

Abstract
Nitration of L-type calcium channels during colonic inflammation impairs phosphorylation by the tyrosine kinase, Src kinase. This results in decreased calcium currents. The purpose of this study was to determine the mechanism of the downregulation of Ca2+ currents in colonic inflammation. In whole cell voltage clamp of mouse single smooth muscle cells, long-duration depolarization produced noninactivating calcium currents that were significantly reduced by the Src kinase inhibitor, protein phosphatase 2 (PP2). Unitary Ba2+ currents were recorded upon repolarization from positive potentials in cell-attached patches of smooth muscle and hCav1.2b-transfected cells to assess the properties of the single channels attributed to the noninactivating open state. Repolarization to −40 mV from 0 mV resulted in single-channel events with conductance of ∼23 pS. The ensemble average of the tail currents from 1,000 sweeps was 337 ± 27 fA in control and 218 ± 49 fA (P < 0.05) in inflamed cells. Neither open-probability nor open-time constants were significantly different between control and inflamed cells. However, the transition to the open state measured as channel availability was significantly reduced from 19 ± 3% to 6.4 ± 1%. Similarly, peak ensemble average current and channel availability were significantly reduced by PP2 and treatment with peroxynitrite in control cells. Mutation of COOH-terminal tyrosine residues in hCav1.2b Chinese hamster ovarian cells also decreased peak ensemble average tail currents and availability. The present findings suggest that the transition of Ca2+ channels to the noninactivating open state is Src kinase dependent. Tyrosine nitration prevents Src-mediated transitions, leading to decreased calcium currents.
Keywords: colitis, trinitrobenzene, single-channel current, nitrotyrosine, tyrosine kinase
the biophysical properties of voltage-gated L-type Ca2+ channels (LTCC), like most other ion channels, are modulated by phosphorylation/dephosphorylation. Although the effects of serine/threonine protein kinases on dihydropyridine-sensitive Ca2+ channels have been extensively studied (for review, see Ref. 7), recent studies also suggest a prominent regulation by tyrosine kinases (1, 9, 23). We (2) and others (18, 20) have previously shown that colonic inflammation markedly reduces whole cell calcium currents of single colonic smooth muscle cells, resulting in decreased colonic motility. The decrease in Ca2+ currents does not appear to be attributable to changes in expression of LTCC, as neither mRNA nor protein expression of these channels is altered during dextran sodium sulfate- or trinitrobenzene (TNBS)-induced experimental colitis in rodent models (16, 18, 26). However, a nonreceptor tyrosine kinase c-Src-mediated regulation of LTCC in colonic smooth muscle cells (12) was impaired during inflammation (16, 26). On the basis of whole cell Ca2+ current recordings and isometric tension experiments in colonic tissue segments, the impaired c-Src kinase regulation of LTCC was suggested to be attributable to inflammation-induced nitration of tyrosine residues within the carboxy terminus of the α subunit of Cav1.2b, preventing tyrosine phosphorylation (26). Nitrated LTCCs are significantly increased in colitis, and site-directed mutation of tyrosine residues (Y1837 and Y2134) within the COOH terminus of hCav1.2b support the concept of Src kinase regulation of LTCC and its impairment following tyrosine nitration (17).
The decrease in the amplitude of the whole cell calcium currents, but not expression of LTCCs, in colitis raises the question of how tyrosine phosphorylation regulates LTCC and the mechanism by which this is altered during inflammation. To address this, we analyzed single channel properties of LTCC from isolated mouse smooth muscle cells. Previous studies defined two distinct parameters of LTCC that may be differentially regulated i.e., modal gating and availability. Whole cell ionic current (I) is a function of both the number of functional channels (n) and their individual properties, namely i (single-channel current amplitude), PO (open probability, i.e., the fraction of time spent in the open state during active sweeps), and Factive (availability i.e., fraction of active sweeps per number of test pulses), where I = n × i × Po × Factive (19). The open probability (Po) inherently measures the characteristics of the gating modes of the individual channel, whereas availability (Factive) defines the frequency with which channels can respond to a voltage stimulus. These two single-channel properties may be differentially regulated by phosphorylation. Reversible protein phosphorylation of the calcium channel induces shifts in the modes of the calcium channel, e.g., by calcium/calmodulin-dependent protein kinase II (CAMKII), thereby increasing the open probability. This shift toward longer opening termed “mode 2” is also sensitive to phosphatase 2A (25). Shift to longer open states (mode 2) is one of the major mechanism by which β-adrenoceptor agonists as well as calcium-channel agonists such as BayK8644 enhance calcium currents. On the other hand, the frequency of channel opening induced by depolarization (availability) may be controlled by a type 1-like phosphatase (32). Studies by Nakayama and colleagues (3, 23, 24) suggest that smooth muscle isoform of the LTCC undergoes a second noninactivating open state upon conditioning depolarization that is sensitive to tyrosine kinase inhibitors. This second tyrosine kinase-sensitive open state, termed O2, has the functional consequence of setting the channel in a noninactivating mode that would allow calcium influx promoting tonic contractions. Here we show that colonic inflammation, tyrosine nitration, and inhibition of c-Src kinase decreases the availability of the O2 state of the smooth muscle LTCC. Mutation of the COOH-terminal tyrosine residues in hCav1.2b (Y1837/2134F) also results in decreased availability. We propose that tyrosine phosphorylation of smooth muscle LTCC regulates the frequency of channel openings upon depolarization. Posttranslational modification by nitration during inflammation hinders tyrosine phosphorylation and hence reduces whole cell calcium currents.
MATERIALS AND METHODS
All animal protocols conformed to the guidelines and approval by the Virginia Commonwealth University Institutional Animal Care and Use Committee.
Induction of colitis.
Colonic inflammation was induced by intracolonic instillation of TNBS (2.5% in 50% ethanol) in male Balb/C mice through a 3.5-French catheter carefully inserted into the rectum. The catheter tip was inserted 4 cm proximal to the anal verge. Mice were carefully held in a vertical position (head down) for 60 s after TNBS instillation to ensure distribution of the TNBS within the entire colon. Control animals received the same volume (100 μl) of vehicle solution. The mice were weighed daily and inspected for diarrhea and rectal bleeding for assessing the disease activity index (DAI), as described previously (13). Briefly, scores were assigned ranging from 0 to 4, with 0 being no weight loss, normal stool consistency and no rectal bleeding, and 4 assigned when the weight loss exceeded 20% of initial weight with gross bleeding. All mice used in this study had a DAI of >3.5. At this level, there is a significant increase in IL-1β (26).
Smooth muscle cells isolated from distal colon tissues were used for electrophysiological experiments 3 days post-TNBS. The time adopted (3 days after TNBS challenge) is consistent with several other studies in mice (5, 8) and correlated well with the DAI, as we have described previously (2, 13).
Cell preparation.
Single smooth muscle cells from mouse distal colon were obtained enzymatically as previously described (12). Chinese hamster ovarian (CHO) cells were maintained in DMEM with 10% fetal bovine serum (Gibco, Grand Island, NY) and 100 U/ml penicillin/streptomycin. Cells were plated on six-well plates and transfected with cDNA encoding α1c-subunit of human jejunal Ca2+ channel (hCav1.2b) (kindly provided by Dr. Farrugia, Mayo Clinic, Rochester, MN) or mutated at specific tyrosine residues (Y1837/2134F), together with β2-subunit and green fluorescent protein, using the GeneJammer transfection reagent (Invitrogen, Carlsbad, CA). Twenty-four hours after transfection, cells were dissociated with trypsin/EDTA and plated again on microscope cover glasses, which were used for patch-clamp recording after 48 h.
Electrophysiology.
Cells were placed in recording chambers perfused with bath solution containing (in mM): 150 KCl, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4 with KOH to set the resting potential at zero current potential of the patch pipette. Single-channel recordings were performed under voltage clamp in the cell-attached configuration. Patch micropipettes with resistances of 7 to 15 MΩ for single-channel measurements and 3–5 MΩ for whole cell experiments were pulled from borosilicate glass capillaries on a Flaming-Brown P97 (Sutter Instruments, Novato, CA) electrode puller. The patch pipette contained (in mM): 100 BaCl2, 75 Tris·HCl, 10 glucose, 10 HEPES, pH 7.3, with CsOH. Unitary Ba2+ currents were recorded at room temperature using EPC-10 patch-clamp amplifier (HEKA, Lambrecht/Pfalz, Germany). Pulse generation and data acquisition were performed using Patchmaster 2.15 software (HEKA). Single-channel Ba2+ currents were examined as tail currents upon repolarization to −40 mV for 300 ms at 0.5 Hz from 0 mV (4 s). The tail currents represent deactivation of the open state of the unitary currents. All of the records were acquired at 5 kHz and filtered at 1 kHz. For whole cell experiments, the patch pipettes contained (in mM): 100 L-aspartic acid, 30 cesium chloride, 1 MgCl2, 5 HEPES, 2 adenosine triphosphate (disodium salt), and 5 EGTA, with pH adjusted to 7.2 using cesium hydroxide, and cells were bathed in HEPES-buffered physiological salt solution, which contained (in mM): 135 NaCl, 5.4 KCl, 0.33 NaH2PO4, 5 HEPES, 1 MgCl2, 10 BaCl2, 5 D-glucose, with pH adjusted to 7.4 by NaOH. Patchmaster data files were converted to axon binary file (ABF) formats using ABF Utility program (Synaptosoft, Fort Lee, NJ) and were analyzed by Clampfit 10.1 (Molecular Devices, Sunnyvale, CA) or Igor Pro 6.0 (WaveMetrics, Portland, OR). Inhibition of c-Src kinase was achieved by pretreatment of the cells with 5 μM of protein phosphatase 2 (PP2) (EMD BioScience, Gibbstown, NJ) for 10 min. For nitration experiments, the cells were perfused with 3-morpholinosydnonimine (10 μM) (Tocris Biosciences, Ellisville, MO) and sodium peroxynitrite (150 μM) (Cayman Chemical, Ann Arbor, MI) for 15 min (26) and washed with HEPES-buffered high K+ solution before the unitary Ba2+ currents were recorded.
Site-directed mutagenesis.
Mutations in human α1c (hCav1.2b) were introduced on specific tyrosine residues by PCR using the site-directed mutagenesis kit (Stratagene, La Jolla, CA) as we have previously described (17).
Data analysis and statistics.
In the analysis of multi-channel recordings, the following assumptions (4, 27) were made in this study: 1) the channels are identical and behave independently; 2) the total number of channels remains constant within the duration of the recording; and 3) the process regulating the gating of channels within a sweep is distribution ergodic with respect to the number of sampling points in each conductance level. Linear leak and capacity currents were digitally subtracted using respective blank sweeps. The maximum number of channels we observed in any given patch was 3. The data analysis method followed in this study was similar to that previously reported (19, 29). The availability (fraction of sweeps containing at least one channel opening, i.e., fraction of active sweeps per total number of test pulses), PO (i.e., fractional occupancy of the open state during active sweeps), and the peak ensemble average current were corrected by the number of channels in the patch (n). Peak current is the maximum of the ensemble average current from one single channel. For multiple-channel patches, n was derived from the maximum current amplitude observed, divided by the unitary current amplitude. Peak current was normalized by division through n. The availability was corrected by the square root method: (1-availabilitycorr) is the nth root of (1-availabilityuncorr) (19). Availabilitycorr and availabilityuncorr are corrected and uncorrected availability, respectively. The corrected PO was calculated on the basis of the corrected number of active sweeps, i.e., total open time (in ms) within all sweeps of the ensemble, divided by (300 ms × n × availabilitycorr × number of test pulses). Openings and closures were identified by the half-height criterion. The number of test pulses in each experiment was between 369 and 1,000 sweeps. All of the single-channel records from each patch were analyzed, collected into histograms of open or closed duration (bin width 1 ms) and fit with exponentials using Clampfit software. The bin width for amplitude histograms was 0.05. Data are presented as the means and SE of n observations. Significance was determined by unpaired t-test or one-way ANOVA analysis and Bonferroni posttest at the level of P ≤ 0.05.
RESULTS
Previous analysis of smooth muscle isoform of LTCC have shown that repolarization from long-duration positive potentials result in single-channel currents that reflect deactivation of the noninactivating O2 open state (24). In whole cell voltage-clamp experiments of guinea pig detrusor muscle, the U shape of the steady-state inactivation curve seen with positive conditioning steps is considered to reflect this O2 state that is sensitive to tyrosine kinases (23). We initially examined whether noninactivating currents on conditioning depolarization were sensitive to the Src kinase inhibitor, PP2, in mouse colonic smooth muscle cells. As shown in Fig. 1A, a noninactivating current is present at the end of a 4-s pulse of a 0-mV step from a holding potential of −60 mV that is significantly reduced by PP2 (5 μM). To further determine whether an O2 state sensitive to Src kinase inhibition could be observed in mouse colonic smooth muscle cells, a two-pulse voltage-step protocol was used to determine the voltage dependence of steady-state inactivation. A variable conditioning pulse was applied for 4 s followed by a 5-ms step to −60 mV and a test pulse to +20 mV for 300 ms. During the conditioning pulse, the calcium currents activated and then inactivated to a steady-state level. The subsequent test pulse activated only the remaining Ca2+ channels. For a conventional time- and voltage-dependent Hodgkin-Huxley conductance, the steady-state inactivation curve describes the relative number of available Ca2+ channels as a function of voltage. This sigmoidal relationship was fit by a Boltzman distribution of the form y = 1 + exp [(V + V0.5/k)]−1, where V0.5 is the voltage at which half the channels are inactivated, and k is the slope factor at this voltage. In 10 mM Ba2+, V0.5 was 6 ± 9 mV (n = 3), k = 8 ± 5 in control and 3 ± 9 mV (n = 3), k = 4 ± 1.5 in the presence of PP2, respectively. Figure 1B (inset) shows that PP2 reduces the availability of the channels when positive conditioning depolarizing potentials are applied. A possible interpretation of this is that calcium channels shift into a tyrosine kinase-sensitive O2 mode at positive potentials.
Fig. 1.
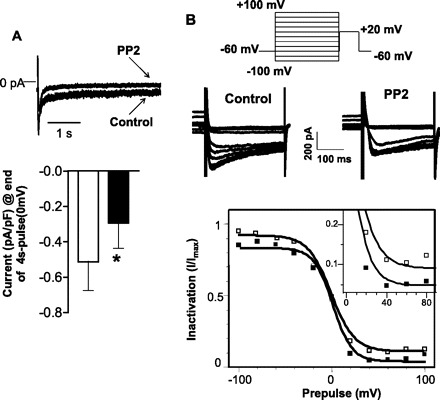
A: Top: raw traces of whole cell patch-clamp recordings before and after inhibition of c-Src kinase by protein phosphatase 2 (PP2) (5 μM) in isolated smooth muscle cells from mouse colon. L-type Ca2+ currents were evoked by depolarizing test pulses from a holding potential of −60 to 0 mV over 4 s. Bottom: comparison of mean amplitude of Ca2+ currents measured at the end of the 4-s depolarizing pulse at 0 mV, normalized to the corresponding cell capacitance. Inhibition of c-Src kinase by PP2 (solid bar) significantly (*P = 0.04) reduced the currents compared with the control (open bar) (data are means ± SE, n = 3; paired t-test). B: voltage dependence of steady-state inactivation of Ca2+ channels was examined over a voltage range of conditioning (−100 to +100 mV) prepulse (10 s) followed by a test pulse of 20 mV. Top: pulse protocol. Middle: representative raw recordings of Ca2+ currents evoked by the test pulse before and after inhibition of c-Src kinase (PP2) (from the last 100 ms of conditioning). Bottom: sigmoidal relationship of the steady-state inactivation to prepulse potential, fit by a Boltzmann distribution. In 10 mM Ba2+, the half maximal inactivation voltage (V0.5) was 6 ± 9 mV with a slope factor (k) of 8 ± 5 mV in control and 3 ± 9 mV, k = 4 ± 1.5 after inhibition of Src kinase (PP2) (n = 3), respectively. Inset: PP2 reduces the availability of channels at positive conditioning potentials (>0 mV).
To determine the single-channel properties and the effects of colonic inflammation on the O2 state, deactivating tail currents were obtained upon repolarization to −40 mV from a holding potential of 0 mV in cell-attached patches. The rationale for this is that the properties of the single deactivating tail currents reflect those of the channels that shift into the O2 mode. Unitary inward currents were recorded over 1,000 sweeps and ensemble average current constructed from control and inflamed colonic smooth muscle cell patches. The peak ensemble current was 337 ± 27 fA (n = 8) in controls and 218 ± 49 fA (n = 5) (P = 0.04) in inflamed cells. Also of note was the lack of significant decay of the current over the 300-ms sweep. Single-channel amplitudes at test potentials from −40 mV to −10 mV were determined from amplitude histograms at each potential and plotted against the test potential to determine channel conductance. Amplitude histograms (Fig. 2B) demonstrated that the mean amplitude of the single-channel events was the same between control (2.4 ± 0.04 pA; n = 6) and inflamed (2.4 ± 0.05 pA; n = 7) cells at −40 mV. The slopes (conductance) of the linear relationships of the means of the corresponding amplitudes and voltage were not different between normal (23 pS, n = 2) and inflamed (27 pS, n = 4) colonic smooth muscle cells. The channels are considered as LTCC because of the typical single-channel conductance of ∼25 pS (31, 33), and unitary currents were absent when nifedipine (1 μM) was included in the patch pipette (data not shown). The decreased peak ensemble average current in the inflamed cells could be due to decreased open probability and/or decreased availability of the channels. A common occurrence that we observed was the presence of a greater number of null sweeps from patches of inflamed colonic smooth muscle cells. An example of this is shown in Fig. 3A of a sweep-by-sweep open probability plot. Over a 1,000 sweeps, significantly greater null sweeps were observed from inflamed cells. The corrected open probability calculated in active sweeps did not differ (P = 0.2) between control (0.29 ± 0.06; n = 6) and inflamed (0.48 ± 0.14; n = 5) cells (Fig. 3B), suggesting that the fraction of time spent in the open state was not affected by inflammation. However, the occurrence of more null sweeps in inflamed cells than normal cells indicated differences in channel availability. Inflammation significantly reduced the channel availability (6.4 ± 1%; n = 5) compared with the control (19 ± 3%; n = 8) cells (P = 0.02) (Fig. 3C).
Fig. 2.
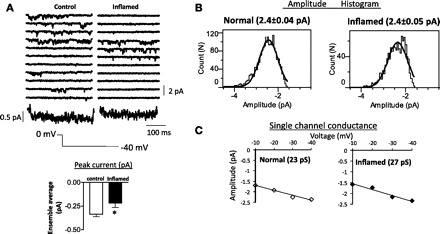
A: typical consecutive recordings showing single L-type Ca2+ channel currents, recorded in smooth muscle cells from control and inflamed mouse colon. Repolarizing command potential of −40 mV was applied for 300 ms every 2 s from a holding potential of 0 mV to examine the single-channel Ba2+ currents representing the deactivation of the open state of L-type Ca2+ channels. The ensemble-averaged currents shown below each set of current traces were calculated from 1,000 sweeps for each membrane patch. Bar graph shows significant decrease in the peak ensemble current during inflammation (means ± SE, n = 5–8, *P = 0.04; unpaired t-test). B: histograms showing amplitude of single L-type Ca2+ channels in control (left) and inflamed (right) at a membrane potential of −40 mV, fitted with a single Gaussian function. C: single-channel conductance was obtained from the current-voltage relationship by plotting unitary current amplitude against membrane potential.
Fig. 3.
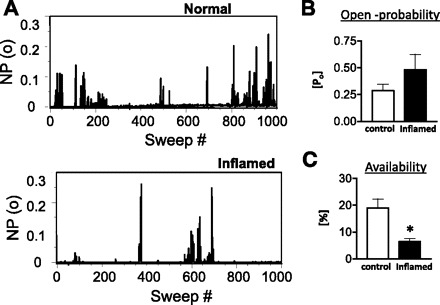
A: an all-sweep open-probability [NP(o)] diary is shown for both normal and inflamed cells. Each bar represents the fraction of time that the channel was open during the 300-ms test pulse. Effects of inflammation on open probability (B) and availability (C) are demonstrated in bar graphs. Inflammation significantly reduced the channel availability (*P = 0.02) without affecting the open probability (P = 0.2). Data are expressed as means ± SE, n = 5–8; unpaired t-test.
Effect of c-Src kinase inhibition and ONOO− treatment on single LTCC properties.
c-Src kinase regulation of LTCC in colonic smooth muscle cells (12) is reported to be impaired during colitis (16, 26) because of tyrosine nitration. To test the possibility that Src kinase affects the transition to the noninactivating mode, single-channel events of deactivating tail currents were analyzed in the presence of PP2. We also tested the effects of peroxynitrite to determine whether nitration of the calcium channel affects channel function in a similar fashion. Amplitude histograms (Fig. 4A) demonstrated that the mean amplitude of the single-channel events after PP2 treatment (2.5 ± 0.1 pA; n = 6) or nitration (2.6 ± 0.1 pA; n = 7) were similar to that of control (2.4 ± 0.04 pA; n = 6 Fig. 2B). However, the mean peak ensemble average currents were significantly (P =0.05) reduced in the presence of PP2 (251 ± 28 fA; n = 6) and after peroxynitrite treatment (212 ± 36 fA; n = 9) compared with that obtained in control (337 ± 27 fA; n = 8 Fig. 2A) (Fig. 4B). The decreased peak ensemble average current after PP2 treatment or nitration could be due to decreased open probability and/or decreased availability of the channels. The corrected open probability calculated in active sweeps in the presence of PP2 was 0.32 ± 0.04 (n = 5) and 0.43 ± 0.1 (n = 8) following nitration (Fig. 4D). These were not significantly different (P > 0.05) from the open probability measured in controls (0.25 ± 0.06; Fig. 3B). However, sweep-by-sweep open probability indicated greater number of null sweeps following nitration and PP2 (Fig. 4C). The corrected channel availability was calculated as 11 ± 1% (n = 6) in the presence of PP2 and 7.3 ± 2% (n = 9) following nitration, which was significantly different from control values (P = 0.006) (Fig. 4E).
Fig. 4.
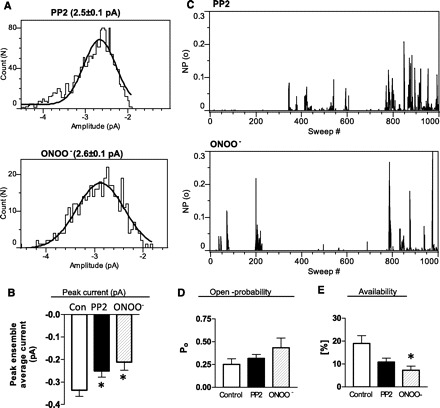
A: histograms showing amplitude of single L-type Ca2+ channels in mouse colon smooth-muscle cells after inhibition of c-Src kinase by PP2 (top) and after tyrosine nitration by 3-morpholinosydnonimine /ONOO− treatment (bottom) at a membrane potential of −40 mV, fitted with a single Gaussian function. Mean amplitudes of events after PP2 or ONOO− were 2.5 and 2.6 pA, respectively. B: bar graph shows significant decrease in the peak ensemble current after c-Src kinase inhibition (PP2) or nitration by ONOO−. C: sweep-by-sweep open-probability plot is shown for both PP2- and ONOO−-treated cells. Each bar represents the fraction of time that the channel was open during the 300-ms test pulse. Effects of c-Src kinase inhibition (PP2) and nitration (ONOO−) of channels on open probability (D) and availability (E) are demonstrated in bar graphs. As seen with inflammation (Fig. 3), ONOO− or PP2 treatment reduced the channel availability without affecting the open probability (data are expressed as means ± SE, n = 6–9, *P <0.05; one-way ANOVA).
Effect of inflammation, Src kinase inhibition and ONOO− treatment on gating kinetics.
The open-time histograms of events in active sweeps from smooth muscle cell patches from all the groups were best fit by double exponential curves reflecting the presence of two open states corresponding to mode 1 and mode 2 gating (Fig. 5A). The time constants of the open-time histograms were as follows: control (τ1 = 1.6 ± 0.2 ms, τ2 = 6.1 ± 0.7 ms; n = 6), inflammation (τ1 = 1.2 ± 0.2 ms, τ2 = 3.9 ± 0.4 ms; n = 6), Src kinase inhibition (τ1 = 1.2 ± 0.2 ms, τ2 = 4.0 ± 0.8 ms; n = 6), and ONOO− treatment (τ1 = 1.2 ± 0.2 ms, τ2 = 4.9 ± 0.8 ms; n = 7). The lack of differences in the open-time constants suggest that modal gating was not affected by inflammation-induced nitration. The closed-time histograms were also best fit by double exponentials reflecting the existence of two different closed states. Both the closed state time constants were not different between any experimental groups (Fig. 5B). The mean closed times were τ1 = 5.1 ± 1 ms, τ2 = 45 ± 8 ms, n = 6 (control); τ1 = 6.4 ± 1 ms, τ2 = 37 ± 3 ms, n = 6 (inflamed); τ1 = 6.7 ± 0.5 ms, τ2 = 48 ± 6 ms, n = 6 (PP2); and τ1 = 7.8 ± 0.7 ms, τ2 = 56 ± 6 ms, n = 7 (ONOO− treatment).
Fig. 5.
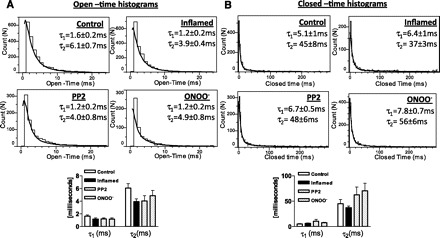
A: open-time histograms were plotted with 1-ms binned duration against the number of events. Open-time distributions were best fit by the sum of two exponentials, and the time constants (τ1 and τ2) from these fits are shown. B: closed-time histograms were plotted with 1-ms binned duration of shut times against the number of events. Closed-time distributions were also best fit by the sum of two exponentials, and the corresponding time constants (τ) from these fits are shown. Bottom: open-time or the closed-time durations were not changed by inflammation, c-Src kinase inhibition, or nitration. Data are expressed as means ± SE, n = 5–7, one-way ANOVA.
Role of tyrosine residues of COOH terminus of hCaV1.2b on single-channel properties.
Previously, we have identified the role of the terminal tyrosine residues in modulating whole cell Ca2+ currents in the human Cav1.2b and demonstrated that the Y2134 and Y1837 are potential residues that, when phosphorylated, bind to the SH2 domain of c-Src kinase, whereas nitration of this residue prevents this interaction (17). In this study, we tested the effect of mutation of the tyrosine residues (Y1837, Y2134) on the availability of the noninactivating state. Figure 6A shows representative raw recordings of single-channel events of the deactivating currents at −40 mV in CHO cells transfected with wild-type, double-mutant (Y1834/2134F) hCaV1.2b, or dominant-negative Src (DN-Src) cotransfected with wild-type hCaV1.2b. The mean peak ensemble average currents were significantly (P = 0.01) different between wild-type (443 ± 60 fA; n = 6), double-mutant (217 ± 44 fA; n = 6), and DN-Src (228 ± 38 fA; n = 9) groups (Fig. 6B). Amplitude histograms (Fig. 6C) demonstrated that the mean amplitude of the single-channel events at −40 mV in Cav1.2b-Y1837–2134 (2.6 ± 0.03 pA; n = 4), in DN-Src (2.4 ± 0.08 pA; n = 9) and wild-type (2.2 ± 0.1 pA; n = 9) were not significantly different. The decreased peak ensemble average current after mutation of tyrosine residues or overexpression of DN-Src could be attributable to decreased open probability and/or decreased availability of the channels. Figure 7A demonstrates that the corrected open probability calculated in active sweeps did not differ (P > 0.05) among these groups: 0.18 ± 0.05 (wild-type, n = 7), 0.32 ± 0.04 (double-mutant, n = 6) and 0.19 ± 0.06 (DN-Src, n = 9). However, along with the occurrence of more null sweeps after mutation of tyrosine residues or overexpression of DN-Src as shown in the sweep by sweep open-probability plot (Fig. 7B), the channel availability was reduced by mutation of tyrosine residues (8 ± 2%, n = 7, P = 0.02) or overexpression of DN-Src (11.6 ± 2%, n = 9; P = 0.06) compared with the wild-type (21 ± 5%; n = 7) group (Fig. 7C). These data suggest that c-Src kinase regulation of LTCC is primarily on regulation of channel availability of the noninactivating mode.
Fig. 6.
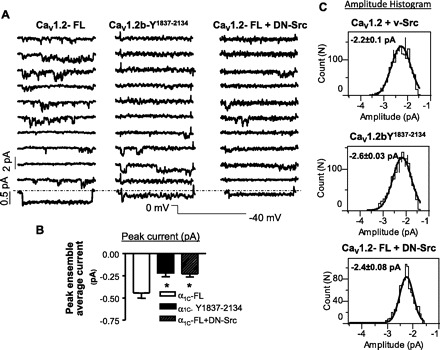
A: representative consecutive traces showing single L-type Ca2+ channel currents, recorded in Chinese hamster ovarian (CHO) cells transfected with CaV1.2-FL (full length) (left), CaV1.2b-Y1837–2134 (middle), or CaV1.2-FL + dominant-negative (DN)-Src (right). Repolarizing command potential of −40 mV was applied for 300 ms every 2 s from a holding potential of 0 mV to examine the single-channel Ba2+ currents representing the deactivation of the open state of L-type Ca2+ channels. The ensemble-averaged currents shown below each set of current traces were calculated from 1,000 sweeps for each membrane patch. B: bar graph shows significant decrease in the peak ensemble currents in CaV1.2b- Y1837–2134 and CaV1.2-FL + DN-Src groups compared with CaV1.2-FL group (means ± SE, n = 5–9, *P = 0.01; one-way ANOVA). C: histograms showing amplitude of single L-type Ca2+ channels in CHO cells transfected with CaV1.2-FL (top), CaV1.2b- Y1837–2134 (middle), or CaV1.2-FL +DN-Src (bottom) at a membrane potential of −40 mV, fitted with a single Gaussian function. The mean amplitudes of the single-channel events were not significantly different between the groups.
Fig. 7.
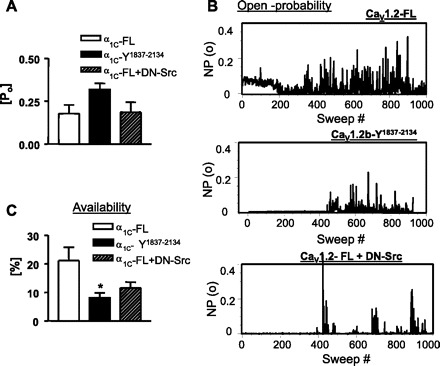
A: all-sweep open-probability plot is shown for CHO cells transfected with CaV1.2-FL (top), CaV1.2b- Y1837–2134 (middle), or CaV1.2-FL +DN-Src (bottom). Each bar represents the fraction of time that the channel was open during the 300-ms test pulse. Effects of mutation of tyrosine residues or cotransfection with DN-Src on open probability (B) and availability (C) are demonstrated in bar graphs. Channel availability was significantly reduced by mutation of tyrosine residues (*P = 0.02) or overexpression of DN-Src (P = 0.06) without affecting the open probability. Data are expressed as means ± SE, n = 5–9; one-way ANOVA.
DISCUSSION
In colonic inflammation, the downregulation of calcium currents has been attributed to either decrease in protein expression (20) or altered regulation of the channels (16). By examining the single-channel properties, the present data suggest that, in TNBS-induced colonic inflammation in mouse, results in altered biophysical properties of the calcium channel such that decrease in the transitions to a second open state may form the basis for reduced amplitude of the ionic current. Studies at the single-channel level discount changes in protein expression during inflammation, consistent with previous findings in rodent models of colonic inflammation (16, 18).
The reduced ensemble average current in inflamed cells confirm results of whole cell voltage-clamp experiments in several ways. First, the peak currents were significantly reduced in the presence of the Src kinase inhibitor and in dominant-negative c-Src-expressing cells. Secondly, tyrosine nitration by peroxynitrite decreased peak amplitude of ensemble average currents similar to previous findings in whole cell voltage-clamp experiments in mouse colon (26). Third, mutations of tyrosine residues 1837 and 2134 within the carboxy terminus resulted in reduced currents. We have previously demonstrated that phosphorylation of these residues are necessary for c-Src association via its SH2 domains (17).
Previous studies in Cav1.2b-transfected cells and in guinea pig detrusor smooth muscle cells suggest that the smooth muscle isoform of the calcium channel undergoes transition from a normal O1 to a second open state, O2, upon positive conditioning depolarizations (3, 24). In this state calcium channels do not, or only very slowly, inactivate during depolarization and deactivate slowly upon repolarization. The conversion to O2 was shown to be sensitive to tyrosine kinase inhibitors in whole cell voltage-clamp experiments (23). In a minimum kinetic scheme (Fig. 8), similar to the computer simulations by Nakayma et al. (23) profiling the U-shaped inactivation at depolarized potentials, the conversion from O1 to O2 may be prevented by tyrosine nitration. This model does not incorporate Ca2+-dependent inactivation process because this mechanism may not be operative when using Ba2+ as a charge carrier. The kinetic scheme would need to be modified to include Ca2+-dependent inactivation. We recorded similar deactivating tail currents when using high (120 mM) Ca2+ as charge carrier (data not shown), suggesting that Ca2+-dependent inactivation does not prevent transition to the O2 state. However, further experiments will be necessary to address the changes in rate constants incorporating inactivation processes. Deactivating tail currents reflect the properties of the open state, and the finding that availability is decreased by inflammation indicates that the transition to the O2 state is also altered by inflammation. It is noteworthy that this transition was observed at modest depolarizations of 0 mV, a membrane potential that is achieved in vivo in physiological settings.
Fig. 8.
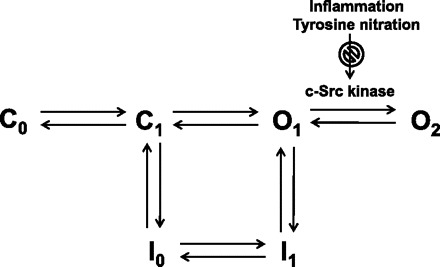
A minimum kinetic scheme illustrating two closed (C1 and C2) and two open states (O1 and O2) that are sequentially linked. Inactivation proceeds from either closed or O1 state, but not from O2. Transition from O1 to O2 is Src kinase dependent and may be downregulated by nitration during colonic inflammation.
Phosphorylation regulates ion channels in several ways. The calcium channels respond to depolarizations by an increase in the frequency of openings (availability) and by shifting of the gating modes toward higher open probability (modes 1 and 2). Agonists such as β-adrenoceptors in the heart affect both the availability and bias to mode 2 gating. There are a large number of studies that establish a role for phosphorylation by protein kinase A in β-adrenergic signaling, which enhances calcium currents by increasing both the availability and open probability of single channels (7). Similarly, facilitation of Ca2+ channels in cardiac myocytes, a mechanism of positive feedback regulation, requires phosphorylation by CAMKII (10) that induces Ca2+ channels to enter mode 2 gating. Our finding that open time constants or open probability within active sweeps were not altered by either inflammation or Src kinase inhibition suggests that modal shift of the open state is not affected; however, the increased null sweeps suggest that tyrosine phosphorylation induces changes in channel availability and that downregulation of Src kinase phosphorylation is the primary mechanism by which current amplitude is altered. Consistent with this, cotransfection with dominant negative c-Src kinase or nitration of tyrosine residues decreased availability.
Tyrosine nitration is one of the hallmark features of inflammatory bowel disease (IBD). Nitric oxide is an important factor in the pathophysiology of IBD, as increased NO production and iNOS activity have been observed in both clinical and experimental IBD (6, 21, 22, 30). Recruitment of mononuclear cells and granulocytes in the mucosa, together with enhanced levels of myeloperoxidase, results in the production of peroxynitrite, a potent oxidant, that incorporates nitro groups on tyrosine residues, resulting primarily in 3-nitrotyrosine. We have previously shown that enhanced nitrotyrosine incorporation of calcium channels from TNBS-treated mouse colon impairs tyrosine phosphorylation and reduces calcium currents and muscle contraction (26). Calcium channels may also be substrates for denitration (15).
Evidence of altered Src kinase regulation of the LTCC has also been noted in patients with atrial fibrillation. In these patients, the density of the α- or β-subunits of the Ca2+ channels as measured by Western blots, and radiolabeled bindings were not decreased (14, 28); however, impaired Src kinase regulations together with an increase in phosphatase activities was suggested to lead to dysregulation of the calcium channel in chronic atrial fibrillation (11). Thus altered tyrosine kinase phosphorylation may be an important universal process contributing to remodeling of LTCC in disease states. It is noteworthy that Src kinase activity was not altered with colonic inflammation (26). Future studies utilizing inside-out patch recordings should provide further insight on the balancing between the kinase and phosphate(s) in the regulation of the smooth-muscle calcium channels.
In summary, the present study provides a mechanistic basis for the decreased calcium currents in colonic inflammation by establishing an alteration in the kinetics of single calcium channel attributable to tyrosine nitration of the calcium channel.
GRANTS
This study was supported by NIH DK046367.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
ACKNOWLEDGMENTS
We thank Dr. Clive Baumgarten for helpful suggestions.
REFERENCES
- 1. Akbarali HI. Signal-transduction pathways that regulate smooth muscle function. II. Receptor-ion channel coupling mechanisms in gastrointestinal smooth muscle. Am J Physiol Gastrointest Liver Physiol 288: G598–G602, 2005 [DOI] [PubMed] [Google Scholar]
- 2. Akbarali HI, Pothoulakis C, Castagliuolo I. Altered ion channel activity in murine colonic smooth muscle myocytes in an experimental colitis model. Biochem Biophys Res Commun 275: 637–642, 2000 [DOI] [PubMed] [Google Scholar]
- 3. Aoyama M, Murakami M, Iwashita T, Ito Y, Yamaki K, Nakayama S. Slow deactivation and U-shaped inactivation properties in cloned Cav1.2b channels in Chinese hamster ovary cells. Biophys J 84: 709–724, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baumgartner W, Hohenthanner K, Hofer GF, Groschner K, Romanin C. Estimating the number of channels in patch-clamp recordings: application to kinetic analysis of multichannel data from voltage-operated channels. Biophys J 72: 1143–1152, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beck PL, Li Y, Wong J, Chen CW, Keenan CM, Sharkey KA, McCafferty DM. Inducible nitric oxide synthase from bone marrow-derived cells plays a critical role in regulating colonic inflammation. Gastroenterology 132: 1778–1790, 2007 [DOI] [PubMed] [Google Scholar]
- 6. Boughton-Smith NK, Evans SM, Hawkey CJ, Cole AT, Balsitis M, Whittle BJ, Moncada S. Nitric oxide synthase activity in ulcerative colitis and Crohn's disease. Lancet 342: 338–340, 1993 [DOI] [PubMed] [Google Scholar]
- 7. Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev 89: 411–452, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Daniel C, Radeke HH, Sartory NA, Zahn N, Zuegel U, Steinmeyer A, Stein J. The new low calcemic vitamin D analog 22-ene-25-oxa-vitamin D prominently ameliorates T helper cell type 1-mediated colitis in mice. J Pharmacol Exp Ther 319: 622–631, 2006 [DOI] [PubMed] [Google Scholar]
- 9. Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, Wilson E. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 281: H1835–H1862, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of l-type calcium channels. Nat Cell Biol 2: 173–177, 2000 [DOI] [PubMed] [Google Scholar]
- 11. Greiser M, Halaszovich CR, Frechen D, Boknik P, Ravens U, Dobrev D, Luckhoff A, Schotten U. Pharmacological evidence for altered Src kinase regulation of I (Ca,L) in patients with chronic atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol 375: 383–392, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Hu XQ, Singh N, Mukhopadhyay D, Akbarali HI. Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J Biol Chem 273: 5337–5342, 1998 [DOI] [PubMed] [Google Scholar]
- 13. Jin X, Malykhina AP, Lupu F, Akbarali HI. Altered gene expression and increased bursting activity of colonic smooth muscle ATP-sensitive K+ channels in experimental colitis. Am J Physiol Gastrointest Liver Physiol 287: G274–G285, 2004 [DOI] [PubMed] [Google Scholar]
- 14. Kamp TJ, Foell JD. l-type Ca2+ channels in atrial fibrillation: wallflowers or a vanishing act. J Mol Cell Cardiol 35: 427–431, 2003 [DOI] [PubMed] [Google Scholar]
- 15. Kang M, Akbarali HI. Denitration of l-type calcium channel. FEBS Lett 582: 3033–3036, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang M, Morsy N, Jin X, Lupu F, Akbarali HI. Protein and gene expression of Ca2+ channel isoforms in murine colon: effect of inflammation. Pflügers Arch 449: 288–297, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Kang M, Ross GR, Akbarali HI. COOH-terminal association of human smooth muscle calcium channel Ca(v)1.2b with Src kinase protein binding domains: effect of nitrotyrosylation. Am J Physiol Cell Physiol 293: C1983–C1990, 2007 [DOI] [PubMed] [Google Scholar]
- 18. Kinoshita K, Sato K, Hori M, Ozaki H, Karaki H. Decrease in activity of smooth muscle l-type Ca2+ channels and its reversal by NF-kappaB inhibitors in Crohn's colitis model. Am J Physiol Gastrointest Liver Physiol 285: G483–G493, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Klein G, Drexler H, Schroder F. Protein kinase G reverses all isoproterenol induced changes of cardiac single l-type calcium channel gating. Cardiovasc Res 48: 367–374, 2000 [DOI] [PubMed] [Google Scholar]
- 20. Liu X, Rusch NJ, Striessnig J, Sarna SK. Down-regulation of l-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology 120: 480–489, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Miampamba M, Sharkey KA. Temporal distribution of neuronal and inducible nitric oxide synthase and nitrotyrosine during colitis in rats. Neurogastroenterol Motil 11: 193–206, 1999 [DOI] [PubMed] [Google Scholar]
- 22. Middleton SJ, Shorthouse M, Hunter JO. Increased nitric oxide synthesis in ulcerative colitis. Lancet 341: 465–466, 1993 [DOI] [PubMed] [Google Scholar]
- 23. Nakayama S, Ito Y, Sato S, Kamijo A, Liu HN, Kajioka S. Tyrosine kinase inhibitors and ATP modulate the conversion of smooth muscle l-type Ca2+ channels toward a second open state. FASEB J 20: 1492–1494, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Nakayama S, Klugbauer N, Kabeya Y, Smith LM, Hofmann F, Kuzuya M. The alpha 1-subunit of smooth muscle Ca(2+) channel preserves multiple open states induced by depolarization. J Physiol 526: 47–56, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ono K, Fozzard HA. Two phosphatase sites on the Ca2+ channel affecting different kinetic functions. J Physiol 470: 73–84, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ross GR, Kang M, Shirwany N, Malykhina AP, Drozd M, Akbarali HI. Nitrotyrosylation of Ca2+ channels prevents c-Src kinase regulation of colonic smooth muscle contractility in experimental colitis. J Pharmacol Exp Ther 322: 948–956, 2007 [DOI] [PubMed] [Google Scholar]
- 27. Schmid R, Seydl K, Baumgartner W, Groschner K, Romanin C. Trypsin increases availability and open probability of cardiac l-type Ca2+ channels without affecting inactivation induced by Ca2+. Biophys J 69: 1847–1857, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schotten U, Haase H, Frechen D, Greiser M, Stellbrink C, Vazquez-Jimenez JF, Morano I, Allessie MA, Hanrath P. The l-type Ca2+-channel subunits alpha1C and beta2 are not downregulated in atrial myocardium of patients with chronic atrial fibrillation. J Mol Cell Cardiol 35: 437–443, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Schroder F, Klein G, Frank T, Bastein M, Indris S, Karck M, Drexler H, Wollert KC. Src family tyrosine kinases inhibit single l-type: Ca2+ channel activity in human atrial myocytes. J Mol Cell Cardiol 37: 735–745, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology 111: 871–885, 1996 [DOI] [PubMed] [Google Scholar]
- 31. Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci 11: 431–438, 1988 [DOI] [PubMed] [Google Scholar]
- 32. Wiechen K, Yue DT, Herzig S. Two distinct functional effects of protein phosphatase inhibitors on guinea-pig cardiac l-type Ca2+ channels. J Physiol 484: 583–592, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshino M, Someya T, Nishio A, Yabu H. Whole-cell and unitary Ca channel currents in mammalian intestinal smooth muscle cells: evidence for the existence of two types of Ca channels. Pflügers Arch 411: 229–231, 1988 [DOI] [PubMed] [Google Scholar]