

Abstract
The NMDA receptor antagonist ketamine can induce a rapid improvement in depressive symptoms that often endures for days after a single intravenous dose. The pharmacodynamic basis for this effect is poorly understood. Using a proton magnetic resonance spectroscopy ([1H]-MRS) method that previously detected a normalization of amino acid neurotransmitter (AANt) content after chronic treatment with conventional antidepressant treatments, we examined whether the acute action of ketamine is associated with alterations in AANt content as well. Ten subjects with major depressive disorder (MDD) received saline, then ketamine in a fixed order, one week apart, under single-blind conditions. Each infusion was associated with three [1H] MRS scans (baseline, 3 hours and 48 hours post-infusion) that measured glutamate, GABA and glutamine within the occipital cortex. Rating scales were administered before, during and after each infusion. The rapid (1 hour) and sustained (at least 7 days) antidepressant effect we observed after ketamine infusion was not associated with either baseline measures of, or changes in, occipital AANt content. Dissociative symptoms were not correlated with changes in depression scores. While our results indicate that changes in occipital AANt content are not a correlate of ketamine’s antidepressant action, this may only apply to the regional and temporal windows of our MRS measurements.
Keywords: major depressive disorder, NMDA antagonist, magnetic resonance spectroscopy, amino acid neurotransmitter
1. Introduction
Innovation in the clinical therapeutics of major depressive disorder (MDD) may be derived from revised pathophysiological models that incorporate the accumulating evidence for a role of dysregulated glutamatergic neurotransmission. Indeed, both preclinical and clinical studies have demonstrated that a variety of glutamatergic compounds have antidepressant effects (Machado-Vieira et al., 2009; Sanacora et al., 2008). In particular, the N-methyl-D-aspartate receptor (NMDAR) antagonist ketamine has emerged as a leading prototype for antidepressant development because it can be both rapidly acting and efficacious in ‘treatment-resistant’ MDD subjects (i.e. those without an adequate clinical response to monoaminergic drugs) (Berman et al., 2000; Mathew et al., 2009; Zarate et al., 2006). Furthermore, the antidepressant effect induced by a single, intravenous administration of ketamine often endures for days, well beyond the transient changes in perception and cognition commonly observed during an infusion of a subanesthetic dose. The identification of how this relatively brief pharmacological blockade of the NMDAR induces a rapid and sustained improvement in depressive symptoms would have important theoretical and practical implications.
Preclinically, NMDAR antagonists induce a variety neurophysiological effects implicated in antidepressant action including the modulation of monoaminergic neurotransmission (Dall’Olio et al., 1999; Paul et al., 1992), the enhancement of both hippocampal neurogenesis (Gould et al., 1997; Keilhoff et al., 2004; Nacher et al., 2003; Nacher et al., 2001) and long-term potentiation (Buck et al., 2006), and increased hippocampal brain-derived neurotrophic factor (BDNF) levels (Garcia et al., 2008). In addition, recent studies suggest that increased alpha-amino-3-hydroxy-5-methyl-4- isoxazole-propionic acid (AMPA)-receptor-mediated synaptic potentiation and subsequent activation of the mTOR signaling pathway initiate the physiological responses that generate antidepressant-like actions in rodent models (Li et al. 2010; Maeng et al., 2008). These studies are in line with a previous report showing that ketamine acutely increases glutamatergic neurotransmission in the prefrontal cortex of rodents (Moghaddam et al., 1997), and collectively, these findings suggest that an acute increase in non-NMDA mediated glutamate neurotransmission is a critical step in ketamine’s mechanism of antidepressant action.
In humans, subanesthetic doses of ketamine appear to acutely increase glutamatergic activity in the prefrontal cortex (Breier et al., 1997; Rowland et al., 2005), however we are not aware of any studies characterizing changes in glutamatergic activity or AANt levels during the window of symptomatic improvement that opens after the acute effects of ketamine have dissipated. Using proton magnetic resonance spectroscopy (1H-MRS), we sought to determine whether measureable alterations in amino acid neurotransmitter (AANt) content are associated with ketamine’s antidepressant activity. Our spectroscopy group has previously identified decreased levels of GABA and increased levels of glutamate in the occipital cortex of adult patients with unipolar depression as compared to levels in healthy controls (Sanacora et al., 2004). In addition, we have found that chronic treatment with either a chemical antidepressant (Sanacora et al., 2002), or electroconvulsive treatment (ECT) (Sanacora et al., 2003), is associated with a normalization of GABA levels. Consequently, changes in AANt levels may represent a neurochemical correlate of antidepressant efficacy that extends to NMDAR antagonists.
The primary aims of this study were to replicate the sustained antidepressant effect in subjects with MDD that has been previously observed after a single ketamine infusion, and to assess for correlations between this clinical response and measures of AANt content in the occipital cortex, the brain region that we have previously shown is associated with abnormal levels in MDD subjects. We predicted that ketamine would reduce scores on depression psychometrics, and that these changes would correlate with an increase in GABA content and alterations in glutamate and glutamine content. Contrary to this prediction, we found that the antidepressant effect of ketamine present at 3 hours and 2 days post-infusion was not associated with changes in AANt content.
2. Methods
2.1 Study Participants
Between November 2004 and October 2006, subjects from the greater New Haven area were recruited through advertisements in local newspapers, on the Internet, and by direct contact with physicians. Men and women (21 to 65 years old) meeting DSM-IV criteria for Major Depressive Disorder, as by determined by the Structured Clinical Interview for DSM-IV (SCID) – Patient Edition (First et al. 2001), who also had a screening Hamilton Depression Rating Scale (HDRS-25, Mazure et al. 1986) score > 25 were eligible. Subjects with a lifetime history of additional DSM-IV Axis I diagnoses, other than substance dependence in sustained full remission or co-morbid anxiety disorders, were excluded. Individuals with stable medical conditions that were without primary CNS effects were included if medication adjustments were not made in the month before study onset. All subjects were free from psychotropic medications for at least 2 weeks before, and during the 2-week experimental period. Low doses of antihistamine (diphenhydramine) therapy were allowed for persistent insomnia. Female subjects were either infertile, or had a negative pregnancy test at screening, and were using an established birth control method. After receiving a description of the study, including the possibility of experiencing distressing changes in mood, cognition and perception of the world and oneself, written informed consent was obtained according to Yale Human Investigation Committee guidelines. One subject was recruited while an inpatient on the Clinical Neuroscience Research Unit (CNRU) of Connecticut Mental Health Center and remained there throughout the study, otherwise, subjects were outpatients that were admitted to the CNRU for one night of observation and ratings after each infusion. All infusions and MRS scans were performed at the Yale Magnetic Resonance Research Center at Yale University School of Medicine.
2.2 Infusion schedule and MRS scan time line
Following at least a 2-week period free of psychotropic medications, subjects received a single 40-minute infusion of 0.9% saline, followed by racemic ketamine hydrochloride (0.5 mg/kg, Ketalar, Monarch Pharmaceuticals, Bristol, Tennessee) one week later, in a single blind, non-counter balanced design. This design was chosen to avoid anticipated carryover effects of ketamine that would complicate the interpretation of data from repeated 1H-MRS measures. Infusions were performed by i.v. infusion pump with subjects lying supine in a hospital bed located in a private room near the spectroscopy suite. Vital signs were measured throughout each infusion session. MRS measurements were made at six time points (Fig 1). The first three scans (MRS 1–3) were associated with the saline infusion, and the second three scans (MRS 4–6) with ketamine. In each group of three MRS sessions, the first scan occurred just prior to the infusion, the second three hours after infusion start, and the third scan was completed 2 days thereafter.
Figure 1.
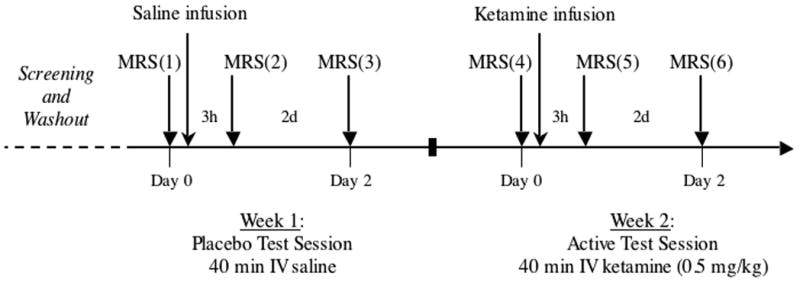
Study Design. MRS (4) was acquired either 7 or 8 days after MRS (1).
2.3 1 H-MRS scanning procedure
Occipital cortex 1H-MRS acquisitions were performed using a slight modification of the J-edited method as previously outlined (Sanacora et al., 2004). Briefly, metabolite levels were measured in the occipital cortex within a 13.5 cc voxel (3.0 × 1.5 × 3.0 cm) box placed across the midline of the brain, centered 1.5 – 2.0 cm from the dura. Cortical GABA content was determined using J-editing, where sub-spectra were acquired with 8K data points over a 410-ms acquisition, a 2.5-second repetition time, and a TE of 68 ms on a 4T Bruker spectrometer at Yale Magnetic Resonance Research Center (MRRC), averaging data in 20-second blocks for 20 minutes. Glutamate and glutamine were measured simultaneously using the unedited subspectra of the J-editing acquisition. For 10 of the studies (5 with placebo, 5 with ketamine), water spectra were collected for quantification relative to tissue water. ANOVA showed no significant effect on creatine/water for group, time (i.e., pre-infusion, immediate post-infusion, or post-infusion two days), or group-by-time. The creatine, therefore, appears as a stable reference in this case.
The unedited subspectrum and the J-edited difference spectrum were fitted simultaneously using a basis set of metabolite spectra measured with the J-editing acquisition sequence. The metabolites fitted were aspartate, GABA, glutamate, glutamine, NAA, NAAG, creatine, phosphocreatine, myoinositol, choline, phosphorylcholine, glycerophosphorylcholine, and scylloinositol. The subspectrum obtained without the editing pulse was fitted simultaneously with the J-edited difference spectrum of GABA (Fig 2). Because of limited resolution in vivo, the results for NAA and NAAG were combined and recorded as NAA, creatine and phosphocreatine were combined and recorded as creatine, and the three choline-containing compounds were combined and recorded as choline. As part of the implementation of the editing method, co-edited macromolecular contamination was evaluated in the GABA spectrum, using two methods: metabolite nulling (Behar, 1994; Rothman et al., 1994; Shen et al., 2004) and frequency switching symmetrically about the coupled macromolecular resonance (Henry et al., 2001), and both cases showed no macromolecular contamination of the resonance. Therefore, the basis set for fitting did not include a macromolecular signal. The level of aspartate, though present in the spectra, was poorly determined at the echo time of 68 ms and was not used.
Figure 2. Representative MR spectra from a single subject.

The data are shown with gray lines, the spectral fits with black lines superimposed on the data, and the residuals are shown immediately beneath in black. (A) Pre-treatment subspectrum obtained without the editing pulse, yielding glutamate, glutamine and metabolites other than GABA. (B) Post-treatment subspectrum. (C) Pre-treatment J-edited difference spectrum, yielding GABA levels. (D) Post-treatment J-edited difference spectrum. The subspectra were fitted only to just beyond the myoinositol resonances, and the difference spectra were fitted only around the GABA resonance.
Uncertainties of individual measurements were determined using a Monte-Carlo analysis, in which the least-squares spectral fits were treated with random Gaussian noise whose standard deviation was equal to that of the raw data and refitted, using 20 repetitions to estimate the standard deviations of the uncertainty for each metabolite measure. For each metabolite, a threshold for rejection was set at twice the average noise-based standard deviation of the respective metabolite. GABA levels whose uncertainties were greater than 11%, glutamine levels whose uncertainties were greater than 20%, and glutamate levels whose uncertainties were greater than 16% were not included in subsequent analysis.
2.4 Psychometric Assessments
The antidepressant effects of ketamine were measured with the Hamilton Depression Rating Scale (HDRS-25; baseline, 60 min, 180 min, 24 hr, 48 hr, 72 hr, 5 days and 7 days after each infusion) and the Beck Depression Inventory (BDI) (Beck AT, 1961) at the same time points as HDRS-25. The 25-item HDRS was chosen as the primary outcome measure secondary to its previously demonstrated sensitivity in measuring the acute effects of ketamine on depressive symptoms (Berman et al., 2000). The HDRS was slightly modified to account for the compressed temporal context of testing and was administered by two un-blinded raters with high inter-rater reliability. Changes in anxiety were measured using the Hamilton Anxiety Rating Scale (HARS; Hamilton 1959) at baseline, 2, 3, 5 and 7 days after each infusion. Perceptual changes were measured using the Clinician Administered Dissociative States Scale (CADSS; Bremner et al. 1998) at baseline, 20, 60, and 80 min after each infusion. The assessments for psychotomimetic effects were performed at baseline, 10, 20, 60, 80, 110, and 210 min after each infusion using four items (unusual thought content, hallucinatory behavior, suspiciousness, conceptual disorganization) of the Brief Psychiatric Rating Scale (BPRS) that constitute a positive symptom subscale (Krystal et al. 1994).
2.5 Statistical Analysis
The primary behavioral outcome variable, HDRS-25 score, was analyzed by a linear mixed effects model using within subjects factors of treatment (placebo vs. ketamine) and time (study time points) and random subject effects. The best-fitting variance-covariance structure was selected according to information criteria. BDI scores, HARS scores, and changes in AANt content were analyzed using similar models. CADSS scores exhibited floor effects and thus, were analyzed using the nonparametric method for longitudinal data by Brunner using the same factors as described above (Brunner et al., 2002). Potential associations between changes in AANt content and changes in HDRS scores were assessed by correlation analysis. In secondary analyses, potential associations between changes in AANt content and changes in HARS, CADSS and BPRS scores were also evaluated. All analyses were considered significant at the alpha=0.05 threshold. Because HDRS scores were our primary outcome measure, the results were not corrected for multiple comparisons. Otherwise, statistical tests were adjusted with a Bonferroni correction (adj P) based on the number of conceptually related tests within each hypothesis (2 tests for psychometric data, 3 tests for our primary analysis of glutamate, GABA and glutamine).
3. Results
3.1 Participant Characteristics
Seventy-three subjects were evaluated after phone screening. Eighteen subjects were considered eligible and subsequently consented. Five subjects were deemed screen failures after providing consent (one from a medical issue; two secondary to substance abuse and two due to inability to wash-off medication). Three subjects withdrew (one based on failure to return before the testing period; one from difficulty with head placement in the scanner, and one from elevated blood pressure during the placebo infusion). Ten subjects [M/F (4/6); mean age 41.7 years (SD 12)] completed the protocol. Four subjects required an antidepressant washout [mean duration 6 weeks (SD 8)], and the rest were un-medicated at the time of recruitment. The mean duration off antidepressants prior to the study entry was 1.4 years (SD 1.78). The mean duration of the current episode was 3.3 years (SD 4.3) and the mean duration of depressive illness was 21.2 years (SD 17.4). The mean lifetime number of depressive episodes was 3.1 (SD 1.7) and the mean number of adequate antidepressant treatments (not including adjunctive medications), as determined by self-report, pharmacy records and prescription histories from prescribing clinicians, was 2.2 (SD 1.9). Eight subjects met criteria for recurrent MDD, and two subjects for a single episode. Nine subjects met criteria for moderate severity and one for severe with mood congruent psychotic features. Five subjects met SCID criteria for co-morbid anxiety disorders (PTSD, social phobia); seven had a lifetime history of anxiety disorder and two subjects met for lifetime substance disorders (one for alcohol abuse and one for both alcohol and substance dependence, both in sustained full remission for many years).
3.2 Effects of ketamine on depressive and anxiety symptoms
Baseline HDRS scores measured prior to the start of the saline (28.4 +/− 2.4) and ketamine (26.8 +/− 4.2) infusions were not significantly different (P > .1). Analysis of HDRS scores over time revealed both a main effect of treatment (F (1,131) = 11.84, P <. 0008) and time (F (7,131) = 2.75, P < .011). Although the pattern of change in HDRS scores did not significantly differ across time under each treatment condition (F (7,131) = 1.43, P = 0.2), post-hoc tests revealed that significant differences in HDRS scores between conditions persisted over time (Fig 3).
Figure 3.
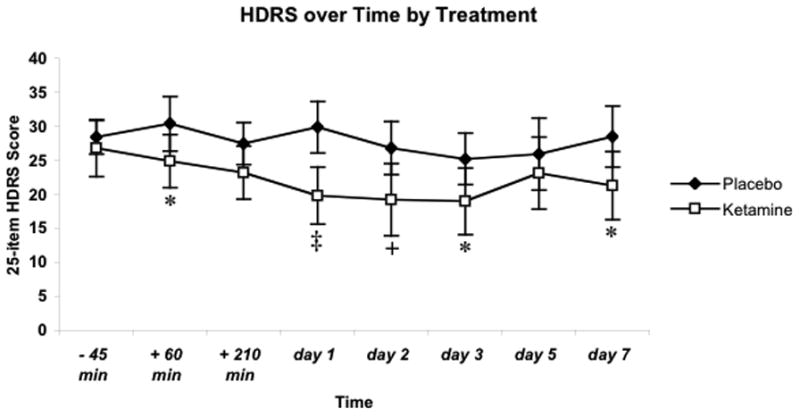
Scores on 25-item Hamilton Depression Rating Score (HDRS) over 7 days after each infusion (n=10). The difference in baseline scores was not significant. Values are represented as mean scores +/− s.e.m. * indicates P<.05; + P<.01; ‡ P<0.001.
The overall effect of ketamine on BDI scores was comparable to those for the HDRS. Mean baseline BDI scores for the saline (26.9 +/− 4.2) and ketamine (24.1 +/− 5) were similar (P > .2). Main effects of treatment (F (1,133) = 9.9, adj P = 0.004) and time (F (1,133) = 2.81, adj P = 0.018), were detected. Although a treatment by time interaction was not observed (F (1,133) = 1.44, adj P = 0.38), post-hoc tests found significant decreases in BDI scores after ketamine treatment at 60 min (P < 0.001), 210 min (P < 0.05), 24 hours (P < 0.01), 48 hours (P < 0.05) and 72 hours (P < 0.05) after treatment, but not at 5 or 7 days.
Because HARS scores at the start of the placebo (15.7 +/− 1.6) and ketamine (12.9 +/− 2.3) infusions were significantly different (F (1,98) = 7.43, P < 0.01), an analysis of the absolute percent change from baseline scores was performed and failed to detect an effect of treatment (F (1,76) = 1.8, adj P > 0.2) or time F (4,76) = .25, adj P >0.8).
3.3 Psychomimetic and dissociative effects of ketamine in MDD subjects
Ketamine had significant effects on a range of psychiatric symptom constructs as determined by significant main effects of treatment (F (1,104) =13.3, P = 0.0004) and time (F (6,104) = 7.47, P < 0.0001), on the total score of the 18-item BPRS. Although a treatment by time interaction was not observed (F (6,104) = 1.32, P = 0.26), post-hoc tests demonstrated that ketamine treatment resulted in lower BPRS scores at 60 min (P < 0.005), 80 min (P < 0.005), 110 min (P < 0.005) and 210 min (P < 0.01). However, an analysis of the BPRS positive symptom subscale using nonparametric K-W tests on data from the 20 min, 60 min and 80 min time points were not significantly different between treatments (all P >.19).
To assess whether ketamine had significant dissociative effects, an analysis of total CADSS scores was performed. While there was no overall effect of treatment (ATS = 0.95, num df = 1, P = .33), a significant interaction between treatment and time (ATS = 4.1, num df = 2.58, P = .01) was observed, explained by increased dissociation at 20 minutes post- ketamine infusion (ATS = 12.5, num df = 1, P = .0004). Changes in CADDS scores at other time points were not significant (all P > .15) and the peak change did not correlate with changes in HDRS scores at any time point.
3.4 Effects of ketamine on vital signs
Analysis of systolic blood pressure detected a main effect of treatment (F (1,103) = 90.2, P < .0001) but not time (F (1,103) = 1.84, P = .1). Although a treatment x time interaction was not observed (F (1,103) = 1.26, P = .28), post-hoc tests confirmed that ketamine significantly increased SBP at 10, 20, 40, 80 and 95 minutes with a mean peak increase of 14.1 mmHg (SD 13.4 mmHg) at 40 minutes. Ketamine was associated with a trend toward increased diastolic blood pressure (F (1,103) = 3.6, P = .06) and had no effect on heart rate (F (1,100) =. 9, P = .35).
3.5 Effect of ketamine on AANt content in the occipital cortex
The standard deviations of individual metabolite measurements, equivalent to the Cramer-Rao lower bounds calculated by the standard LCModel program (Provencher, 1993), had the following mean ± variance: GABA = 0.038 ± 0.044, glutamate = 0.380 ± 0.454, glutamine = 0.144 ± 0.156, NAA = 0.033 ± 0.040, myoinositol = 0.063 ± 0.066, creatine = 0.052 ± 0.060, choline = 0.020 ± 0.022. As shown in Fig 4, 1H-MRS measurements of glutamate, GABA and glutamine content in the occipital cortex were highly reproducible across scans. However, a main effect of treatment was not observed on measures of glutamate (F (1,39) = 2.11, adj P = 0.4), GABA (F (1,36) = 0.72, adj P > .99), or glutamine (F (1,39) = 0.12, adj P >.99). In addition, significant main effects of time (all P > 0.09) or a treatment x time interaction (all P > 0.47) were not detected. Secondary analyses of ketamine’s effect on all other metabolites (see Methods) and, of the glutamine/glutamate ratio, were also without main effects.
Figure 4.

[ 1H]-MRS measurements of amino acid neurotransmitter content in the occipital cortex of MDD subjects before and after ketamine infusion. Values represent mean values of the ratio of each metabolite to creatine (Cr). error bars = +/− s.e.m.
To determine whether the antidepressant effect we observed at 2 days was associated with alterations in AANt content at that time point, correlation analyses of changes in HDRS scores and changes in AANt content were performed. Results indicated that changes in glutamate (r = 0.36, adj P > .99), GABA (r = −0.23, adj P > .99), and glutamine (r = .77, adj P = 0.21) content were not significantly correlated with depressive symptoms. Because it was also possible that the clinical effect at 2 days might be associated with earlier changes in AANt content (3 hours after infusion), secondary correlation analyses were performed. Again, associations between glutamate (r = 0.52, adj P = 0.54), GABA (r = 0.17, adj P > .99) and glutamine (r = 0.38, adj P > .99) and changes in HDRS scores on day 2 were not significant.
3.6 Correlation of baseline occipital amino acid content and clinical response
Baseline measures of glutamate, GABA and glutamine were not correlated with changes in HDRS scores at any time point.
4. Discussion
The antidepressant effect of ketamine reported here replicates two important features described in the previous controlled studies that used identical infusion parameters (40-minute infusion of racemic ketamine at 0.5 mg/kg) (Berman et al., 2000; Zarate et al., 2006). First, the improvement in symptoms was rapid, as measured by significant decreases in HDRS and BDI scores one hour post-infusion. Second, the antidepressant effect persisted for up to one week as measured by a significant decrease in HDRS scores at day 7. Also consistent with previous reports, we found that ketamine induced perceptual alterations during the infusion, as measured by the CADSS, which dissipated prior to the emergence of antidepressant effects. The absence of a correlation between peak perceptual changes and changes in HDRS scores in this study suggests that ketamine’s antidepressant effect is not dependent on the induction of a dissociative state, a possibility consistent with the finding that the rapid antidepressant effect of the NR2B subunit-selective receptor antagonist traxoprodil is also not dependent on a dissociative reaction in research subjects (Preskorn et al., 2008). Consequently, it is likely that NMDA receptor antagonists constitute a class of drugs whose antidepressant efficacy is independent of their dissociative potential.
In contrast to the significant changes on CADDS scores, we did not observe changes in the BPRS positive symptom subscale. This finding is consistent with the report of negligible effects on BPRS positive symptom scores in the majority of MDD subjects treated with the same ketamine dosing parameters used in this study (Mathew et al., 2009), but contrary to the significant effect reported by Berman (2000). Additional studies, such as those examining the relationship between pharmacokinetic variability or genetic variation in NMDA subunits within specific clinical populations, and clinical responses, are needed to clarify the basis for such contradictory findings. Overall, ketamine treatment was well tolerated and was without persisting negative effects. Although ketamine induced a statistically significant rise in systolic blood pressure, it was without clinical significance in this study.
The second objective of this study was to characterize the effects of ketamine on occipital AANt content as measured by 1H-MRS. Because we have previously shown that a subset of MDD subjects have low GABA and elevated glutamate content in the occipital cortex, and furthermore, that GABA alterations appear to normalize after successful treatment with either chemical antidepressants (Sanacora et al., 2002) or ECT (Sanacora et al., 2003), we hypothesized that decreased depression scores (reflecting clinical improvement) following ketamine treatment would correlate negatively with a change in GABA levels, and positively with a change in glutamate levels. We did not observe any such change in AANt content following ketamine treatment, and correlations between clinical response and cortical AANt content were not observed. Moreover, we found no evidence to support the possibility that baseline occipital cortex AANt levels can serve as a predictive biomarker of clinical response to ketamine.
The small sample size of this study limits our ability to draw firm conclusions regarding the effects of ketamine on occipital GABA and glutamate content. However, because this study used a very similar MRS methodology and sample size to those that previously detected relatively large effect sizes on occipital GABA content after treatment with SSRIs (d = 1.3) (Sanacora et al., 2002) and ECT (d = 2.6) (Sanacora et al., 2003), we believe the small effect size at 3 hours (d = .21), and 2 days (d = .28) post-infusion indicates that ketamine does not produce the same magnitude of effect at these early time points as compared to the effect of chronic treatment with SSRIs or ECT. However, it is also possible that the absence of experimental support for our hypotheses is due to baseline differences in AANt profiles between study populations, as there was no control group to confirm that either the GABA or glutamate levels were abnormal in this group of MDD subjects. Although there is no specific reason to assume this cohort of depressed subjects is markedly different from those enrolled in the earlier studies, until appropriate controls are utilized, the possibility that our negative findings are secondary to investigating a subgroup of depressed subjects that were without baseline AANt abnormalities cannot be excluded.
Additional limitations of this study are notable. The 1H-MRS method used in this study can only measure static levels of total tissue amino acid content, and may thereby be insensitive to dynamic changes in other aspects of AANt neurotransmission that are induced by ketamine, such as changes in glutamate efflux or turnover. However, because a previous study using 1H-MRS did detect significant changes in glutamine content in the anterior cingulate cortex of healthy subjects during ketamine infusion (Rowland et al., 2005), it is possible that we may have missed significant changes in AANt content that developed at earlier time points, or within different brain regions, following ketamine treatment. Future imaging studies on the neurobiological mechanisms underlying ketamine’s antidepressant effect may benefit from dynamic measures of AANt function and the assessment of other brain regions.
In summary, while we replicated the rapid antidepressant-like effects of ketamine, we did not observe ketamine-induced changes in AANt content in the occipital cortex at several time points associated with clinical improvement. Further studies on the neurochemical basis of NMDAR antagonists’ antidepressant activity are warranted given that the rapid and robust improvement in symptoms they can induce in ‘treatment-resistant’ depressed subjects may reflect unidentified brain mechanisms that contribute to both treatment resistance and antidepressant efficacy.
Acknowledgments
We would like to thank Ms. Yael Levin and Ms. Jessica Howell for their assistance on this project, and Ms. Lisa Roach for her assistance with data management and manuscript preparation. This study was supported by the Yale/Pfizer imaging alliance and from NIH 5K02MH076222-05 (GS). Additional funding was provided by R01 DA021785 (GM) and K02 AA13430 (GM). Dr. Sanacora has received consulting fees form AstraZeneca, Bristol-Myers Squibb, Evotec, Eli Lilly & Co., Johnson & Johnson, Roche, Novartis and Sepracor Inc. He has also received additional grant support from AstraZeneca, Bristol-Myers Squibb, Merck & Co., Roche, and Sepracor Inc. In addition, he is a co-inventor with Dr. Krystal on a filed patent application by Yale University (PCTWO06108055A1) concerning the use of glutamate modulating drugs as antidepressants and anxiolytics. Dr. Krystal has received consulting and advisory fees from Abbott Laboratories, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Co., Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceuticals, Lohocla Research Corporation, Merz Pharmaceuticals, Pfizer Pharmaceuticals, SK Holdings Co., Ltd, Takeda Industries, Teva Pharmaceutical Industries, Ltd., and Transcept Pharmaceuticals. He has also received additional research support from the Janssen Research Foundation (provided drug and some study support to the Department of Veterans Affairs), and holds Exercisable Warrant Options with Tetragenex Pharmaceuticals (value less than $100). Dr. Krystal’s relevant patents and inventions are related to 1) Patent #5,447,948. September 5, 1995. Dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia, and 2) Intranasal Administration of Ketamine to Treat Depression (pending). M. Fasula and Drs. Valentine, Mason, Watzl, Gomez, and Pittman are without potential conflicts of interest as related to the content of this report.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beck AT, WC, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:53–63. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- Behar K, Rothman DL, Spencer DD, Petroff OAC. Analysis of macromolecule resonances in 1H NMR spectra of human brainMagn Reson Med. Magn Reson Med. 1994;32:294–302. doi: 10.1002/mrm.1910320304. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biological Psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154(6):805–811. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- Brunner E, Domhof S, Langer F. Nonparametric analysis of longitudinal data in factorial experiments. New York, NY: John Wiley and Sons; 2002. [Google Scholar]
- Buck N, Cali S, Behr J. Enhancement of long-term potentiation at CA1-subiculum synapses in MK-801-treated rats. Neurosci Lett. 2006;392(1–2):5–9. doi: 10.1016/j.neulet.2005.08.054. [DOI] [PubMed] [Google Scholar]
- Dall’Olio R, Gaggi R, Bonfante V, Gandolfi O. The non-competitive NMDA receptor blocker dizocilpine potentiates serotonergic function. Behav Pharmacol. 1999;10(1):63–71. doi: 10.1097/00008877-199902000-00006. [DOI] [PubMed] [Google Scholar]
- Garcia LS, Comim CM, Valvassori SS, Reus GZ, Barbosa LM, Andreazza AC, Stertz L, Fries GR, Gavioli EC, Kapczinski F, Quevedo J. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):140–144. doi: 10.1016/j.pnpbp.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. Journal of Neuroscience. 1997;17(7):2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry PG, Dautry C, Hantraye P, Bloch G. Brain GABA editing without macromolecule contamination. Magn Reson Med. 2001;45(3):517–520. doi: 10.1002/1522-2594(200103)45:3<517::aid-mrm1068>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Keilhoff G, Bernstein HG, Becker A, Grecksch G, Wolf G. Increased neurogenesis in a rat ketamine model of schizophrenia. Biological Psychiatry. 2004;56(5):317–322. doi: 10.1016/j.biopsych.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Manji HK, Zarate CA. The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology and Therapeutics of Mood Disorders. Neuroscientist. 2009 doi: 10.1177/1073858409336093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Murrough JW, Aan Het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol. 2009:1–12. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. Journal of Neuroscience. 1997;17(8):2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacher J, Alonso-Llosa G, Rosell DR, McEwen BS. NMDA receptor antagonist treatment increases the production of new neurons in the aged rat hippocampus. Neurobiology of Aging. 2003;24(2):273–284. doi: 10.1016/s0197-4580(02)00096-9. [DOI] [PubMed] [Google Scholar]
- Nacher J, Rosell DR, Alonso-Llosa G, McEwen BS. NMDA receptor antagonist treatment induces a long-lasting increase in the number of proliferating cells, PSA-NCAM-immunoreactive granule neurons and radial glia in the adult rat dentate gyrus. European Journal of Neuroscience. 2001;13(3):512–520. doi: 10.1046/j.0953-816x.2000.01424.x. [DOI] [PubMed] [Google Scholar]
- Paul IA, Trullas R, Skolnick P, Nowak G. Down-regulation of cortical beta-adrenoceptors by chronic treatment with functional NMDA antagonists. Psychopharmacology (Berl) 1992;106(2):285–287. doi: 10.1007/BF02801986. [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30(6):672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Rothman D, Behar K, Petroff OA. Improved quantitation of shor TE 1H NMR human brain spectra by removal of short T1 macromolecule resonances. Soc Magn Reson, 2nd Meeting; 1994. p. 47. [Google Scholar]
- Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162(2):394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu Y-T, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of GABA and glutamate in major depression. Archives of General Psychiatry. 2004;61(7):705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Ciarcia JJ, Ostroff RB, Krystal JH. Increased occipital cortex GABA concentrations following electroconvulsive therapy in depressed patients. American Journal of Psychiatry. 2003;160(3):577–579. doi: 10.1176/appi.ajp.160.3.577. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. American Journal of Psychiatry. 2002;159(4):663–665. doi: 10.1176/appi.ajp.159.4.663. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7(5):426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Yang J, Choi IY, Li SS, Chen Z. A new strategy for in vivo spectral editing. Application to GABA editing using selective homonuclear polarization transfer spectroscopy. J Magn Reson. 2004;170(2):290–298. doi: 10.1016/j.jmr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]