

Abstract
OBJECTIVE: Complete Freund's Adjuvant (CFA) is known to arrest autoimmune diabetes development in non-obese diabetic (NOD) mice. However, CFA alone cannot induce effective remission in diabetic NOD mice. Previously, we reported that anti-CXC chemokine ligand 10 (CXCL10) antibody can promote beta-cell proliferation in NOD mice. In the present study, we aimed to examine whether anti-CXCL10 plus CFA treatment can effectively reverse autoimmune diabetes development. METHODS: Systemic supply of anti-CXCL10 antibody by CXCL10 DNA vaccination in combination with CFA injection was performed in new-onset diabetic NOD mice. Remission rate of diabetes, histological characteristics of residual insulitis lesions, residual beta-cell mass, and regulatory T cell population in local pancreas were examined. RESULTS: A high frequency of diabetes reversal was observed after combination treatment with anti-CXCL10 plus CFA. In mice showing diabetes reversal, residual beta-cell mass was significantly increased, and some beta-cells were in a proliferative state. Although systemic cytokine profiles were unaffected, the frequency of "hybrid regulatory T cells", i.e. regulatory T cells expressing CXCR3, was significantly increased in local pancreatic lesions. This was possibly associated with the regulation of anti-islet autoimmunity. CONCLUSIONS: Anti-CXCL10 plus appropriate immune adjuvant therapy arrested, and reversed, type 1 diabetes development.
Keywords: type 1 diabetes, NOD mouse, Complete Freund's Adjuvant, regulatory T cell, DNA accination, CXC chemokine ligand 10, beta-cell proliferation
Abbreviations: Ab - antibody; APC - antigen-presenting cell; arIPb - anti-rat IP-10 Ab; BCG - Bacillus Calmette-Guerin; CAG - CMV immediate enhancer/β-actin; CD4 - cluster of differentiation 4 (glycoprotein expressed on lymphocytes); cDNA - complementary DNA; CFA - Complete Freund's Adjuvant; CMV - cytomegalovirus; CXC - C-X-C motif chemotactic cytokine; CXCL10 - C-X-C motif chemokine ligand 10; CXCR3 - CXC chemokine receptor 3 (mediate the biological activities of chemokines); CY - cyclophosphamide; DAB - diaminobenzidine; DNA - deoxyribonucleic acid; ELISA - enzyme-linked immunosorbent assay; FCS - fetal calf serum; FoxP3 - forkhead box P3 (transcriptional activator); GrB - granzyme B; HBSS - Hanks' balanced salt solution; H&E stain - hematoxylin and eosin stain; HRP - horseradish peroxidase; Ig - immunglobulin; IgG - immunglobulin G; IFN-γ - interferon gamma; IL - interleukin; IP-10 - interferon gamma-induced protein 10 kDa (also known as CXCL10); Ki-67 protein - protein detected by monoclonal antibody anti-Ki-67 (proliferation marker); LADA - latent autoimmune diabetes; NIH - National Institutes of Health; NOD - non-obese diabetic; OCT - optimal cutting temperature; OD - optical density; pCAGGS plasmid - CAG promoter plasmid (monocistronic vector under the control of the CAG promoter, gives higher levels of transgene than conventional CMV and β-actin promoters); Pdx-1 - pancreas duodenum homeobox 1; PFA - paraformaldehyde; RPMI-1640 medium - Roswell Park Memorial Institute 1640 (cell culture medium for leukocytes); SCID - severe combined immunodeficiency (mouse model without functional T and B cells); T1D - type 1 diabetes; TGF-β - transforming growth factor beta; Th1- T-helper 1; TMB - tetramethylbenzidine; TNF - tumor necrosis factor; Treg cell - regulatory T cell
Introduction
Type 1 diabetes (T1D) is considered to be a T cell-mediated autoimmune disease characterized by selective destruction of pancreatic β-cells, resulting in insulin deficiency and hyperglycemia [1]. Many studies using the non-obese diabetic (NOD) mouse have suggested that T-helper 1 (Th1) cells are critical in the development of autoimmune β-cell injury [2, 3].
CXC chemokine ligand 10 (CXCL10), also known as interferon-γ inducible protein 10 (IP-10), is a Th1-related chemokine. It serves as a signaling molecule, and is produced by several types of cells (monocytes, endothelial cells, fibroblasts, etc.) in response to interferon (IFN)-γ.
CXCL10 is a potent chemotactic agent that directs the migration of lymphocytes expressing chemokine (C-X-C motif) receptor 3 (CXCR3), a receptor for CXCL10. Also, it promotes Th1-type immune responses [4]. Recent studies revealed CXCL10 to be involved in the pathogenesis of T1D. We and others have demonstrated that high risk T1D patients have elevated serum CXCL10 levels [5, 6]. These levels are also increased in adult patients with latent autoimmune diabetes (LADA) [7], but to a lesser extent.
In animal models, we have shown anti-CXCL10 antibody administration to suppress the development of cyclophosphamide (CY)-induced diabetes in NOD mice [8]. Also, in vivo induction of anti-CXCL10 antibody by CXCL10 DNA vaccination suppressed spontaneous development of diabetes in NOD mice [9]. Also, other research groups have reported that blockade of CXCL10-CXCR3 interaction could suppress the development of insulitis in a virus-induced diabetes model [10, 11]. These studies have demonstrated that suppression of diabetes development is possible in different animal models. However, there are no reports of hyperglycemia reversal in established autoimmune diabetes, through administration of CXCL10 blockade.
CXCL10 is expressed in residual β-cells of insulitis lesions in NOD mice [12] and human T1D patients [13, 14]. Moreover, Rhode et al. reported that constitutive CXCL10 expression in β-cells accelerated autoimmune diabetes development in a virus-induced diabetes model [15]. These findings suggested a close association between β-cell-specific localization of CXCL10 and pathogenesis of T1D. Recently, CXCL10 was found to directly impair function, or induce apoptosis, of β-cells [13]. We have shown that CXCL10 has an anti-proliferative effect on β-cells. Also, blocking CXCL10-CXCR3 interaction by anti-CXCL10 antibody, promoted β-cell proliferation in pre-diabetic NOD mice, which contributed to the prevention of autoimmune diabetes development [8, 9, 16]. Thus, blocking CXCL10 action could be a strategy for reversing hyperglycemia in established autoimmune diabetes, via amelioration of β-cell dysfunction, prevention of β-cell apoptosis, and/or promotion of β-cell proliferation.
CFA and Bacillus Calmette-Guerin (BCG) contain mycobacterial cell wall components. Also, they exert an immune adjuvant effect protecting NOD mice from diabetes onset [17, 18, 19]. CFA was identified as an inducer of tumor necrosis factor (TNF)-α production by antigen-presenting cells (APCs) [20]. Temporarily, it can eliminate diabetogenic T cells in NOD mice, via induction of apoptosis mediated by TNF-α [20]. This effect contributes to the inhibition of spontaneous diabetes development in NOD mice. Moreover, it was recently reported that CFA treatment can induce regulatory T (Treg) cells in pancreatic lymph nodes of diabetic NOD mice [21]. Therefore, CFA may be an essential inducer of Treg cells in the inhibition of autoimmune diabetes.
However, in most studies, CFA treatment alone has failed to reverse hyperglycemia in NOD mice with new-onset diabetes [22, 23]. Therefore, we hypothesized that a combined intervention with anti-CXCL10 plus CFA can promote remission of autoimmune diabetes. We assumed that induction of residual β-cell proliferation, together with regulation of anti-islet autoimmunity, promotes remission of autoimmune diabetes. Therefore, in an effort to effectively reverse hyperglycemia in autoimmune diabetes, we treated new-onset diabetic NOD mice with CFA in combination with CXCL10 DNA vaccination, to induce anti-mouse CXCL10 antibody production, and to promote the reversal of hyperglycemia in vivo [9].
Materials and methods
Mice
Eight week old female NOD mice were purchased from CLEA Japan Inc. (Tokyo, Japan). They were kept under specific pathogen-free conditions in the animal facility of Keio University School of Medicine. Urinary glucose analysis was started at 12 weeks of age, and performed weekly using Tes-tape (Shionogi, Osaka, Japan). Blood glucose levels were determined using Glutest-Ace (Sanwa Kagaku, Nagoya, Japan) when glycosuria was detected. The onset of diabetes was defined by blood glucose levels above 250 mg/dl in two consecutive measurements taken 48 h apart. All experiments using mice were approved by, and performed according to, the guidelines of the animal committee of Keio University.
Plasmid vectors
Plasmid pCAGGS-CXCL10 was constructed by inserting CXCL10 cDNA into rat. In the CXCL10 cDNA, one amino acid (number 92) of its deduced amino acid sequence was replaced by that of mouse CXCL10 [24]. The replacement was performed into the multiple cloning site of the pCAGGS expression vector [9]. Previously, we obtained a monoclonal antibody (Ab), anti-rat IP-10 Ab (arIPb), by immunizing mice with rat CXCL10. This Ab neutralizes rat and mouse CXCL10 [24, 25]. The expression capacity of the resulting pCAGGS-CXCL10 plasmid DNA was confirmed by transient transfection into 293 cells. The transcript was detected in 293 cells, and in the culture medium, by western blotting using arIPb [9] (data not shown).
CFA injection and DNA vaccination
New-onset diabetic NOD mice were anesthetized with pentobarbital 2 days after diabetes onset. Then, 50 μl CFA (Sigma-Aldrich, St. Louis, MO, USA) conjugated with an equal volume of saline (final concentration 0.5 mg/ml) was injected into each foot pad (total 100 μl/mouse). Simultaneously, 50 μg pCAGGS-CXCL10, or pCAGGS plasmid DNA (control plasmid DNA), were injected into the bilateral tibialis anterior muscles (total of 100 μg plasmid DNA/mouse) using an electroporation method, and an insulin syringe with a 27-gauge needle [9, 26]. For electroporation, a pair of electrode needles with a 5-mm gap was inserted into the muscle to encompass the DNA injection sites, and electric pulses were delivered using an electric pulse generator. Three pulses of 100 V each were delivered to each injection site at a rate of one pulse per second. Each pulse had a duration of 50 ms. Then, three pulses of the opposite polarity were applied [9, 26].
DNA vaccination was repeated two weeks later. After treatment, blood glucose levels were followed over time. Two consecutive non-fasting measurements below 200 mg/dl were considered as a reversal of hyperglycemia. If blood glucose levels were again above 250 mg/dl by 10 weeks after first DNA vaccination, the mice were finally considered to be diabetic.
Measurement of anti-mouse CXCL10 antibody in sera of NOD mice treated with DNA vaccination plus CFA injection
A direct ELISA was used to determine the anti-mouse CXCL10 antibody level in sera of NOD mice, as reported previously [9]. Briefly, recombinant mouse CXCL10 (Pepro Tech, London, UK) was used to coat a 96-well ELISA plate (Nunc, Roskilde, Denmark) at a concentration of 50 ng/well. Diluted serum (1:8) from treated mice was added to the plate. Sheep anti-mouse Ig horseradish peroxidase (HRP)-conjugated antibody (Amersham Pharmacia Biotech, UK) was used as a labeled antibody. Tetramethylbenzidine (TMB; Pharmingen, San Diego, CA, USA) was used as a soluble HRP substrate. Antibody level was expressed as optical density (OD) value.
Evaluation of insulitis and measurement of residual islet area
The pancreas was removed from each mouse, fixed in 10% formaldehyde, and embedded in paraffin. Thin sections at five levels, at least at 150 μm centers, were cut for H&E staining to evaluate islet-infiltrating immune cells by light microscopy. At least 30 islets from each mouse were analyzed by two independent observers. Histology was classified as follows: islet free of insulitis, peri-insulitis, intra-insulitis, and contracted islet. Concurrently, residual islet area in the insulitis lesion was defined as an area of islet cell region free of infiltrating immune cells. This was determined in square micrometers using ImageJ software (NIH, USA), and H&E stained pancreatic tissue section.
Immunohistological analyses
Insulin, glucagon, and forkhead box P3 (FoxP3) staining was performed using pancreatic tissue, or a pancreatic lymph node, which was removed from each mouse, fixed in 10% formaldehyde, and embedded in paraffin. Thin sections of these tissues were prepared, being cut at 100 μm centers. The cut samples were deparaffinized, and stained with anti-insulin antibody (Oriental Yeast Co., Tokyo, Japan), anti-glucagon antibody (Nichirei Co., Tokyo, Japan), or anti-mouse FoxP3 antibody (eBioscience, San Diego, CA, USA). This was followed by sequential incubations with the appropriate biotinylated secondary antibodies, avidin-biotin complex and H2O2-diaminobenzidine (DAB).
The pancreas was removed from each mouse, inflated with OCT compound, and snap-frozen in liquid nitrogen, to perform pancreas duodenum homeobox 1 (Pdx-1) and Ki-67 staining. Four percent paraformaldehyde (PFA)-fixed 6-μm fresh frozen pancreatic tissue sections, were incubated with anti-Ki-67 antibody (Invitrogen, Carlsbad, CA, USA), and anti-mouse Pdx-1 antibody (Trans Genic Inc., Kumamoto, Japan). This was followed by Alexa-568-labeled anti-rat IgG, and Alexa-488-labeled anti-rabbit IgG, respectively (Molecular Probes Inc., Eugene, OR, USA).
Flow cytometric analysis
Lymphocyte staining was performed using the following antibodies:
- FITC-conjugated anti-mouse CD4 monoclonal antibody (H129.19, PharMingen, San Diego, CA, USA),
- PE-conjugated anti-mouse CD8 monoclonal antibody (53-6.7, PharMingen),
- FITC-conjugated anti-mouse T cell receptor (TCR)-β monoclonal antibody (H57-597, PharMingen),
- PE-conjugated anti-mouse CD11b monoclonal antibody (M1/70, PharMingen),
- FITC-conjugated anti-mouse CD45R/B220 monoclonal antibody (RA3-6B2, PharMingen), and
- PE-conjugated anti-mouse CXCR3 monoclonal antibody (R&D Systems, Minneapolis, MN, USA).
Cells were fixed, and permeabilized, with Foxp3 staining buffer (eBioscience), for intercellular staining. Permeabilized cells were stained with FITC-conjugated anti-mouse Foxp3 monoclonal antibody (FJK-16s, eBioscience). The prepared cells were analyzed with Epics Altra (Coulter, Hialeah, FL, USA).
Semi-quantitative real-time PCR
Total RNA was extracted from a pancreatic lymph node or the spleen using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA). DNase treatment was performed according to manufacturer instructions. Extracted RNA was reverse transcribed using Not I-d(T)18 primer, and a first-strand cDNA synthesis kit (GE Healthcare Bio-Sciences, Piscataway, NJ, USA), according to manufacturer instructions. Semi-quantitative RT-PCR was carried out for IFN-γ, IL-10, IL-4, transforming growth factor (TGF)-β, FoxP3, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; internal control), using an ABI Prism 7700 sequence detector (PE Applied Biosystems, Foster City, CA, USA) and assay-on-demand real-time PCR kits (PE Applied Biosystems). All reactions were performed using TaqMan Universal MasterMix (PE Applied Biosystems). The obtained mRNA level was expressed in relation to the amplified GAPDH PCR product from the same sample: (sample PCR product/GAPDH PCR product) x constant.
Adoptive transfer of splenocytes
Ten weeks after first DNA vaccination, splenocytes were harvested from NOD mice treated with CXCL10, or control DNA vaccination in combination with CFA injection. Then, 1 x 106 splenocytes were transferred into female NOD-SCID mice (n = 8 per group). Blood glucose levels were measured twice weekly, thereafter.
Isolation of mononuclear cells in local pancreas
Mononuclear cells that locally infiltrated the pancreas were isolated using a modification of our previously reported method [27]. In brief, mice were killed, and pancreatic lymph nodes, easily detectable by their size and shape, were discarded. Next, the common bile duct was clamped at its entrance to the duodenum, and cannulated under a dissecting microscope, using a 26-gauge needle. Approximately 2.5 ml of cold Hanks' balanced salt solution (HBSS) (Sigma-Aldrich) containing collagenase XI (1 mg/ml; Sigma-Aldrich) were injected into the common bile duct.
The distended pancreas was removed and incubated in a 50 ml plastic tube for 15 min in a 37°C water bath. Cold HBSS was added to stop the digestion. After vortexing the tube for 15 sec, it was centrifuged at 320 g for 5 min. The supernatant was discarded, and the pellet was washed 3 times with RPMI-1640 (Life Technologies, Grand Island, NY, USA) containing 2% fetal calf serum (FCS) (Life Technologies). The suspension, including mononuclear cells, was filtered through a 70 μm nylon mesh cell strainer (BD Bioscience, Bedford, MA, USA), and was then used for flow cytometric analysis.
Statistical analyses
Results are presented as means ± SEM. After application of the adoptive transfer system, log-rank test was used to compare the remission of diabetes, and diabetes development. Comparisons between the two groups were made using the Mann-Whitney U test. P-values less than 0.05 were considered statistically significant.
Results
Anti-mouse CXCL10 antibody was induced in sera from mice treated with CXCL10 DNA vaccination plus CFA injection
To determine whether anti-mouse CXCL10 antibody was induced in pCAGGS-CXCL10-treated NOD mice, we determined the level of serum anti-mouse CXCL10 antibody at 8 to 10 weeks after first CXCL10 DNA vaccination, using direct ELISA. As shown in supplementary Figure A1 (see Appendix), CXCL10 vaccination using pCAGGS-CXCL10 plus CFA injection (CXCL10 DNA group) induced anti-mouse CXCL10 antibody in the sera of most treated NOD mice. The antibody levels were the same as those found in our previous study [9]. On the other hand, anti-mouse CXCL10 antibody was not induced in any NOD mice receiving gene transfer of pCAGGS-control plus CFA (control DNA group).
Figure A1. Anti-mouse CXCL10 antibody was induced by CXCL10 DNA vaccination.
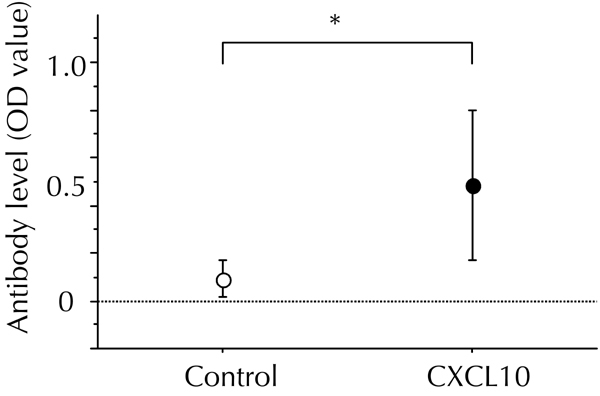
In combination with CFA treatment, the first DNA vaccination with pCAGGS-CXCL10 (CXCL10 DNA vaccination), or pCAGGS control plasmid DNA (control DNA vaccination), was performed in new-onset diabetic NOD mice. Each DNA vaccination was repeated two weeks later. 8 to 10 weeks after the DNA vaccination, a direct ELISA was performed to measure the OD values of anti-mouse CXCL10 antibody in sera. The OD values in the control DNA group (n = 9) and the CXCL10 DNA group (n = 9) were 0.09 ± 0.04 and 0.49 ± 0.10, respectively. * p < 0.05 by Mann-Whitney U test.
CXCL10 DNA vaccination in combination with CFA injection reversed hyperglycemia in new-onset diabetic NOD mice
We performed CXCL10, or control, DNA vaccination in combination with CFA injection in new-onset diabetic NOD mice, and followed blood sugar levels in the two groups. As shown in Figure 1 (A, B, and D), a much higher frequency of hyperglycemia reversal was observed in the CXCL10 DNA than in the control DNA group (69.2% (18/26) to 18.2% (4/22)), by 10 weeks after first DNA vaccination. CXCL10 DNA vaccination without CFA treatment produced a low frequency of hyperglycemia reversal (10%, 1/10) (Figure 1 (C and D).
Figure 1. CXCL10 DNA vaccination in combination with CFA injection effectively reversed hyperglycemia in new-onset diabetic NOD mice.
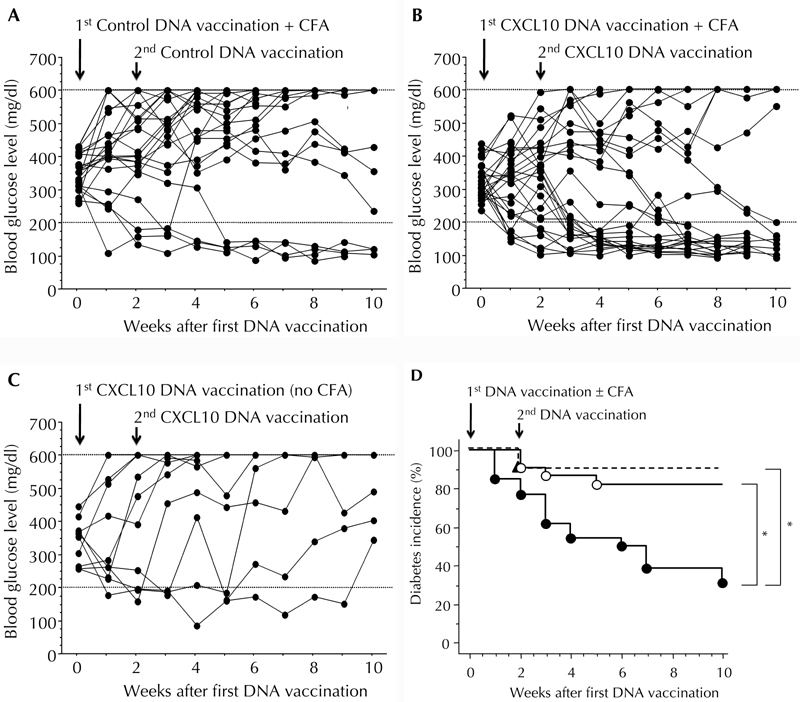
A, B, C: Time courses of blood glucose levels in NOD mice after the indicated treatments. A: Mice treated with control DNA vaccination plus CFA injection (n = 22). B: Mice treated with CXCL10 DNA vaccination plus CFA injection (n = 26). C: Mice treated with CXCL10 DNA vaccination alone (n = 10). D: Kaplan-Meier plot of cumulative incidence in diabetes remission after the indicated treatments. Closed circles and solid lines: Mice treated with CXCL10 DNA vaccination plus CFA injection. Open circles and solid lines: Mice treated with control DNA vaccination plus CFA injection. Closed triangles and dotted lines: Mice treated with CXCL10 DNA vaccination alone. * p < 0.05 by the log-rank test.
Residual islet size was increased in NOD mice with hyperglycemia reversal
To study the characteristics of residual islets in NOD mice showing reversal of hyperglycemia, pancreatic sections were H&E stained. In comparison with new-onset diabetic NOD mice, or established diabetic NOD mice, residual islet mass was significantly increased in CXCL10 DNA group mice showing diabetes reversal 8 to 10 weeks after the first DNA vaccination (Figure 2 (A-E)). Moreover, CXCL10 DNA-treated mice with reversed diabetes exhibited only peri-insulitis, or no insulitis, in more than half of the residual islets (Figure 2F).
Figure 2. Increased residual islet areas in CXCL10 DNA group with reversal of hyperglycemia.
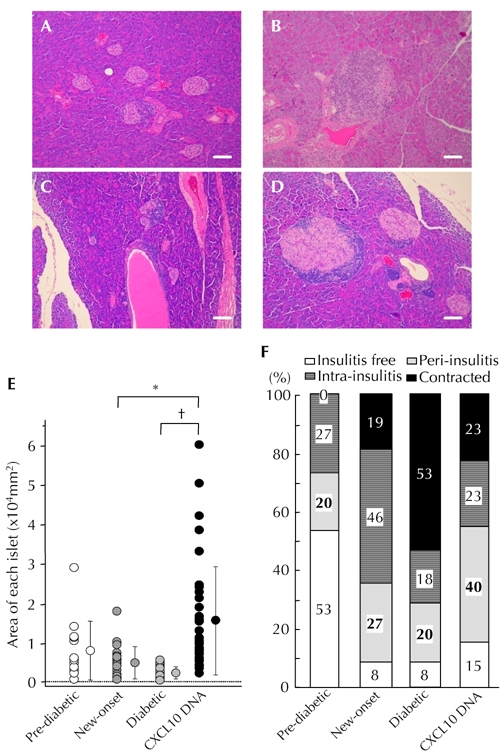
A-D: HE staining. Representative islet lesions in pre-diabetic NOD mice (A), new-onset diabetic NOD mice (B), established diabetic NOD mice in control DNA group (C), and CXCL10 DNA group with reversal of hyperglycemia (D). E: Islet areas in the four groups were measured and plotted. Residual islet areas were larger in mice with reversal of hyperglycemia from the CXCL10 DNA group. F: Degrees of insulitis in the four groups. Each group consisted of 8 mice, and at least 30 islets from each mouse were observed by two independent observers. More than half of the residual islets in the CXCL10 DNA group with reversal of hyperglycemia showed peri-insulitis or no insulitis (D). * p < 0.05, † p < 0.01 by Mann-Whitney U test. Scale bars: 80 μm.
α-cells were scattered within residual islets in NOD mice with hyperglycemia reversal
To clarify structural changes in the residual islets of NOD mice showing hyperglycemia reversal, immunohistochemical staining was performed for insulin and glucagon (Figure 3). Unexpectedly, histological examination revealed that glucagon-positive cells were scattered within the residual islets in CXCL10 DNA group mice exhibiting diabetes reversal at 8 to 10 weeks after first DNA vaccination (Figure 3D). Considering that α-cells are generally localized around insulitis lesions in new-onset diabetic NOD mice (Figure 3F), the distinct pattern of α-cell localization, shown in Figure 3D, raised the possibility of deregulated proliferation of residual β-cells in the process of diabetes reversal.
Figure 3. Alpha-cells scattered within residual islets in the group of CXCL10 DNA-treated mice with reversal of hyperglycemia.
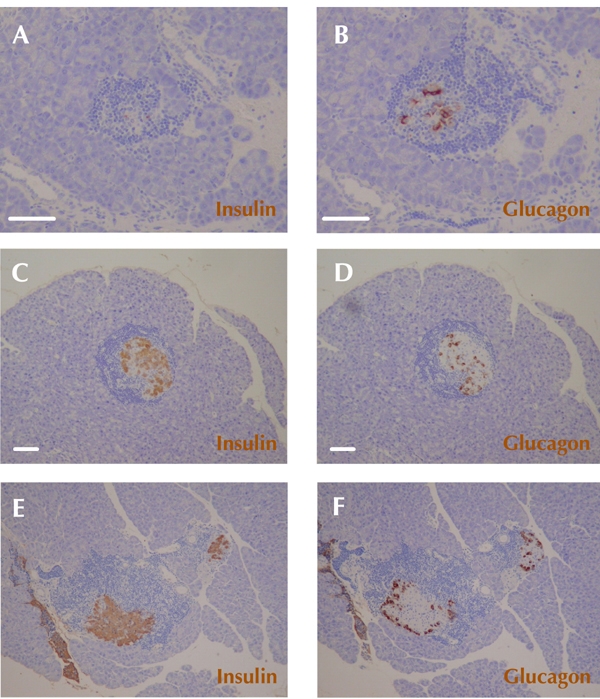
Immunohistochemical staining of insulin (A, C, E) and glucagon (B, D, F) was performed. A and B: Representative insulitis lesions in established diabetic NOD mice in the group of control DNA mice. C and D: Representative insulitis lesions in the group of CXCL10 DNA-treated mice with reversal of hyperglycemia 8 to 10 weeks after first DNA vaccination. Glucagon-positive cells (α-cells) are scattered within remaining islets (D). E and F: Representative typical distribution patterns of α-cells and β-cells in insulitis lesions of a new-onset diabetic NOD mice. Scale bars: 80 μm.
Proliferating β-cells were common in NOD mice showing diabetes reversal
Next, we evaluated the proliferative state of residual β-cells in NOD mice showing diabetes reversal. We performed immunohistochemical double-staining of Pdx-1, a key gene for mature β-cells, and Ki-67, a marker of cell proliferation, 8 to 10 weeks after first DNA vaccination. As shown in Figure 4, a higher frequency of Ki-67-positive islet cells was observed in CXCL10 DNA group mice showing diabetes reversal. Also, most Ki-67-positive cells inside residual insulitis lesions were positive for Pdx-1, indicating a portion of the residual β-cells to be in a proliferative state.
Figure 4. Increased residual β-cells in a proliferative state in the group of CXCL10 DNA-treated mice with reversal of hyperglycemia.
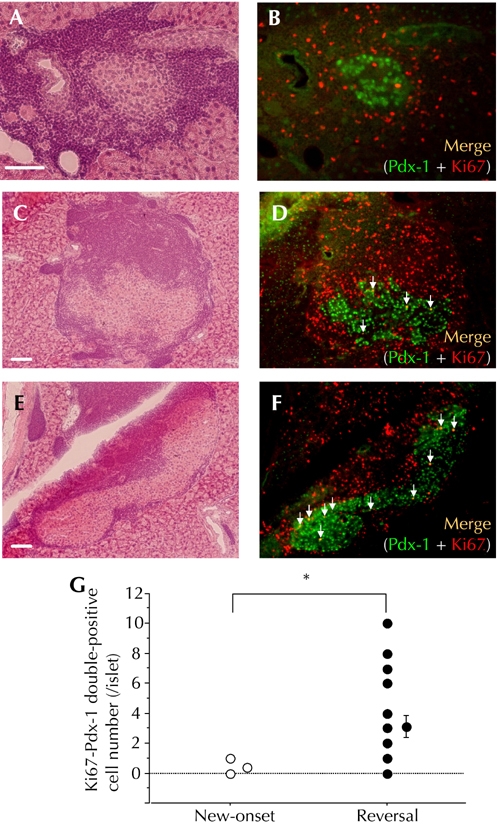
HE staining (A, C, E) and immunohistochemical double staining of Pdx-1 and Ki67 (B, D, F) were performed. A and B: Representative insulitis lesion in a new-onset diabetic NOD mouse. C, D, E, and F: Two representative insulitis lesions in a CXCL10 DNA vaccinated mouse with reversal of hyperglycemia at 8 to 10 weeks after first DNA vaccination. White arrows indicate Pdx-1 and Ki67 double-positive islet cells (C, D: intra-insulitis, E, F: peri-insulitis). G: The numbers of Pdx-1 and Ki67 double-positive islet cells per islet were counted and plotted. Open circles: new-onset diabetic NOD mice (n = 6, total of 24 islets). Filled circles: CXCL10 DNA-treated mice with reversal of hyperglycemia (n = 8, total of 24 islets). * p < 0.01 by Mann-Whitney U test. Scale bars: 80 μm.
Systemic cytokine profile was not affected and anti-islet autoimmunity persisted in NOD mice with hyperglycemia reversal
Flow cytometric analysis showed reduced CD4+ and CD8+ T cells in the spleens of mice with reversal of hyperglycemia in the CXCL10 DNA group, compared to control DNA group mice (supplemental Figure A2, see Appendix). However, there were no significant differences between the two groups in mRNA expression levels of cytokines (IFN-γ, IL-10, IL-4), or Treg-related factors (FoxP3, TGF-β) in the spleen (supplementary Figure A3, and data not shown). This suggested that autoimmune regulation did not play a crucial role in the process of hyperglycemia reversal, at least not in the periphery.
Figure A2. Lymphocyte populations in the spleen were affected by CXCL10 DNA vaccination in combination with CFA injection.
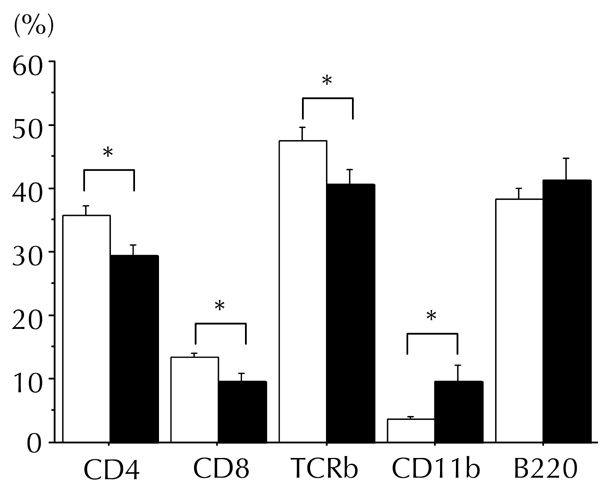
Populations of splenic lymphocytes (CD4+ cells, CD8+ cells, whole T cells (TCR-β), macrophages (CD11b), and B cells (B220)) were measured by flow cytometry 8 to 10 weeks after first DNA vaccination. Counts of splenic CD4+, CD8+, and whole T cells were slightly reduced in the group of CXCL10 DNA-treated mice with reversal of hyperglycemia (filled bar, n = 7), as compared to the control DNA group (open bar, n = 7). In contrast, splenic macrophage count was increased. Cell populations are presented as the ratio of each cell count to that of gated lymphocytes. * p < 0.05 by Mann-Whitney U test.
Figure A3. Splenic cytokines and FoxP3 mRNA expression levels were not affected by CXCL10 vaccination in combination with CFA injection.
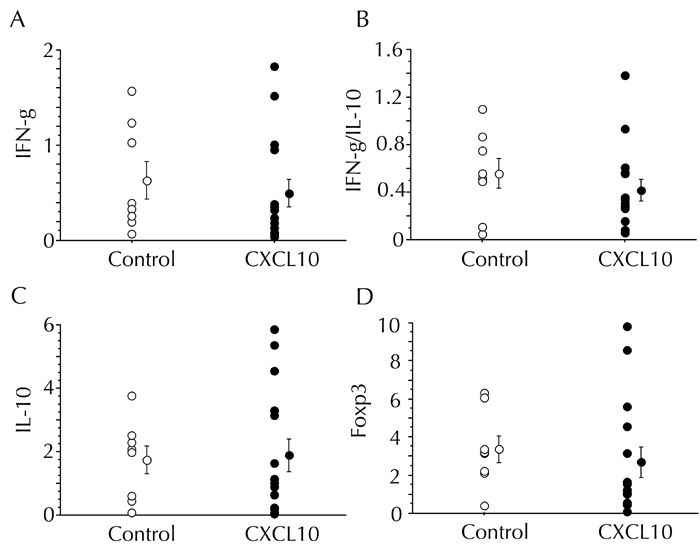
The expression levels of IFN-γ, IL-10, and FoxP3 mRNAs in the spleen were measured by semi-quantitative real-time PCR 8 to 10 weeks after first DNA vaccination. The control DNA group (n = 8) and the CXCL10 DNA group (n=12) are indicated by open and filled circles, respectively. A, C, and D: Data are presented in arbitrary units. B: The ratio of IFN-γ to IL-10 mRNA expression levels is presented.
To investigate whether diabetogenic T cells circulated in the periphery of CXCL10 DNA group mice with hyperglycemia reversal, adoptive transfer of splenocytes from each group into NOD-SCID mice was performed. Essentially, the incidence of diabetes development in recipients of splenocytes from the CXCL10 DNA group with reversal of hyperglycemia, was equivalent to that in recipients of splenocytes from the control DNA group without reversal of hyperglycemia (supplemental Figure A4, see Appendix). This indicated that anti-islet autoimmunity persisted in CXCL10 DNA group mice showing hyperglycemia reversal.
Figure A4. Anti-islet autoimmunity persists in the spleens of mice with reversal of hyperglycemia in the CXCL10 DNA group.
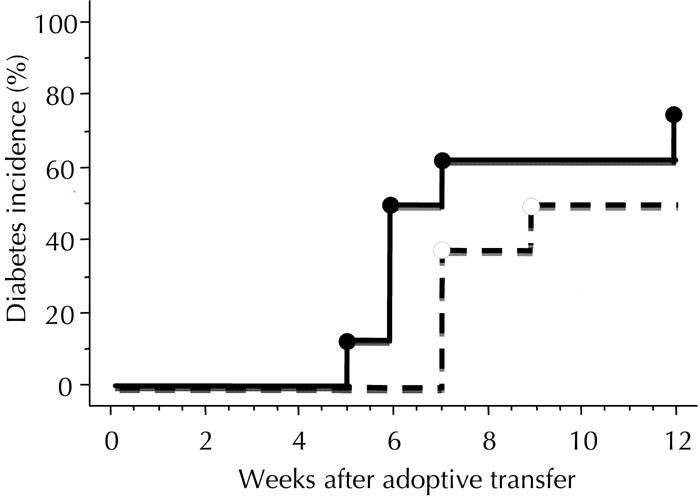
Adoptive transfer of whole splenocytes from the CXCL10 DNA group with reversal of hyperglycemia, or from control DNA group without reversal, was performed in female NOD-SCID mice 8 to 10 weeks after the first DNA vaccination. The incidence of diabetes development was investigated. The control DNA group (n = 8) and the CXCL10 DNA group (n = 8) are indicated by dotted and solid lines, respectively. There was no significant difference in diabetes incidence between the two groups, calculated by log-rank test.
Local pancreatic "hybrid regulatory T cell" frequency was increased in NOD mice with hyperglycemia reversal
To investigate the involvement of Treg cells in CXCL10 DNA group mice with reversal of hyperglycemia, we determined the population of FoxP3+CD4+ cells in NOD mice at 4 weeks after first DNA vaccination. We found that there were no significant differences in splenic, pancreatic lymph node, or local pancreatic Treg cell populations between the CXCL10 DNA group and the control DNA group (Figure 5G).
Figure 5. Increased local hybrid Treg cell population in the pancreas of mice in the CXCL10 DNA group.
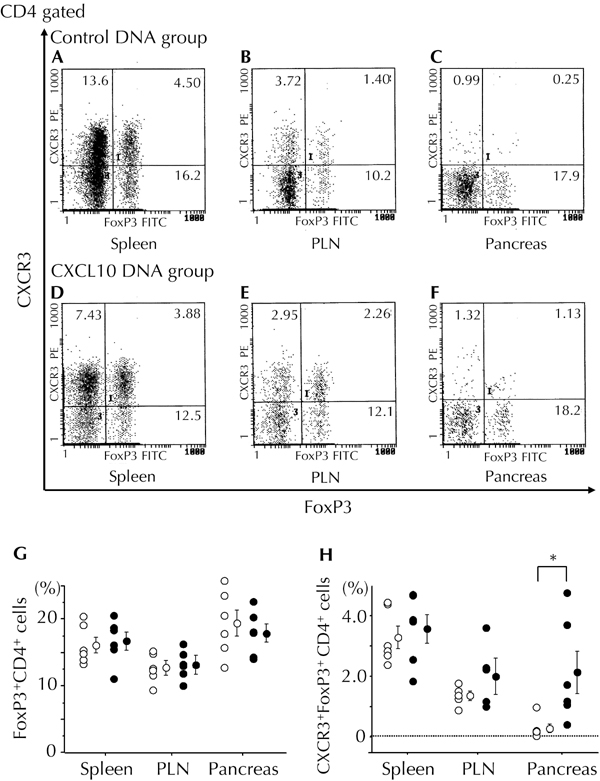
Treg cell-related populations in the spleen (A and D), pancreatic lymph nodes (B and E), and local pancreatic tissue (C and F) were measured by flow cytometry 4 weeks after first DNA vaccination. Representative flow cytometric data from the control DNA group (A-C) and the CXCL10 DNA group (D-F) are shown. G and H: The ratio of FoxP3+CD4+ cells to CD4+ cells (G) and the ratio of CXCR3+FoxP3+CD4+ (hybrid Treg) cells to CD4+ cells (H) are shown. Open circles: control DNA group (n = 6). Filled circles: CXCL10 DNA group (n = 6). * p < 0.05 by Mann-Whitney U test. PLN: pancreatic lymph node.
CXCR3+FoxP3+CD4+ cells have very recently attracted attention as a distinct subset of Treg cells, referred to as “hybrid Treg cells”. They are reported to be induced by CXCR3-FoxP3+CD4+ regulatory T cells (i.e. usual Treg cells) under Th1-type immune-skewed local conditions in vivo [28]. To investigate the involvement of hybrid Treg cells in the process of hyperglycemia reversal in mice from the CXCL10 DNA group, we analyzed this cell population in spleen, pancreatic lymph nodes, and local pancreatic tissues 4 weeks after first DNA vaccination. Intriguingly, the local pancreatic population of hybrid Treg cells was almost 10-fold higher in the CXCL10 DNA group than in the control DNA group, a very significant difference (Figure 5 (C, F, and H)). In contrast, there was no significant difference in the incidence of hybrid Treg cells in spleen, or pancreatic lymph nodes, between the CXCL10 DNA group and the control DNA group (Figure 5 (A, B, D, E, and H)).
Discussion
CXCL10 DNA vaccination, in combination with CFA injection, reversed hyperglycemia in new-onset diabetic NOD mice (Figure 1 (B and D)). In this study, we aimed to elucidate the mechanism underlying the remission of autoimmune diabetes. We focused on the induction of residual β-cell proliferation, and on the regulation of anti-islet autoimmunity.
The onset of overt hyperglycemia in T1D is believed to be due to severe β-cell loss, i.e. residual β-cells are virtually destroyed early in the disease process. However, our previous study in NOD mice revealed that overt hyperglycemia can be caused by β-cell dysfunction rather than severe β-cell loss, at least in the early disease process [29]. Also, we found that severe β-cell destruction can appear late in the autoimmune process [30]. In typical new-onset T1D patients, residual functional β-cell mass is not necessarily eliminated, and may even be larger than was previously thought [31, 32]. Residual β-cell mass should not be neglected as it represents a target for interventions aimed at prevention, and possibly a cure, of T1D.
β-cell dysfunction early in the disease process can be caused by pro-inflammatory cytokines, and chemokines, secreted from islet-infiltrating immune cells or islet cells. It was recently reported that CXCL10 is produced by β-cells stimulated by IFN-γ and TNF-α, and that CXCL10 directly affects β-cells, leading to β-cell dysfunction or apoptosis [13]. In NOD mice, pro-inflammatory cytokines are abundant in insulitis lesions [33], which possibly contributes to the induction of CXCL10 in β-cells. Previously, we showed that CXCL10 is expressed in residual β-cells within insulitis lesions [12], which could contribute to β-cell dysfunction including impaired β-cell proliferation [8, 9, 13]. Moreover, our previous studies demonstrated that CXCL10 neutralization suppressed autoimmune diabetes development via accelerated residual β-cell proliferation [8, 9]. Therefore, we hypothesized that blocking CXCL10 action in insulitis lesions might be critical to achieve remission of autoimmune diabetes.
In the present study, we used the method known as DNA vaccination [34, 35]. CXCL10 DNA vaccination was performed using in vivo gene transfer of rat CXCL10-expressing plasmid DNA. Due to a difference in the amino acid sequence between mouse and rat CXCL10, rat CXCL10 protein produced in the injected muscles was identified as a foreign material by the immune systems of mice. This resulted in the in vivo induction of anti-CXCL10 antibody in mice.
Previously, we showed that anti-CXCL10 antibody produced in the periphery of NOD mice treated with rat CXCL10 DNA vaccination, reacts with rat CXCL10 and mouse CXCL10 [9]. Similarly, the present study detected anti-CXCL10 antibody as anti-mouse antibody (supplementary Figure A1, see Appendix), thus implying the efficacy, and reproducibility, of our CXCL10 vaccination method. It remains to be shown whether the in vivo induced anti-mouse CXCL10 antibody in our system can neutralize mouse CXCL10. Previously, to address this issue, we established a hybridoma producing neutralizing antibodies against mouse CXCL10 via immunization of mice with recombinant rat CXCL10 protein [24, 25]. The results obtained suggested that a portion of the anti-CXCL10 antibodies induced by rat CXCL10 DNA vaccination could neutralize mouse CXCL10. Although, further study is needed to clarify the details of this neutralizing action.
In NOD mice showing reversal of hyperglycemia, residual islet mass was increased (Figure 2 (D and E)), and some residual β-cells were positive for Ki-67 in their nuclei (Figure 4 (D, F, and G)). This means that some portion of the residual β-cells were in a proliferative state. Surprisingly, there was a scattered distribution of α-cells within residual islets (Figure 3D). Generally, α-cells are distributed in the periphery of the insulitis area, and are spared in the natural course of insulitis development in NOD mice (Figure 3F). Therefore, this unexpected observation suggests that residual β-cells proliferated in a non-physiological manner during the process of diabetes remission.
CFA is reported to have potency to eliminate anti-islet autoimmunity by inducing apoptosis of effector T cells [22]. However, this effect is considered to be temporary [22]. In the present study, CFA injection in combination with control DNA vaccination induced hyperglycemia reversal only in a small portion of new-onset diabetic NOD mice (Figure 1A and D). This has already been demonstrated by several investigations using CFA injection alone [22, 23]. Considering that diabetes reversal in control DNA mice observed within a few weeks of first treatment (Figure 1A and D), it seems unlikely that reversal by CFA alone is associated with β-cell proliferation. In fact, proliferating β-cells (Pdx-1 and Ki-67 double positive cells) were not observed in control group mice with hyperglycemia reversal (data not shown). Instead, it seems that hyperglycemia reversal by CFA alone was mainly provided by improvement of β-cell function. Also, it seemed that the anti-diabetic effect of CFA may be dependent on the remaining mass of β-cells, which could have contributed to the amelioration of hyperglycemia. The details of this process are still not clear.
On the other hand, despite the persistence of diabetogenic immune cells (supplemental Figure A4, see Appendix), mice in the CXCL10 DNA group showed efficient reversal of hyperglycemia, and maintained near-normal blood sugar levels. This suggested an immunoregulatory mechanism operating at the local level in pancreatic tissues. In fact, the FoxP3-positive cell population was increased in both insulitis lesions and pancreatic lymph nodes CXCL10 DNA group mice, long after reversal of hyperglycemia. This was in association with elevated expression of TGF-β in pancreatic lymph nodes (supplemental Figure A5, see Appendix). However, contrary to our expectations, most of the Treg cells (FoxP3+CD4+ cells) did not differ between the CXCL10 DNA group and the control DNA group, in the early stage after first DNA vaccination (Figure 5G). Therefore, this study was focused on a distinct subset of Treg cells.
Figure A5. FoxP3-positive cells persisted in both insulitis lesions and pancreatic lymph nodes in the group of CXCL10 DNA-treated mice long after reversal of hyperglycemia.

Immunohistochemical staining of FoxP3 was performed more than 10 weeks after the first DNA vaccination. A and B: Representative insulitis lesion in the control DNA group without reversal of hyperglycemia. C and D: Representative insulitis lesion in the CXCL10 DNA group with reversal of hyperglycemia. E, F, and G: Representative pancreatic lymph node in the control DNA group without reversal of hyperglycemia (E), and in the CXCL10 DNA group with reversal of hyperglycemia (F and G). H: FoxP3 and TGF-β mRNA expression levels in pancreatic lymph nodes were measured in control DNA group mice without reversal of hyperglycemia (open circles, n = 3), and in CXCL10 DNA group mice with reversal of hyperglycemia (filled circles, n = 3) by semi-quantitative real-time PCR. Scale bars: 80 μm.
There are reports describing an association between CFA injection and Treg cell induction in local pancreatic lesions of NOD mice [21, 36]. Interestingly, Qin et al. reported that granzyme B (GrB)-producing CD4 cells were induced in NOD mice injected with CFA, leading to protection against autoimmune diabetes [36]. According to their study, these CD4 cells are a Treg cell subset that can induce apoptosis of diabetogenic effector lymphocytes via a GrB-dependent mechanism. Recently, it was proposed that a portion of GrB-producing Treg cells may be hybrid Treg cells that express CXCR3 on their surfaces. These hybrid Treg cells were derived from CXCR3-negative Treg cells, via a T bet-dependent mechanism in response to Th1-type immunity [28]. In our study, local pancreatic hybrid Treg cell frequency was significantly increased in NOD mice showing hyperglycemia reversal (Figure 5H). Therefore, CFA may play an important role in the induction of hybrid Treg cells in response to Th1-type immunity in insulitis lesions.
It remains to be shown whether there is a direct association between anti-CXCL10 antibody and the increase in pancreatic hybrid Treg cell frequency. However, residual β-cell proliferation triggered by anti-CXCL10 antibody may lead to release of islet-associated antigens, and persistence of Th1-type anti-islet autoimmunity in insulitis lesions, which in turn may contribute to the local expansion of hybrid Treg cells in the pancreas. This possible relation could be an important aspect in autoimmune diabetes, and merits further study. Whether hybrid Treg cells play a direct role in anti-islet autoimmunity remains uncertain. Our CXCR3-deficient NOD mice showed acceleration of diabetes development (unpublished data), suggesting that hybrid Treg cells may be essential to the regulation of anti-islet autoimmunity.
As we reported previously, CXCL10 DNA vaccination suppressed diabetes development in NOD mice through enhanced β-cell proliferation without affecting the degree of insulitis, or the systemic and local cytokine profiles, in the accelerated phase of diabetes development [9]. These findings suggest that anti-mouse CXCL10 antibody, induced by CXCL10 DNA vaccination, has the ability to induce β-cell proliferation. It does not seem to affect the chemotaxis of CXCR3-positive T cells, which are usually considered to be Th1 cells, towards CXCL10-producing tissues. Also, our study demonstrated that CXCL10 DNA vaccination did not affect systemic cytokine profiles (supplemental Figure A3,), or anti-islet autoimmunity (supplemental Figure A4, both in the Appendix). This suggested that CXCL10 DNA vaccination itself did not contribute directly to the induction or local distribution of Treg cells including hybrid Treg cells.
CFA was expected to induce apoptosis of diabetogenic effector T cells and to promote Treg cell activity in NOD mice. However, CXCL10 DNA vaccination plus CFA treatment showed persistence of anti-islet autoimmunity in the spleen (supplemental Figure A4). Consequently, we speculated that Treg cell activity promoted by CFA injection was essential for the reversal of hyperglycemia in CXCL10 DNA group mice, as CXCL10 DNA vaccination without CFA treatment produced only a low frequency of hyperglycemia reversal (Figure 1 (C and D)). Previous reports demonstrated that CFA treatment has a strong potency to suppress the development of autoimmune diabetes in NOD mice via the development of Treg cells, especially in pancreatic lymph nodes [21, 36]. In our study, hybrid Treg cells were significantly increased in the local pancreas, but not in the spleen, of mice from the CXCL10 DNA group (Figure 5H). These findings suggested that CFA-induced anti-islet autoimmune tolerance was performed mainly at the local pancreas, but not in the periphery. This may have contributed to the survival of a portion of diabetogenic effector cells in the spleen of CXCL10 DNA group mice, despite CFA injection. However, this is a matter of speculation. Further studies are required to reveal details.
In conclusion, we have demonstrated that CXCL10 DNA vaccination in combination with CFA injection could provide a strategy for treating autoimmune diabetes. Unfortunately, whilst CFA is an excellent immune regulator of anti-islet autoimmunity in NOD mice, it cannot be applied to humans because of its toxicity. Therefore, in its present form, translation to clinic is not realistic. However, humanized CXCL10 neutralizing antibody is now being tested in an ongoing clinical trial. Possibly, CFA might be replaced by another immune therapy, such as anti-CD3 antibody [37, 38], anti-CD20 antibody [39], or Bacillus Calmette-Guerin vaccination [40]. Anti-CXCL10 antibody in combination with another immune therapy may offer a novel future strategy for T1D. We hope that our findings provide a strong foundation for the development of such a combination therapy.
Disclosures (conflict of interests statement): The authors report no conflict of interests.
Acknowledgments
This work was supported by Keio Gijuku Academic Development Funds (YO), and the Nateglinide Memorial Toyoshima Research and Education Fund (YO).
References
- 1.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis, and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 2.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD, and related strains. Adv Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- 3.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 4.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10, and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184(3):963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimada A, Morimoto J, Kodama K, Suzuki R, Oikawa Y, Funae O, Kasuga A, Saruta T, Narumi S. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care. 2001;24(3):510–515. doi: 10.2337/diacare.24.3.510. [DOI] [PubMed] [Google Scholar]
- 6.Nicoletti F, Conget I, Di Mauro M, Di Marco R, Mazzarino MC, Bendtzen K, Messina A, Gomis R. Serum concentrations of the interferon-gamma-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed type I diabetes mellitus patients, and subjects at risk of developing the disease. Diabetologia. 2002;45(8):1107–1110. doi: 10.1007/s00125-002-0879-5. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki R, Shimada A, Maruyama T, Funae O, Morimoto J, Kodama K, Oikawa Y, Kasuga A, Matsubara K, Saruta T, Narumi S. T-cell function in anti-GAD65(+)diabetes with residual beta-cell function. J Autoimmun. 2003;20(1):83–90. doi: 10.1016/s0896-8411(02)00093-8. [DOI] [PubMed] [Google Scholar]
- 8.Morimoto J, Yoneyama H, Shimada A, Shigihara T, Yamada S, Oikawa Y, Matsushima K, Saruta T, Narumi S. CXC chemokine ligand 10 neutralization suppresses the occurrence of diabetes in nonobese diabetic mice through enhanced beta cell proliferation without affecting insulitis. J Immunol. 2004;173(11):7017–7024. doi: 10.4049/jimmunol.173.11.7017. [DOI] [PubMed] [Google Scholar]
- 9.Shigihara T, Shimada A, Oikawa Y, Yoneyama H, Kanazawa Y, Okubo Y, Matsushima K, Yamato E, Miyazaki J, Kasuga A, Saruta T, Narumi S. CXCL10 DNA vaccination prevents spontaneous diabetes through enhanced beta cell proliferation in NOD mice. J Immunol. 2005;175(12):8401–8408. doi: 10.4049/jimmunol.175.12.8401. [DOI] [PubMed] [Google Scholar]
- 10.Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Holländer GA, Piali L. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med. 2002;8(12):1414–1420. doi: 10.1038/nm1202-792. [DOI] [PubMed] [Google Scholar]
- 11.Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol. 2003;171:6838–6845. doi: 10.4049/jimmunol.171.12.6838. [DOI] [PubMed] [Google Scholar]
- 12.Shigihara T, Oikawa Y, Kanazawa Y, Okubo Y, Narumi S, Saruta T, Shimada A. Significance of serum CXCL10/IP-10 level in type 1 diabetes. J Autoimmun. 2006;26(1):66–71. doi: 10.1016/j.jaut.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 13.Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, Strieter RM, Oberholzer J, King CC, Maedler K. CXCL10 impairs beta cell function, and viability in diabetes through TLR4 signaling. Cell Metab. 2009;9(2):125–139. doi: 10.1016/j.cmet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka S, Nishida Y, Aida K, Maruyama T, Shimada A, Suzuki M, Shimura H, Takizawa S, Takahashi M, Akiyama D. et al. Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: a mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes. 2009;58(10):2285–2291. doi: 10.2337/db09-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhode A, Pauza ME, Barral AM, Rodrigo E, Oldstone MB, von Herrath MG, Christen U. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol. 2005;175(6):3516–3524. doi: 10.4049/jimmunol.175.6.3516. [DOI] [PubMed] [Google Scholar]
- 16.Shimada A, Oikawa Y, Yamada Y, Okubo Y, Narumi S. The role of the CXCL10/CXCR3 system in type 1 diabetes. Rev Diabet Stud. 2009;6:81–84. doi: 10.1900/RDS.2009.6.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadelain MW, Qin HY, Lauzon J, Singh B. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes. 1990;39:583–589. doi: 10.2337/diab.39.5.583. [DOI] [PubMed] [Google Scholar]
- 18.McInerney MF, Pek SB, Thomas DW. Prevention of insulitis, and diabetes onset by treatment with complete Freund's adjuvant in NOD mice. Diabetes. 1991;40:715–725. doi: 10.2337/diab.40.6.715. [DOI] [PubMed] [Google Scholar]
- 19.Harada M, Kishimoto Y, Makino S. Prevention of overt diabetes, and insulitis in NOD mice by a single BCG vaccination. Diabetes Res Clin Pract. 1990;8:85–89. doi: 10.1016/0168-8227(90)90017-n. [DOI] [PubMed] [Google Scholar]
- 20.Ryu S, Kodama S, Ryu K, Schoenfeld DA, Faustman DL. Reversal of established autoimmune diabetes by restoration of endogenous beta cell function. J Clin Invest. 2001;108:63–72. doi: 10.1172/JCI12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian B, Hao J, Zhang Y, Tian L, Yi H, O'Brien TD, Sutherland DE, Hering BJ, Guo Z. Upregulating CD4+CD25+FOXP3+ regulatory T cells in pancreatic lymph nodes in diabetic NOD mice by adjuvant immunotherapy. Transplantation. 2009;87(2):198–206. doi: 10.1097/TP.0b013e3181933261. [DOI] [PubMed] [Google Scholar]
- 22.Kodama S, Davis M, Faustman DL. The therapeutic potential of tumor necrosis factor for autoimmune disease: a mechanistically based hypothesis. Cell Mol Life Sci. 2005;62:1850–1862. doi: 10.1007/s00018-005-5022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okubo Y, Shimada A, Kanazawa Y, Shigihara T, Oikawa Y, Imai T, Miyazaki J, Itoh H. Hyperplastic islets observed in "reversed" NOD mice treated without hematopoietic cells. Diabetes Res Clin Pract. 2008;79(1):18–23. doi: 10.1016/j.diabres.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Tamaru M, Nishioji K, Kobayashi Y, Watanabe Y, Itoh Y, Okanoue T, Murai M, Matsushima K, Narumi S. Liver-infiltrating T lymphocytes are attracted selectively by IFN-inducible protein-10. Cytokine. 2000;12(4):299–308. doi: 10.1006/cyto.1999.0560. [DOI] [PubMed] [Google Scholar]
- 25.Yoneyama H, Narumi S, Zhang Y, Murai M, Baggiolini M, Lanzavecchia A, Ichida T, Asakura H, Matsushima K. Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J Exp Med. 2002;195(10):1257–1266. doi: 10.1084/jem.20011983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oikawa Y, Shimada A, Kasuga A, Morimoto J, Osaki T, Tahara H, Miyazaki T, Tashiro F, Yamato E, Miyazaki J, Saruta T. Systemic administration of IL-18 promotes diabetes development in young nonobese diabetic mice. J Immunol. 2003;171(11):5865–5875. doi: 10.4049/jimmunol.171.11.5865. [DOI] [PubMed] [Google Scholar]
- 27.Faveeuw C, Gagnerault MC, Lepault F. Isolation of leukocytes infiltrating the islets of Langerhans of diabetes-prone mice for flow cytometric analysis. J Immunol Methods. 1995;187:163–169. doi: 10.1016/0022-1759(95)00180-i. [DOI] [PubMed] [Google Scholar]
- 28.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis, and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamada S, Irie J, Shimada A, Kodama K, Morimoto J, Suzuki R, Oikawa Y, Saruta T. Assessment of beta cell mass, and oxidative peritoneal exudate cells in murine type 1 diabetes using adoptive transfer system. Autoimmunity. 2003;36(2):63–70. doi: 10.1080/0891693031000079266. [DOI] [PubMed] [Google Scholar]
- 30.Shimada A, Charlton B, Taylor-Edwards C, Fathman CG. Beta-cell destruction may be a late consequence of the autoimmune process in nonobese diabetic mice. Diabetes. 1996;45:1063–1067. doi: 10.2337/diab.45.8.1063. [DOI] [PubMed] [Google Scholar]
- 31.Pipeleers D, Chintinne M, Denys B, Martens G, Keymeulen B, Gorus F. Restoring a functional beta-cell mass in diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):54–62. doi: 10.1111/j.1463-1326.2008.00941.x. [DOI] [PubMed] [Google Scholar]
- 32.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 33.Yoon JW, Jun HS. Autoimmune destruction of pancreatic beta cells. Am J Ther. 2005;12:580–591. doi: 10.1097/01.mjt.0000178767.67857.63. [DOI] [PubMed] [Google Scholar]
- 34.Shedlock DJ, Weiner DB. DNA vaccination: antigen presentation, and the induction of immunity. J Leukoc Biol. 2000;68:793–806. [PubMed] [Google Scholar]
- 35.Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–870. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- 36.Qin HY, Mukherjee R, Lee-Chan E, Ewen C, Bleackley RC, Singh B. A novel mechanism of regulatory T cell-mediated down-regulation of autoimmunity. Int Immunol. 2006;18:1001–1015. doi: 10.1093/intimm/dxl035. [DOI] [PubMed] [Google Scholar]
- 37.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 38.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S. et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352(25):2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 39.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM. et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen HF, Klingensmith GJ, Jensen P, Simoes E, Hayward A, Chase HP. Effect of Bacillus Calmette-Guerin vaccination on new-onset type 1 diabetes. A randomized clinical study. Diabetes Care. 1999;22:1703–1707. doi: 10.2337/diacare.22.10.1703. [DOI] [PubMed] [Google Scholar]