control studies is a crucial first step [Zondervan and Cardon 2007]. Subsequently, it is critical to avoid association between case-control status (or other phenotypes) and any variables that may affect genotyping quality. Variables that may affect DNA sample quality include tissue collection date, storage and shipping conditions, tissue type, DNA extraction method, extraction batch, and study site. Factors that may affect genotyping process quality are reagent batches, instrumentation and processing batches (such as plate effects). Therefore, to avoid confounding, we recommend balancing case-control status across experimental factors and randomizing the order of processing and the plate positions of samples. We also recommend the use of at least five duplicate sample pairs to assess the overall accuracy of genotyping and at least 30 pairs for SNP filtering. The duplicates should be selected to represent the overall quality and tissue source of the study samples. Family trios as additional control samples are useful for detecting SNPs with poor clustering via Mendelian error detection. Rigorous sample tracking systems should be employed to avoid sample identity problems, since a large fraction of sample switches are not detectable.
In the past few years, despite the phenomenal increase in the number of published GWAS and in the number of replicated associations for complex human diseases [Hirschhorn 2009; Manolio, et al. 2008], much of the genetic variation in disease traits remains unexplained by SNP associations detected so far. One possible explanation is that the variance is due to a large number of SNPs with small effects, in which case many more loci might be detected as power increases with increasing sample size. Studies with very large sample sizes, on the order of tens of thousands of subjects, have more power, but are also more likely to be affected by experimental errors. Therefore, QC/QA of genotypic data in GWAS will continue to be an important aspect of human genetics research.
Supplementary Material
Acknowledgments
Funding support has been provided through the NIH Genes, Environment and Health Initiative. Additional funding sources for projects are: (1) Addiction, PI Laura J. Bierut, U01HG004422, NIAAA: U10AA008401, NIDA: P01CA089392, R01DA013423; (2) T2D, PI Frank B. Hu, U01HG004399; and (3) Lung Cancer, PI Neil Caporaso, NIH GEI: HG-06-033-NCI-01 and the Intramural Research Program of National Institutes of Health, National Cancer Institute, Division of Cancer Epidemiology and Genetics. Genotyping was performed at the Broad Institute of MIT and Harvard, with funding support from the NIH GEI (U01HG04424), and Johns Hopkins University Center for Inherited Disease Research, with support from the NIH GEI (U01HG004438) and the NIH contract ‘High throughput genotyping for studying the genetic contributions to human disease’ (HHSN268200782096C). The GENEVA Coordinating Center receives support from U01 HG 004446 (PI Bruce S Weir). Assistance with data cleaning was provided by the National Center for Biotechnology Information.
LJ Bierut and JP Rice are inventors on the patent "Markers for Addiction" (US 20070258898) covering the use of certain SNPs in determining the diagnosis, prognosis, and treatment of addiction. Dr. Bierut served as a consultant for Pfizer Inc. in 2008.
Footnotes
Web Resources
GENEVA: http://www.genevastudy.org
Illumina BeadStudio software: http://www.illumina.com
netCDF: http://www.unidata.ucar.edu/software/netcdf
Genome coverage software: http://www.well.ox.ac.uk/~carl/gwa/cost-efficiency/
References
- Barrett JC, Cardon LR. Evaluating coverage of genome-wide association studies. Nat Genet. 2006;38(6):659–662. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- Broman KW. Cleaning genotype data. Genet Epidemiol. 1999;17 Suppl 1:S79–S83. doi: 10.1002/gepi.1370170714. [DOI] [PubMed] [Google Scholar]
- Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361(9357):598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, et al. Replicating genotype-phenotype associations. Nature. 2007;447(7145):655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- Clayton DG, Walker NM, Smyth DJ, Pask R, Cooper JD, Maier LM, Smink LJ, Lam AC, Ovington NR, Stevens HE, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet. 2005;37(11):1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19(7):1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis MC, Agrawal A, Cole JW, Hansel NN, Barnes KC, Beaty TH, Bennett SN, Bierut LJ, Boerwinkle E, Doheny KF, et al. The gene, environment association studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet Epidemiol. 2010 doi: 10.1002/gepi.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. An exact test for randomness of mating. J. Genet. 1954;52:631–635. [Google Scholar]
- Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360(17):1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40(10):1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39(10):1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118(5):1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Rodriguez LL, Brooks L, Abecasis G, Ballinger D, Daly M, Donnelly P, Faraone SV, Frazer K, Gabriel S, et al. New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet. 2007;39(9):1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Miyagawa T, Nishida N, Ohashi J, Kimura R, Fujimoto A, Kawashima M, Koike A, Sasaki T, Tanii H, Otowa T, et al. Appropriate data cleaning methods for genome-wide association study. J Hum Genet. 2008;53(10):886–893. doi: 10.1007/s10038-008-0322-y. [DOI] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008 doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16(9):1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov A, Below JE, Tikhomirov A, Konkashbaev A, Nicolae D, Cox NJ. Differential bias in genotype calls between plates due to the effect of a small number of lower DNA quality and/or contaminated samples. Genetic Epidemiology. 2008;32:676. [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2006 ISBN 3-900051-07-0, URL http://www.R-project.org.
- Simon-Sanchez J, Scholz S, Fung HC, Matarin M, Hernandez D, Gibbs JR, Britton A, de Vrieze FW, Peckham E, Gwinn-Hardy K, et al. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet. 2007;16(1):1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- Tian C, Plenge RM, Ransom M, Lee A, Villoslada P, Selmi C, Klareskog L, Pulver AE, Qi L, Gregersen PK, et al. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4(1):e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J. Bayesian Methods for Examining Hardy-Weinberg Equilibrium. Biometrics. 2009 doi: 10.1111/j.1541-0420.2009.01267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Wang Z, Li Q, Wacholder S, Hunter DJ, Hoover RN, Chanock S, Thomas G. Population substructure and control selection in genome-wide association studies. PLoS ONE. 2008;3(7):e2551. doi: 10.1371/journal.pone.0002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler A, Konig IR, Thompson JR. Biostatistical aspects of genome-wide association studies. Biom J. 2008;50(1):8–28. doi: 10.1002/bimj.200710398. [DOI] [PubMed] [Google Scholar]
- Zondervan KT, Cardon LR. Designing candidate gene and genome-wide case-control association studies. Nat Protoc. 2007;2(10):2492–2501. doi: 10.1038/nprot.2007.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.