

Abstract
The wealth of kinetic and structural information makes inorganic pyrophosphatases (PPases) a good model system to study the details of enzymatic phosphoryl transfer. The enzyme accelerates metal-complexed phosphoryl transfer 1010-fold: but how? Our structures of the yeast PPase product complex at 1.15 Å and fluoride-inhibited complex at 1.9 Å visualize the active site in three different states: substrate-bound, immediate product bound, and relaxed product bound. These span the steps around chemical catalysis and provide strong evidence that a water molecule (Onu) directly attacks PPi with a pKa vastly lowered by coordination to two metal ions and D117. They also suggest that a low-barrier hydrogen bond (LBHB) forms between D117 and Onu, in part because of steric crowding by W100 and N116. Direct visualization of the double bonds on the phosphates appears possible. The flexible side chains at the top of the active site absorb the motion involved in the reaction, which may help accelerate catalysis. Relaxation of the product allows a new nucleophile to be generated and creates symmetry in the elementary catalytic steps on the enzyme. We are thus moving closer to understanding phosphoryl transfer in PPases at the quantum mechanical level. Ultra-high resolution structures can thus tease out overlapping complexes and so are as relevant to discussion of enzyme mechanism as structures produced by time-resolved crystallography.
Inorganic pyrophosphatases (PPases) catalyze one of the oldest and most common reactions in cells and provide a good system for detailed analysis of enzymatic phosphoryl transfer from polyphosphate to water. The kinetics are well characterized (1, 2) and high-resolution structures are available along the reaction pathway (3). The enzyme accelerates hydrolysis of metal complexed inorganic pyrophosphate by 1010 compared with the uncatalyzed reaction (1)—but for PPases, as for enzymes in general, the exact source of rate enhancement remains unclear.
The original model of catalysis suggested that the mechanism proceeded in four steps with all steps after substrate binding partially rate-determining (1). The nucleophile, which is generated by coordinating a water molecule (Onu) to two metal ions and which is further strengthened by donating a hydrogen bond to D117, is one key to pyrophosphate hydrolysis in PPases. In addition, the substrate pKa is adjusted by extensive coordination to charged atoms (positively charged side chains and M2+; ref. 3).
Our most recent solution studies (P. Halonen, unpublished data; refs. 2 and 4) indicate that the enzyme-substrate complex (EM2:MPPi or EM2:M2PPi) undergoes isomerization during the catalytic cycle (Scheme S1; ref. 4). In addition, fluoride inhibition studies (4) are consistent with structural studies (3, 5) suggesting that the nucleophile is coordinated to D117.
Scheme 1.

We earlier determined the structure of complexes A and E (Scheme S1), but now have direct structural information on the mechanistically key intermediates C and D, as well as much higher resolution information on E. PPase is specifically inhibited by fluoride ion, which blocks the C to D conversion (4). We report the 1.9-Å crystal structure of the fluoride-inhibited substrate complex and the 1.15-Å structure of the product complex of yeast PPase. Based on these structures we suggest that low barrier hydrogen bonds (LBHBs), proton tunneling, and active site preorganization are important in rate enhancement in at least this class of metal-dependent phosphoryl transferases.
Materials and Methods
Crystallization and Data Collection.
Protein purification and crystallization (3) and flash-freezing (5) for PPase:Pi2 were as described. The 20–1.5-Å data were collected at λ = 0.891 Å and data from 2.5–1.15 Å from a second crystal at λ = 0.901 Å at Deutsches Elektronen Synchrotron (DESY). Frames were integrated and scaled in hkl (6). PPase:FPPi crystals were grown in 19 μl sitting drops containing 30 mM Mes, pH 6.0, 1 mM MnCl2, 5 mM NaF, 1 mM NaPPi, and 16% 2-methyl-2,4-pentanediol (MPD) and equilibrated against 18 or 20% MPD. The final crystals were grown by macro seeding into 44-μl drops. The crystals were mounted with the above solution plus 29% MPD and frozen in liquid nitrogen. Data to 1.9 Å resolution were collected from a single crystal at λ = 1.0039 Å, T = 100 K at DESY and integrated as above.
Refinement.
The initial phasing of the PPase:Pi2 structure was from 1wgj. B factors were set to 15 and the noncrystallographic symmetry (NCS) restrained with a weight of 500. The packing of the two subunits was adjusted by rigid body refinement (7). At 3.0 Å resolution, 6 Mn atoms were added, the structure refined (7) and manually rebuilt (8). Peaks >4σ were analyzed and waters, phosphates, and the missing Mn2+ added. At 2.3 Å (Rw = 23.4%), grouped B refinement was started and at 2.25-Å NCS weights lowered to 300. Individual temperature factor (B-factor) refinement followed by one round of simulated annealing from 1,000 K gave Rf/Rw of 23.4/20.2% for 8.0–1.7-Å data.
Refinement then continued with the conjugate gradient least squares option in shelx-97 (9) with default effective standard deviations of 0.015, 0.1, 0.01, and 0.025 to restrain protein chemistry. The refinement was done against all I by using the SWAT bulk solvent correction, but the Rf set of reflections was as above, with Rf/Rw on F of 28.9/21.3%. Refinement to 1.5 Å resolution and addition of restrained anisotropic B-factors lowered Rw to 17.6%. Thirty new waters were added per cycle and those with Uij > 0.8 were rejected. At 1.15 Å resolution and Rf/Rw = 18.3/15.3%, anisotropic restraints were released for Mn2+, the structure was refined against all data including the Rf set of reflections, and floating hydrogens were added to all protein residues except H30 Nδ1 in subunits A and B and H87 Nδ1 in A because they cannot be protonated (data not shown). In the last rounds, the BUMP restraint was removed to allow D117 to adjust optimally to the electron density map. The refinement converged to r = 11.01% (Table 1).
Table 1.
X-ray statistics
| Data collection | PPase:Pi2 | PPase:FPPi |
|---|---|---|
| Rmerge (I) (last shell) | 7.5% (36.3%) | 8.1% (24.7%) |
| Resolution | 8.0–1.15 Å | 8.0–1.9 Å |
| Independent observations (multiplicity) | 212 495 (3.6) | 53 366 (4.5) |
| Completeness (last shell) | 85.4% (75.5%) | 97.6% (92.6%) |
| I/σI (last shell) | 11.95 (3.0) | 12.58 (4.9) |
| Refinement | ||
| Reflections |F| > 2 σ (F) | 87 817 | |
| Reflections I (I > 4σ I) | 205 805 (154 122) | |
| Rwork/Rfree (F > 4σ F) | 13.6% (11.0%) | 15.5/18.2% |
| Non H atoms | 5769 | 5261 |
| Multiple conformations | 202 (3.5%) | 106 (2.0%) |
| Waters | 1021 | 648 |
| Protein B-factors | 13.8 Å2 | 9.4 Å2 |
| Phosphate B-factors | 8.8 Å2 | 9.0 Å2 |
| Mn2+ B-factors | 8.7 Å2 | 7.8 Å2 |
| rmsd from target values | ||
| Bond lengths | 0.011 Å | 0.007 Å |
| Bond angles | 0.026 Å | 1.5° |
| Rigid-bond ADPs | 0.05 Å2 | |
| Approximate isotropic ADPs | 0.108 Å2 |
Rmerge = Σ | Ih − 〈I〉 |/Σ Ih, where Ih is the measured and 〈I〉 the average intensity of reflection hkl. Rfree (7) and Rwork are conventional crystallographic R-factors (R = Σ ∥ Fobs | − | Fcalc ∥/Σ | Fobs |), where Fobs is the observed and Fcalc the calculated structure factor of reflection hkl. B-factors were analyzed with moleman (G. Kleywegt, unpublished program) and values for rms deviations are from shelxpro for PPase:Pi2 and CNS for PPase:FPPi. ADP, anisotropic displacement parameter.
The PPase:FPPi structure was refined much as above with NCS restraints (7, 10), starting from the 1.5-Å PPase:Pi2 structure, with B-factors set to 15, and including Mn2+ and the 100 waters with the lowest B-factors. Pyrophosphate was added at 3.5 Å; the two subunits were refined and, at 2.4 Å, water molecules were added. The resolution was increased to 1.90 Å. At this point, the water between M1 and M2 was changed to F−, waters added, individual B-factor refinement started, and NCS restraints released. An alternate P1 Pi could be added to the B active site and refined so that the occupancy (Pi plus PPi) at the P1 site was 100%. Finally, a B:P2 Pi was added at the same occupancy as B:P1 Pi and Na+ was added to A-PPase:FPPi. The final Rf/Rw are 18.2/15.5% (Table 1).
Results
Refinement and Selection of Structures for Comparison.
The product complex (3) of yeast PPase (PPase:Pi2) has been refined at 1.15 Å resolution (R = 11.0%), and the fluoride-inhibited substrate complex (PPase:FPPi) at 1.9 Å resolution (Rf/Rw = 18.2/15.5%) (Table 1). The names reflect the differences in active site contents: each additionally contains four Mn2+ (M1–M4). M1 and M2 bind before substrate, and M3 and M4 as part of substrate (3). As before (3), the two product phosphates are P1 (leaving group) and P2 (electrophile) and the same names are used for the two phosphorous atoms on pyrophosphate.
Both structures (PPase:Pi2 and PPase:FPPi) have two independent subunits per asymmetric unit (A and B—e.g., A-PPase:Pi2 and B-PPase:Pi2) and both are very similar to the PPase:Pi2 complex collected at −15°C (1wgj; ref. 3). As previously noted (5), the two independent subunits A and B from a single crystal structure superimpose less well on each other than do A subunits between different structures (e.g., 0.16 Å between A-PPase:Pi2 and A-PPase:FPPi vs. 0.36 Å between A- and B-PPase:Pi2). This is due to crystal contacts to the subunit B active site, which confound comparisons between A and B subunits. We therefore focus mainly on the A-subunit active sites.
The key differences among the A-subunit structures pertain to the precise chemical mechanism of the enzyme and are principally the presence of multiple conformations in the active site. In A-PPase:Pi2, the P1 and P2 phosphates, M3 and E58, N116, and D117 have two conformations, A-PPase:Pi2up and A-PPase:Pi2down, named for the orientation of P2 in the active site (Fig. 1). The A-PPase:Pi2down conformation corresponds to our postulated “immediate hydrolysis product” (3), but was not visible in 1wgj because the largest change, 1.74 Å, is not resolvable at 2.0 Å. We have thus directly visualized the stable intermediates around the transition state. Unusual interactions around D117 suggest a key role for this residue in catalysis.
Figure 1.

Three superimposed A-active-site structures. The PPase:FPPi structure is in gray, the PPase:Pi2down structure is in yellow and the PPase:Pi2up structure is in orange, showing three separate conformations for the PPi/Pi atoms. All H2O except the nucleophile Onu are omitted for clarity. The figure shows the similarities between substrate and product binding; the P2up conformation mimics substrate binding, but hydrolyzed P1 moves away. The inline direction from F or Onu toward P2 is always preserved.
The Structure of the A-PPase:FPPi Complex Active Site.
In this structure, the pyrophosphate seems to be partially degraded. In B-PPase:FPPi, the occupancy of the P1 and P2 sites is best modeled as 80% PPi and 20% Pi, but A-PPase:FPPi is best modeled as 100% PPi. The active site shows evidence for PPi and an “associated electron density peak” between M1 and M2 (see Figs. 5 and 6, which are published as supplemental data on the PNAS web site, www.pnas.org). We identify the peak as the tight-binding fluoride ion that inactivates the enzyme (11) for three reasons. First, the major conformation of D117 Oδ2 is 0.67 Å further away from this peak than in A-PPase:Pi2 (Fig. 1) because of the unexpected presence of an Na+ between the F− and Oδ2. The Na+ explains the (Fo–Fc) electron density peak within 2.6 Å of F− and four other oxygen ligands (Fig. 2a). This would be expected if F− replaced H2O, with concomitant loss of hydrogen bond donors and increase in negative charge. Second, fluoride inhibition data (12) suggest that F− binds at this position, and, third, in the D117E variant (5) a carboxylate oxygen of E117 also binds here.
Figure 2.

Geometry in A-PPase active sites. Coordination in: (a) F-PPi; (b) P1; (c) P2down; (d) P2up. Key distances discussed in the text are shown. Atoms are colored by B-factors: dark blue, < 5Å2; light blue, 5–8 Å2; purple, 8–11 Å2; magenta, 11–15 Å2; red, >15 Å2. Hydrogen bonds are color-coded by length, with red for potential LBHBs. P2(O2) in c and d is believed to remain coordinated to M1 and M4; the labeling of the other oxygens in P2down and P2up is arbitrary. Metal coordination is shown in green and a gray line shows a distance, but without coordination or H-bonding.
In both subunits, the D117 side chain has two conformations, but the D117down conformation is less pronounced than in the A:PPase:Pi2 active site (see below): the difference between Oδ1up and Oδ1down is only 1.23 Å. (In B-PPase:FPPi, both D117 and N116 have two conformations, albeit at low occupancy.) Our ultra-high resolution PPase:Pi2 structure allowed us to identify the alternate conformations in PPase:FPPi that would otherwise have been uninterpretable.
Very few other changes are seen in the active site compared with the our original product complex structure (3). The backbone atoms of the residues coordinating the P1 (leaving group) side of the substrate hardly move at all, but there are small side chain adjustments. Mn2P2O7, one of the forms of substrate, has 18 lone pairs, of which six are coordinated to metal and nine form hydrogen bonds, six to protein and three to waters (Fig. 2a). The tightest interactions are to Y93 Oη (2.51 Å), Y192 Oη, and R78 Nη1 (Fig. 2a). The short hydrogen bond to Y93 may provide some of the “steric crowding” that is adduced as part of the mechanism of rate enhancement by LBHBs (13).
The Up and Down Conformations in the A-PPase:Pi2 Active Site.
The current A-PPase:Pi2up conformation is essentially the same as our earlier structure 1wgj. However, residues E58, N116, D117, M3, and both phosphates in the 1.15-Å A-PPase active site have similar (minor) occupancies, ranging from 30–35% on the ligand atoms and 43–45% on side chains (Fig. 2 b–d). These form the A-PPase:Pi2down complex linked by similar occupancies and by the conformation of the P2 phosphate, which points “down” toward the base of the active site (Fig. 1). To form A-PPase:Pi2up from A-PPase:Pi2down, the P2 phosphate rotates “up” so that one of its oxygens is in approximately the same position as the bridging O in the A-PPase:FPPi complex (Fig. 1). This could occur by preserving the coordination of P2(O2) to M4 and M1, as they are mobile enough to allow the rotation (3), and by losing coordination to M2. M3up is coordinated to P1, P2, and E58, whereas M3down is toward the exit of the active site and is coordinated only to P1 (Fig. 1); we now assign M3 as the “regulatory metal ion” (14).
Importantly, the conformations in
A-PPase:Pi2down and
A-PPase:Pi2up are mutually
incompatible; a mixture is sterically impossible. Steric clashes
include two clear conformations for the N116 C⩵O, separated by 1.66
Å at the O, and two overlapped conformations for the D117
CO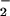 (Fig. 3; Fig.
5b in supplemental data). The P2down
conformation is obviously sterically incompatible with the presence of
Onu (Fig. 1), and the A-PPase P1 phosphate also
has two conformations (and additional, unmodellable disorder);
P1down O3 and P2up O1
(Figs. 1 and 2b) are 1.97 Å apart, again incompatible.
(Fig. 3; Fig.
5b in supplemental data). The P2down
conformation is obviously sterically incompatible with the presence of
Onu (Fig. 1), and the A-PPase P1 phosphate also
has two conformations (and additional, unmodellable disorder);
P1down O3 and P2up O1
(Figs. 1 and 2b) are 1.97 Å apart, again incompatible.
Figure 3.
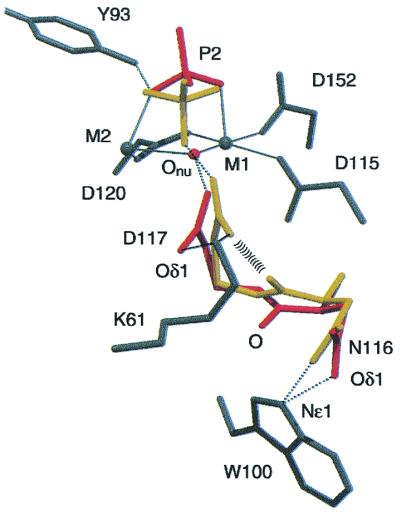
Nucleophile generation. Residues in A-PPase:Pi2 are color
coded by the conformation in which they occur. Red, up conformation;
yellow, down conformation; gray, in both conformations. Metal
coordination is shown with solid and H-bonding with dotted lines. As
shown, the backbone 116C⩵O pushes the D117
CO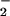 toward Onu.
toward Onu.
In the A-PPase:Pi2down
conformation, there are eight interactions with the P2 phosphate (Fig.
2b). P2 should be HPO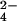 , with ten lone
pairs (five coordinated by M1-M4), one H-bond donor, presumably P2(O1)
to Y93, and two H-bond acceptors (from D117 and K56). The
D117Oδ2down–P2(O4) distance is 2.55 Å, short
enough to be a potential LBHB (15). In the
A-PPase:Pi2up conformation there
are two short interactions, between P2(O1) and P1(O3), and between
P2(O4) and Y93 (Fig. 2 c and d). The latter does
not exist in P2down, probably because M1 and M2
each coordinate two of the P2down oxygens,
pulling P2down(O1) away from Y93 (Fig.
2c).
, with ten lone
pairs (five coordinated by M1-M4), one H-bond donor, presumably P2(O1)
to Y93, and two H-bond acceptors (from D117 and K56). The
D117Oδ2down–P2(O4) distance is 2.55 Å, short
enough to be a potential LBHB (15). In the
A-PPase:Pi2up conformation there
are two short interactions, between P2(O1) and P1(O3), and between
P2(O4) and Y93 (Fig. 2 c and d). The latter does
not exist in P2down, probably because M1 and M2
each coordinate two of the P2down oxygens,
pulling P2down(O1) away from Y93 (Fig.
2c).
Intriguingly, in both A and B subunits the P1(P-O4) and P2(P-O2) bonds appear to have more double bonding character than the other P-O bonds. In both cases, there is more electron density between the atoms than for the other P-O bonds, which appears at this resolution as a tube (Fig. 6 in supplemental data). Such localization of the double bond has been observed in crystalline and fixed environments of hydrated phosphate (16) but, to the best of our knowledge, this is the first time it has been seen in an enzyme active site.
Mechanistic Implications.
The A-PPase:Pi2down conformation corresponds to the “immediate product” intermediate D in Scheme S1, as the nucleophilic water is now attached to P2, but still coordinated to M1 and M2. Therefore, it is worth examining the changes between A-PPase:Pi2down and A-PPase:FPPi, and A-PPase:Pi2down and A-PPase:Pi2up for changes that may have mechanistic implications.
The most surprising feature of
A-PPase:Pi2down concerns the
interactions centered on N116 (Fig. 3). The distance between N116
Odown and D117 Oδ1down is
2.00 Å. This strained interaction appears to be coupled to the motion
of Oδ2 toward Onu. The changes start from the
H-bond between W100 Nɛ1 and N116 Oδ1. In the up conformation the
H-bond is bent, but in the down conformation N116 Oδ1 moves so that
the H-bond shortens, straightens, and strengthens (Fig. 3). This
creates steric conflict with N116 Oup, so that
N116 C⩵O rotates up (Fig. 3) to the N116 Odown
position, and the change propagates along the backbone so that D117
Cαup moves to the
Cαdown position. The resultant change in
backbone angles favors D117 side chain rotation toward
Onu to form an LBHB between D117 Oδ2 and
Onu (see Discussion). This H-bond is
most probably from nucleophilic water (donor) to D117
CO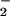 (acceptor); the issue should be definitively
settled by the neutron diffraction structure currently underway.
(acceptor); the issue should be definitively
settled by the neutron diffraction structure currently underway.
P1 makes eight interactions in A-PPase:Pi2down—four are H-bonds to protein, two are H-bonds to water, and two are to M2+ (Fig. 2b). P1 is pulled away from its position in the A-PPase:FPPi complex by hydrogen bonds to the side chains of R78 and K193: when the A-PPase:FPPi and A-PPase:Pi2down structures are superimposed, the distance between the P1 phosphorus atoms is 1.3 Å, whereas the distance between the P2 phosphorus atoms is 0.8 Å. The side chains, but not the main chains, of R78, K193, K198, and E148 adjust to this motion (Fig. 1).
Discussion
The essentially complete coordination of lone pairs in substrate and product complexes argues for an associative rather than a dissociative mechanism. If the mechanism were dissociative, the charge at P2 would be reduced in the transition state compared with the ground state, resulting in reduced coordination in the A-PPase:FPPi and A-PPase:Pi2down structures, which bracket the transition state, as compared with the A-PPase:Pi2up structure. In fact, we see similar P2 coordination in all three structures. Our results are thus consistent with model studies (vide infra) showing that metal-coordinated anions attack phosphate monoester dianions via an associative mechanism (17). By contrast, neutral water attack on phosphate monoester dianions proceeds via a dissociative mechanism, both in solution and on enzymes (17–19).
The structures presented here are completely consistent with our earlier proposal that Onu is the nucleophile that directly attacks P2 (3), and not with other suggestions (20); the pyrophosphate conformation allows direct inline attack of Onu on P2 (Fig. 4). The inline direction is preserved even as the nucleophile moves (compare the FPPi and Pi2up conformations in Figs. 1 and 4). The O-P2-F angle is 175.1° in subunit A and 179.3° in subunit B, and the conformation of P2 in the A-PPase:Pi2down is as predicted (ref. 3; Figs. 1 and 2).
Figure 4.

Angles around F/Onu/P2(O4) in A-PPase FPPi, P2down, and P2up complexes. The F-P-O angle is consistent with Sn2 attack, but the angles around the O are not close to tetrahedral.
An Extended Mechanism for Pyrophosphatase.
A number of lines of evidence suggest, however, that the original pyrophosphatase hydrolysis scheme is not sufficient. First, the two conformations captured in the 1.15-Å structure of A-PPase:Pi2 correspond to intermediates D and E in Scheme S1, compared with only one such intermediate in the original scheme. Second, we now have strong evidence for two substrate complexes (B and C in Scheme S1), based on studies of S. cerevisiae inorganic pyrophosphatase (Y-PPase) fluoride inhibition (4), pH-dependence of catalysis (2), and single turnover kinetics (P. Halonen, unpublished data). What inferences, therefore, can be drawn about B and C based on the structures described here?
In the PPase:Mn2 complex (pdb 1wgi; A in Scheme S1), M1 and M2 are separated by a two-water molecule bridge (3) but they are separated by a single fluoride ion in the PPase:FPPi (C-analogue) complex. If the structure of PPase:FPPi closely mimics C, when does the two-water bridge (A) collapse to a one-water bridge (C)? We suggest that complex B in Scheme S1 contains a two-water bridge between M1 and M2; substrate binds to the two-water bridge structure, which then dehydrates. Similarly, the difference between D and E, as observed here, is the rehydration of the nucleophile binding site. Our proposal thus creates a pleasing symmetry in the reaction mechanism: B is the hydrated version of substrate complex C, and E is the hydrated version of product complex D. It also explains why the pKa of the ionizable group controlling the B to C transition is greater than 7, but the pKa for the C to D transition is 5.9 (2). The former pKa would correspond to ionization of one of the water molecules in the two-water molecule bridge leading to loss of the other water molecule. Its pKa would be much less than 16, but not as low as the pKa of Onu coordinated to two metal ions, which is 5.9 (2).
Sources of Catalytic Power in Pyrophosphatase.
What more general lessons about enzyme catalytic power can be drawn? Enzyme catalytic power is in essence due to transition state stabilization: transition state is bound tighter than substrate or product. For an enzyme like PPase that accelerates a reaction 1010-fold, enzyme should bind transition state 1010 times tighter than substrate or product. This requires either “passive binding”—molecular recognition through hydrogen bonding, etc.—or “dynamic binding,” where special interactions between the catalyst and substrate occur during catalysis (21). There is, however, considerable disagreement about the nature of these “special interactions.”
One proposal is orbital steering: reacting groups are placed to maximize the overlap of the highest occupied molecular orbital on the nucleophile and the lowest unoccupied molecular orbital on the electrophile (22). A second proposal is that symmetrical, extremely strong hydrogen bonds form (LBHBs, with X . . . Y distances of 2.4–2.55 Å and ΔH 15–20 kcal/mol; ref. 15). A third, formally related suggestion is that ground-state proton tunneling occurs (23); in such a model, ground-state vibrational forces distort a protein to the point that the “reactant” and “product” curves intersect, allowing rapid tunneling of the proton or hydrogen from reactant to product. A fourth proposal is that substrate binding and transition state binding may not be separable, and so the key element is preorganization of the active site to ensure efficient passage across the transition state (maximization of the transmission factor; refs. 24 and 25). We now examine the structures presented here to see whether they allow any distinction between these proposals.
Orbital Steering.
Orbital steering appears unlikely in PPase because, even though the Obridge-P-F angles in PPase:FPPi are very close to 180°, none of the angles around Onu or F are close to tetrahedral, as would be expected for precise orbital alignment (Fig. 4).
Low Barrier Hydrogen Bonds.
As mentioned above, Onu, with a pKa of 5.9 (2), could form an LBHB with D117 Oδ2 because the pKa of D117 may be as high as 6.25 (2), possibly due to its interaction with the backbone carbonyl of N116. In the A-PPase:Pi2down active site, the P2-O4 to D117 Oδ2 distance is 2.55 Å and, in the A-PPase:Pi2up active site, the Onu-D117 Oδ2 distance is 2.75 Å. The distance from D117-Oδ2down to Onu would be only 2.34 Å. Even though this calculation mixes different active site conformations, it shows that D117 Oδ2 approaches the nucleophile very closely.
The strained conformation in the A-PPase:Pi2down active site, set up by the rigid W100 (see above), further supports this model. The 2.00-Å contact between D117 Oδ1down and N116 Odown may sterically compress D117 Oδ2 toward Onu (Fig. 3). A similar mechanism has been proposed for mandelate racemase and other enzymes where dynamic binding during the transition state allows efficient proton transfer (13).
No direct role can be ascribed to the short hydrogen bonds between Y93Oη and P2 oxygens in the A-PPase:FPPi and A-PPase:Pi2up complexes. The Y93F variant has a kcat 8% of that of wild-type S. cerevisiae inorganic pyrophosphatase (Y-PPase; ref. 14) and its role may be to provide additional compression in the active site.
Active Site Preorganization.
Canon et al. (25) and Warshel (24) suggested that the primary way in which enzymes achieve rate enhancement is by reducing the organizational work for both substrate and environment to reach the transition state: i.e., there is no substantial difference between substrate and transition state binding. We see some evidence to support such a mechanism of rate enhancement for PPase. Neither PPi nor the immediate product A-PPase:Pi2down fit perfectly. Using the presence of short (and presumably strong) H-bonds as the criterion, the P2up conformation (in PPase:FPPi and PPase:Pi2up) fits better into the P2-binding site, and the P1up conformation (only in PPase:Pi2up) fits better into the P1-binding site (Fig. 2). The only conformation between PPase:FPPi and PPase:Pi2down that might simulate the up-conformation at both P1 and P2 is the transition state, consistent with preorganization of the active site.
In addition, the lack of substantial backbone movement between substrate and product in the ligands binding P1 may help achieve rapid catalysis. Like the shock absorbers on a car, the side chains may isolate the rest of the protein from the active site “protein quake” [cf. myoglobin (26)], thus allowing the active site to respond faster. Consistent with this, motion in the active site is split in two. Between A-PPase:FPPi and A-PPase:Pi2down, only side chains move in the P1 site but the P1 phosphorus moves by 1.3 Å; but main chain and side chain movement occurs in the P2 site, where the P2 phosphorus moves only 0.8 Å. Conversely, between A-PPase:Pi2down and A-PPase:Pi2up there is only slight motion at P1, but main chain motion at P2.
Some of the vibrational motions in the active site, as evidenced by the anisotropic B-factors, also appear to be arranged to achieve efficient catalysis. This ground-state effect may enhance the rate of reaction. For instance, in A-PPase:Pi2up the major anisotropic motion of Onu is in the M1 and D117 Oδ2 directions. In addition, D117 Oδ2up shows anisotropic motion toward D117 Oδ2down.
Ground-State Proton Tunneling.
Ground-state-based proton tunneling from Onu to D117 may also play a role, as has been proposed for hydrogen tunneling in methylamine dehydrogenase (23). The motion described above compresses D117 Oδ2up toward the catalytically active D117 79 δ2down conformation, thus allowing efficient tunneling between Onu and D117 and generation of the nucleophile. This argument is in some ways formally analogous to the suggestion that an LBHB forms between D117 Oδ2 and Onu; in both models, rate enhancement is due to compression of the heavy atoms in the hydrogen bond.
These structures provide a framework to test current hypotheses of enzymological rate enhancement in PPases, lending support to LBHBs and ground-state effects in catalysis but not to orbital steering. They also demonstrate that significant mechanistic observations can be made from ultra-high resolution structures of enzyme substrate/product complexes. For a nonallosteric enzyme like PPase, the structures are as relevant as those obtained by time-resolved crystallography because a mixture of mechanistically relevant conformations can be found on the enzyme. We are thus moving closer to our goal of understanding phosphoryl transfer in PPases at the quantum mechanical level.
Supplementary Material
Acknowledgments
We thank R. Andersen, M. Merckel, T. Kajander, and P. Vahakoski. This work was supported by the Academy of Finland (Grant 4310 to A.G. and R.L.; 42979 to P.H.; and a training grant to V.T.), and the European Union TMR/LSF to European Molecular Biology Laboratory Hamburg Outstation (ERBFMGECT980134).
Abbreviations
- PPases
pyrophosphatases
- LBHB
low-barrier hydrogen bond
- B-factor
temperature factor
- NCS
noncrystallographic symmetry
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org [1e9g and r1e9gsf (PPase:Pi2) and 1e6a and r1e6asf (PPase:FPPi)].
References
- 1.Baykov A A, Cooperman B S, Goldman A, Lahti R. In: Progress in Molecular and Subcellular Biology. Schröder H C, Müller W E G, editors. Vol. 23. Heidelberg: Springer; 1999. pp. 127–150. [DOI] [PubMed] [Google Scholar]
- 2.Belogurov G A, Fabrichniy I P, Pohjanjoki P, Kasho V N, Lehtihuhta E, Turkina M V, Cooperman B S, Goldman A, Baykov A A, Lahti R. Biochemistry. 2000;39:13931–13938. doi: 10.1021/bi000895s. [DOI] [PubMed] [Google Scholar]
- 3.Heikinheimo P, Lehtonen J, Baykov A, Lahti R, Cooperman B S, Goldman A. Structure. 1996;4:1491–1508. doi: 10.1016/s0969-2126(96)00155-4. [DOI] [PubMed] [Google Scholar]
- 4.Baykov A A, Fabrichniy I P, Pohjanjoki P, Zyryanov A B, Lahti R. Biochemistry. 2000;39:11939–11947. doi: 10.1021/bi000627u. [DOI] [PubMed] [Google Scholar]
- 5.Tuominen V, Heikinheimo P, Kajander T, Torkkel T, Hyytiä T, Kapyla J, Lahti R, Cooperman B S, Goldman A. J Mol Biol. 1998;284:1565–1580. doi: 10.1006/jmbi.1998.2266. [DOI] [PubMed] [Google Scholar]
- 6.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 7.Brünger A T. X-PLOR, A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1987. , Version 3.1. [Google Scholar]
- 8.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 9.Sheldrick G M, Schneider T R. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 10.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J-S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 11.Baykov A A, Bakuleva N P, Nazarova T I, Avaeva S M. Biochim Biophys Acta. 1977;481:184–194. doi: 10.1016/0005-2744(77)90150-4. [DOI] [PubMed] [Google Scholar]
- 12.Pohjanjoki, P., Fabrichniy, I. P., Kasho, V. N., Cooperman, B. S., Goldman, A., Baykov, A. A. & Lahti, R. (2000) J. Biol. Chem., in press. [DOI] [PubMed]
- 13.Frey P A, Cleland W W. Bioorg Chem. 1998;26:175–192. [Google Scholar]
- 14.Pohjanjoki P, Lahti R, Goldman A, Cooperman B. Biochemistry. 1998;37:1754–1761. doi: 10.1021/bi971771r. [DOI] [PubMed] [Google Scholar]
- 15.Cleland W W, Frey P A, Gerlt J A. J Biol Chem. 1998;273:25529–25532. doi: 10.1074/jbc.273.40.25529. [DOI] [PubMed] [Google Scholar]
- 16.Mighell A D, Smith J P, Brown W E. Acta Crystallogr B. 1969;25:776–780. [Google Scholar]
- 17.Cleland W W, Hengge A C. FASEB J. 1995;9:1585–1594. doi: 10.1096/fasebj.9.15.8529838. [DOI] [PubMed] [Google Scholar]
- 18.Admiraal S J, Herschlag D. Chem Biol. 1995;2:729–739. doi: 10.1016/1074-5521(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 19.Admiraal S J, Schneider B, Meyer P, Janin J, Veron M, Deville-Bonne D, Herschlag D. Biochemistry. 1999;38:4701–4711. doi: 10.1021/bi9827565. [DOI] [PubMed] [Google Scholar]
- 20.Harutyunyan E H, Kuranova I P, Vainshtein B K, Höhne W E, Lamzin V S, Dauter Z, Teplyakov A V, Wilson K S. Eur J Biochem. 1996;239:220–228. doi: 10.1111/j.1432-1033.1996.0220u.x. [DOI] [PubMed] [Google Scholar]
- 21.Kirby A J. Angew Chem Int Ed Engl. 1996;35:707–724. [Google Scholar]
- 22.Mesecar A D, Stoddard B L, Koshland D E., Jr Science. 1997;277:202–206. doi: 10.1126/science.277.5323.202. [DOI] [PubMed] [Google Scholar]
- 23.Sutcliffe M J, Scrutton N S. Philos Trans R Soc London A. 2000;358:367–386. doi: 10.1098/rsta.2000.0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warshel A. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 25.Cannon W R, Singleton S F, Benkovic S J. Nat Struct Biol. 1996;3:821–833. doi: 10.1038/nsb1096-821. [DOI] [PubMed] [Google Scholar]
- 26.Ansari A, Berendzen J, Bowne S F, Frauenfelder H, Iben I E, Sauke T B, Shyamsunder E, Young R D. Proc Natl Acad Sci USA. 1985;82:5000–5004. doi: 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}