

Abstract
The aim of this study was to delineate the temporal and spatial sequence of STAT1 and STAT3 activation during development of GVHD following fully Major Histocompatibility (MHC)-mismatched allogeneic Bone Marrow Transplantation (BMT). Activation of inflammatory signaling pathways in GVHD target organs was assessed by western blotting, phospho-flow cytometry and electromobility shift assays (EMSA). Development of GVHD was associated with significant expansion of phospho[p]-STAT1 and p-STAT3 expressing CD4+ T cells and CD8+T cells. GVHD-specific STAT3/STAT1 activation preceded activation of Nuclear Factor-κB (NF-κB) and Mitogen Activated Protein Kinase (MAPK) and was associated with subsequent induction of STAT1 or STAT3-dependent inflammatory gene expression programs (e.g. expression of IRF-1, SOCS1, IL-17). Our studies may help to establish a functional hierarchy of the signaling events leading to the development of GVHD and could be helpful in designing new molecularly targeted treatment approaches for GVHD.
Keywords: GVHD, STAT1, STAT3, Inflammation, BMT
1. INTRODUCTION
The pathobiology of GVHD is a complex process which involves conditioning therapy-induced tissue damage, creation of a proinflammatory environment and host antigen presenting cell (APC)-dependent activation of donor T cells. Eventually, donor cells infiltrate GVHD target organs leading to tissue damage. Tissue infiltration is controlled by inflammation-induced expression of adhesion molecules, chemokines, and chemokine receptors on donor T cells and parenchymal target organs [1, 2].
We and others have shown that GVH responses can stay confined within the lymphohematopoietic system, if donor T cells are administered in an environment devoid of any inflammation [3–6]. Understanding the molecular events controlling the inflammatory response may help to develop novel approaches for treatment of GVHD. Small molecule inhibitors targeting either Histone deacetylase [7, 8] [9, 10], the proteasome [11, 12] or IKK [13] may have the capacity to prevent the development of GVHD while maintaining Graft versus Leukemia (GVL). Despite mRNA gene expression profiling of GVHD target tissues [14, 15], inflammatory signaling pathways controlling the different stages of GVHD have remained elusive. We have recently identified STAT1 activation[7] as a very early event in the development of GVHD. We have therefore sought to further elucidate the complex network of signaling pathways during the early phases of GVHD in relation to STAT1. Indeed, we demonstrated that prolonged activation of STAT1 and STAT3 are dominant events occurring in host GVHD target organs, the spleen and also splenic T cells. We observed a differential pattern of NF-kB activation between spleen and GVHD parenchymal target organs. Furthermore, we demonstrated that activation of MAPK pathway occurred more in response to conditioning-induced inflammation than as a consequence of GVHD induction except for ERK1,2 activation, which was transiently induced during induction of GVHD. Our studies should provide the conceptual framework to identify molecular targets for the treatment of GVHD.
2. MATERIALS AND METHODS
2.1 Animals
Female C57BL/6 ([H2b], B6) and BALB/c [H2d] mice were purchased from Jackson Laboratories (Bar Harbour, ME) and used between 8 – 12 weeks of age. All mice were housed in autoclaved microisolator environments, provided with sterile water and irradiated food ad libitum. All manipulations were performed in a laminar flow hood. All experiments were conducted in accordance with the local IACUC guidelines.
2.2 Lethal conditioning and induction of GVHD
GVHD was induced in the fully MHC-mismatched strain combination BALB/c →B6. Recipients were lethally irradiated with 10.75 Gy and were reconstituted within 4 hours with a single intravenous inoculum of 5×106 allogeneic or syngeneic bone marrow cells. GVHD was induced by co-injection of allogeneic spleen cells (4 ×107). To avoid bias from cage-related effects, animals in different groups were randomized between cages.
2.3 Western blotting
Extraction of proteins from GVHD target organs for immunoblotting was performed as previously described using a modified RIPA buffer[16]. Antibodies against p-STAT1(Tyr701), total STAT1, p-STAT3(Tyr705), STAT3, p-ERK,1,2 p-IkB, p-MEK1,2, p-JNK, p-p65 and GAPDH, were purchased from Cell Signaling Inc. (Beverly, MA).
2.4 Clinical GVHD assessment and histopathology
GVHD morbidity was assessed using a standard scoring system, which is based on summation of five criteria scores: percentage of weight change, posture, activity, fur texture, and skin integrity [17].
For histopathological analysis of GVHD target tissues, samples were collected from liver and small and large intestines, and were fixed in 7% formalin. Formalin-preserved tissue samples were embedded in paraffin, cut into 5-μm-thick sections, and stained with hematoxylin and eosin. GVHD-specific histomorphologic changes were assessed using the following scoring system (0–3): luminal sloughing of cellular debris/cross section (0= none, 1 = 1, 2 = 2, 3 = 3 or more); crypt cell apoptosis (0 = none, 1= 1–6/10 crypts, 2 = > 6/10 crypts, 3= >6+ confluent/10 crypts); Lamina Propria (LP) infiltrate (0 = none, 1 = some, 2 = clusters, 3 = aggregates/follicles). Slides were coded without reference to mouse strain or treatment and were systematically examined by a pathologist (A.S.).
2.5 Immunohistochemistry
Immunohistochemistry was performed according to standard protocols in the UPCI tissue pathology core laboratory. Briefly, slides were deparaffinized and hydrated with D/W following standard protocols. Endogenous peroxidase was blocked with 3% Hydrogen peroxide (Fisher Scientific, Pittsburgh, PA) for 10 minutes, and slides were subjected to heat-induced antigen retrieval using Dako (Dako North America, Carpinteria, CA) Citrate buffer in a decloaking chamber (Biocare Medical, Concord, CA). After washing with TBS and blocking with background eraser blocking reagent (Biocare) for 10 min., slides were incubated with the appropriate antibodies. After washing, slides were stained with HRP-a-rabbit and DAB chromogen (Dako). Immunohistochemical STAT1 and p-STAT1 staining was assessed using the following scoring system (0–3): 0 = none, 1 = focal, 2 = patchy, 3 = diffuse).
2.6 Cytokine multiplex analysis
The LabMap serum/supernatant assays were performed in 96-well microplate format according to a protocol provided by the manufacturer (BioSource International, Camarillo, CA). A filter-bottom, 96-well microplate (Millipore, Billerica, MA) was blocked for 10 minutes with PBS/BSA. To generate a standard curve, fivefold dilutions of appropriate standards were prepared in supernatant culture medium. Serum samples were analyzed using the mouse cytokine/chemokine Lincoplex assay (Lincoresearch, St. Charles, MO). Analysis of experimental data was performed using five-parametric-curve fitting.
2.7 Transcription factor (TF) multiplex analysis
Nuclear and cytoplasmic extracts were prepared from GVHD tissues samples using the Pierce N-PER (Pierce Biotechnology, Rockford, IL) kit according to the manufacturer’s recommendation. Detection of DNA-transcription complexes was performed using Marligen (Ijamsvilee, MD) Muliplex Transcription factor Assay.
2.8 Electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared as described before [18]. For electrophoretic mobility shift analysis, 5 μg of nuclear protein were incubated in 10 mM Tris (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 10 mM DTT, 1 mM EDTA, 5% glycerol, 1 μg of BSA, and 1 μg of poly(dI-dC) with 20,000 cpm of radioactive 32 P-labeled probe for 15 minutes at room temperature. This was followed by separation of the DNA-protein complexes on 4% acrylamide gels. The following oligonucleotides (NF-kB site of the H2K gene) were annealed to complementary strands, resulting in double-stranded probes: 5 AGC-TCA-GGG-CTG-GGG-ATT-CCC-CAT-CTC-CAC-AGG-3, 5-AGC-TCC-TGT-GGA-GAT-GGG-GAA-TCC-CCA-GCC-CTG-3 as previously described. Incubation in the presence of excess concentrations of cold oligonucleotides served as specificity controls.
2.9. ELISA
IL-17 specific Quantikine ELISA was purchased from R&D (Minneapolis, MN) and performed according to the manufacturer’s brochure.
2.10 Microarray analysis
RNA was extracted using TriZol from snap frozen liver samples from three animals per group. RNA from each group was pooled. RNA amplification and Gene expression profiling using PIQOR Immunology Microarrays was performed by Memorec Biotec Inc. (Cologne, Germany) as described by the manufacturer. The immunology antisense microarray set comprises a total of 1076 key genes for immune response, cell death, extracellular matrix, and signal transduction. For analysis of differential gene expression liver RNA from animals with GVHD was compared to RNA extracted from syngeneic controls. Each gene of interest was spotted in quadruplicates on the array. The mean fold expression in comparison to syngeneic control and its standard deviation are presented.
2.11 Real-time PCR analysis
For the determination of mRNA levels of IRF-1 and SOCS1, total RNA was isolated using the Mini RNA isolation II kit (QIAGEN) according to the manufacturer’s instructions. Total RNA was converted to cDNA using the Superscript III Reverse Transcriptase (RT) (Invitrogen, Carlsbad, California). qRT-PCR was performed on ABI Prism 7700 Sequence detection System (Applied Biosystems, Foster City, CA). PCR was carried out with SYBR Green PCR master mix (ABI) using 1ul cDNA in a 25ul final reaction mixture (15min at 95°C; 40 cycles of 15 sec at 95°C, 60 sec at 60°C and 10min at 79°C). The average of threshold cycle (CT) for each gene was determined from triplicate reactions, and data were analyzed by taking the difference between mean threshold PCR cycle value for target and control gene (ΔCT). Target gene expression was normalized to β-actin using the ΔCT value. This was then calibrated to the control sample in each experiment to give the ΔΔCT value, where the control had a ΔΔCT value of 0. The fold target gene expression, compared with the calibrator value, is given by the formula 2-ΔΔCT. The following primers sets were used: IRF-1 sense 5′ ATGCCAATCACTCGAATGCGGA 3′, antisense 5′ GGCTGCCACTCAGACTGTTCAA 3′, β-actin sense 5′ GAA ATC GTG CGT GAC ATC AAA G 3′, antisense 5′ TGT AGT TTC ATG GAT GCC ACA G 3′. SOCS1 expression and GAPDH as endogenous control were studied using the Applied Biosystems Taqman® Gene Expression Assay system.
2.12 Cell selection procedures
Pan T cells were selected from spleen cells by negative selection (no-touch preparation) using MACS beads (Miltenyibiotec, Bergisch Gladbach, Germany) according to the manufacturer’s recommendation. A purity of ≥90% was achieved after selection.
2.13 Flow cytometry
Spleen cells were fixed immediately after harvesting with prewarmed BD Phosflow Lyse/Fix buffer for 10 minutes at 37°C. Cells were then permeabilized for 30 min on ice with 1ml of BD Phosflow Perm Buffer III, washed twice with BD Pharmingen Stain Buffer and stained. The following antibodies were used for two-color immunfluorescence staining: PE-CD4 (RM4-5), PE-CD8 (53–6.7), Alexa Fluor® 488 labeled phosphorylated STAT1 (pY701, clone 4a), STAT3 (pY705, clone 4/P-STAT3) and the appropriate istotype control antibodies. All antibodies were from BD Pharmingen (San Diego, CA).
2.14 Statistical analysis
Statistical analyses were performed using the Mann-Whitney test for nonparametric data. A p-value of< 0.05 was considered to be significant.
3. RESULTS
3.1 Systemic expression of STAT1 and STAT3-activating cytokines during GVHD
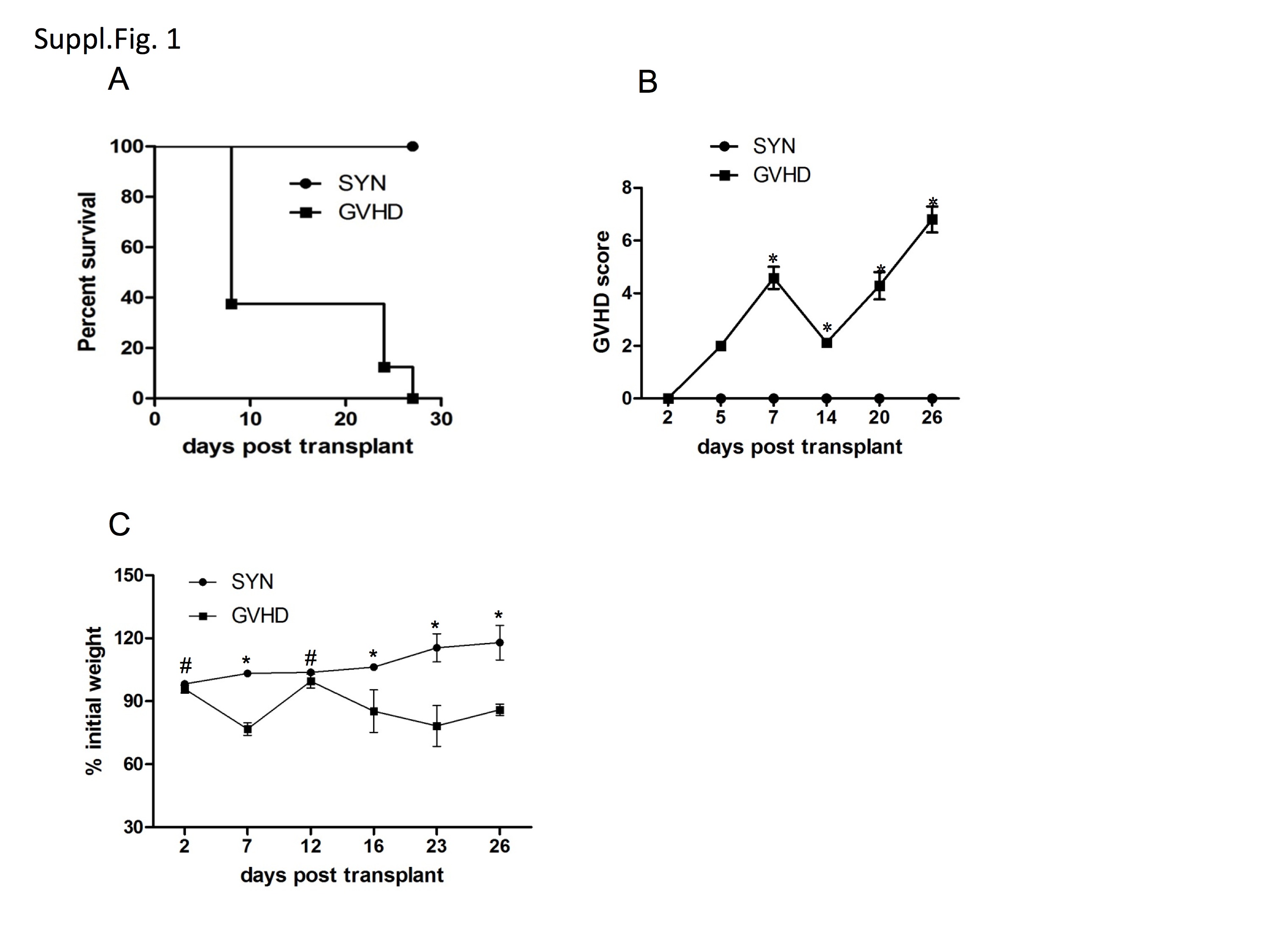
GVHD was induced in the fully MHC-mismatched strain combination BALB/c →B6 following lethal irradiation. Similar to prior results from us and other investigators we observed a biphasic course of GVHD development (Suppl. Fig. 1) with an initial early weight loss and mortality followed by a short recovery which was then followed by a second phase GVHD morbidity and mortality. We hypothesized that STAT1 and STAT3 activation may be due to systemic action of cytokines signaling through these pathways stimulated by the early cytokine storm. We therefore studied serum levels of the most important activators of STAT1 (IFN-γ) and STAT3 (IL-6 and IL-10) at different time points during GVHD and in syngeneic controls. IFN-γ and IL-6 became detectable on day +3 and peaked on day +6 post-BMT followed by a steep decrease on day +13. As shown in Figure 1a, IFN-γ levels were significantly increased in GVHD animals in comparison to syngeneic controls at all time points tested. IL-6 levels were significantly elevated above syngeneic controls on days +6 and +13. In contrast, IL-10 became detectable on day +6. Whereas IFN-γ and IL-6 levels dropped significantly from day +6 to day +13 in GVHD animals, there was no significant difference in IL-10 levels between day +6 and +13 in animals with GVHD. This indicates a significant reduction in IL-6/IL-10 ratio in animals with GVHD between day+6 and day +13 post BMT (20,77+3.78 [SEM] vs. 1,95+ 0.65 [SEM], p =0.02 Mann-Whitney test).
Figure 1. Systemic cytokine levels and splenic signaling pathway activation.
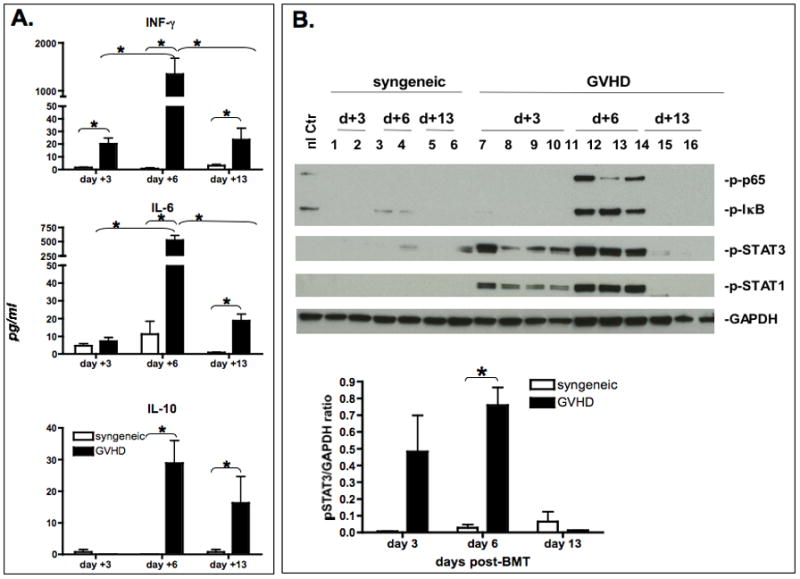
A. Serum was collected from syngeneic controls (open bars) or animals with GVHD (black bars) on day +3, +6 and +13 post-BMT. Serum was analyzed using Lincoplex multiplex assay. Bars represent the mean (n=4) ± Standard Error of Mean (SEM). Statistical significance was determined by Mann Whitney test. Asterisks denote significant differences (p< 0.05). Data are representative of one out of three independent experiments.
B. Inflammatory signaling pathway expression in the spleen. GVHD was induced using the BALB/c → B6 strain combination following total body irradiation. Untreated normal B6 and recipients of syngeneic bone marrow (B6→B6) served as controls. Spleens were harvested on day +3, +6 and +13 post-BMT. Protein was extracted and analyzed by western blotting for STAT1, p-STAT1 (Tyr701), STAT3 and p-STAT3 (Tyr705), p-IkB, p-p65(Ser536) expression. Each lane represents one experimental animal. Two – four animals were studied per group. 40ug total protein was loaded onto each lane. Densitometry results of p-STAT1 and p-STAT3 expression normalized to GAPDH are presented. Data are representative of one out of three independent experiments. Peak activation of pSTAT1 in comparison to syngeneic controls occurs on day +6 (GVHD vs. syngeneic control p=0.013).
3.2 Activation of STAT1 and STAT3 in the spleen
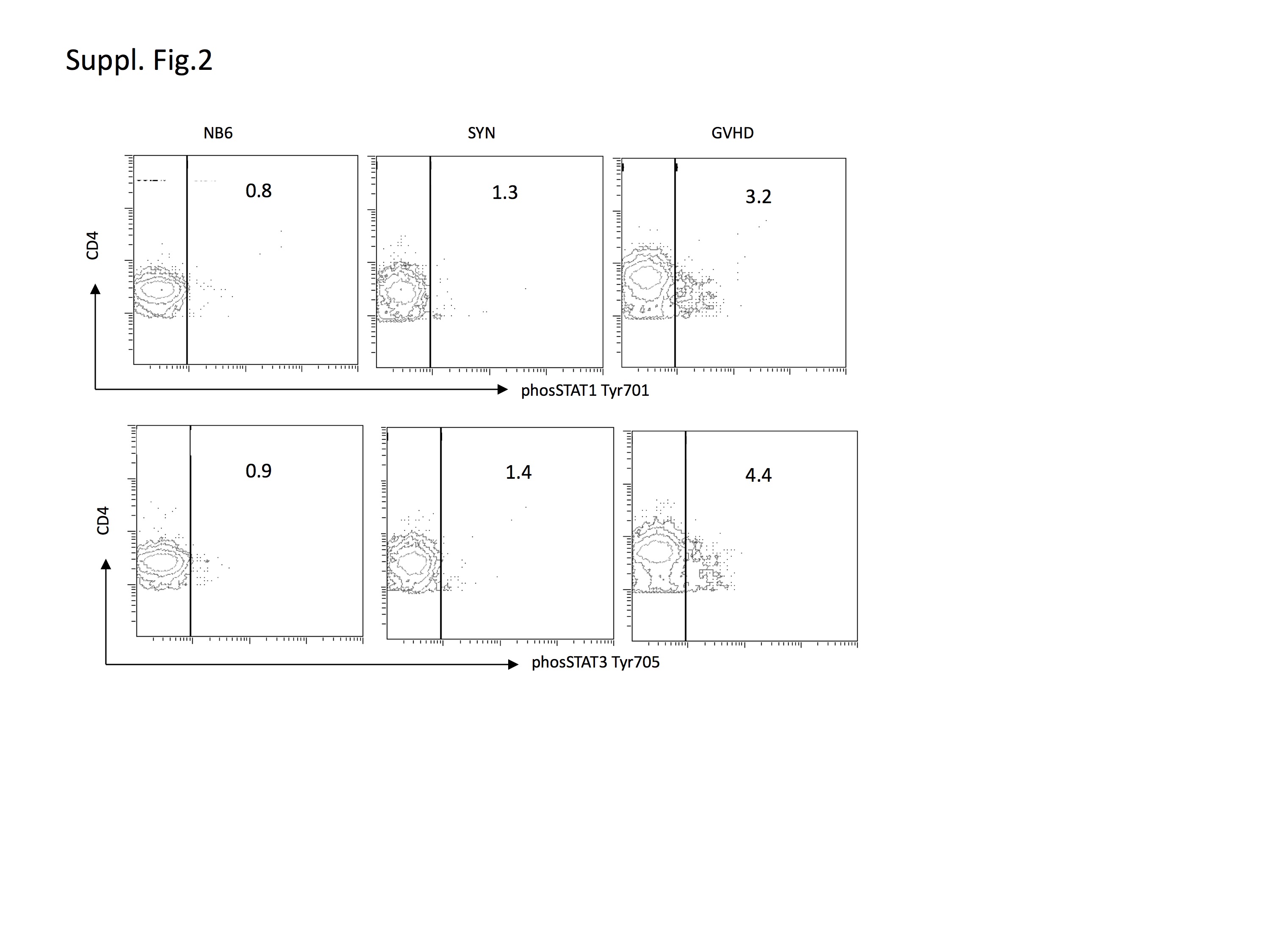
We next evaluated the temporal and spatial sequence of STAT1 activation during GVHD in relation to STAT3 and other critical inflammatory signaling pathways. Based on the relevance of STAT1 and STAT3 in controlling inflammation and T cell polarization, we studied the activation of STAT1 and STAT3 in total cell lysates of spleen tissues as an example of a secondary lymphoid organ and also splenic T cells in comparison to other major inflammatory signaling pathways. Rapid induction and phosphorylation of STAT1 and STAT3 occurred in a GVHD-specific pattern peaking on day +6 (Figure 1b), in contrast to syngeneic controls, which did not show any significant activation. Activation of STAT1 and STAT3 could be detected as early as day +1 post-BMT in the spleen. Separate experiments were set up to further delineate the expression/activation of STAT1 and STAT3 in splenic T cells. Due to the expected insufficient protein yield from selected splenic T cell populations from individual animals on days +3, we negatively selected splenic T cells from pooled splenocytes (5–10 animals) using no-touch preparation. Cell lysates were prepared from these enriched T cell populations (Purity >90%) and studied by western blot. Similar to total spleen lysates, STAT1 and STAT3 phosphorylation was significantly increased over syngeneic controls in freshly isolated splenic T cells on day +3. (Figure 2a). Next, we analyzed STAT1 and STAT3 phosphorylation in CD4+ and CD8+ T cells using flow cytometry (Figure 2b, Suppl.Fig. 2) following induction of GVHD or in syngeneic controls. A significant expansion of pSTAT1(Tyr701) and pSTAT3(Tyr705) expressing CD4+ and CD8+ T cells could be observed (Figure 2b) during induction of GVHD in the spleen in comparison to syngeneic controls.
Figure 2. Inflammatory signaling pathways in splenic T cells.
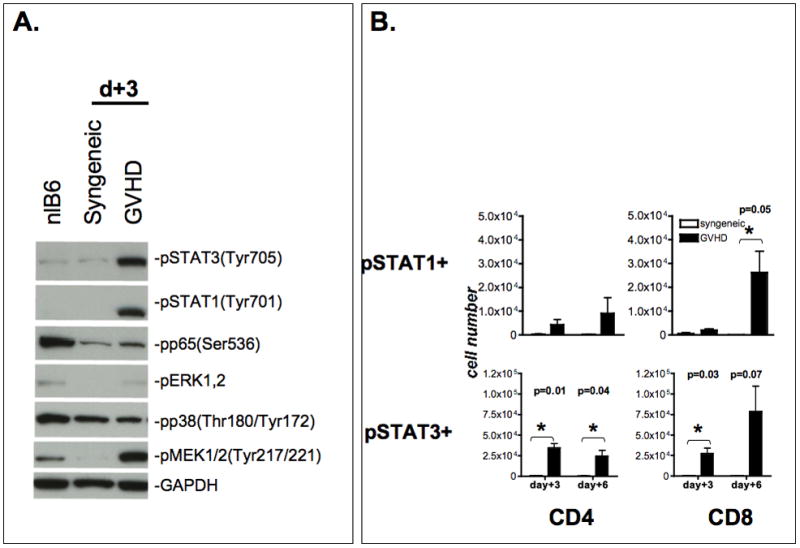
A. Inflammatory signaling pathways in splenic T cells. Animals were sacrificed on day +3 post-BMT. Normal B6, syngeneic controls and GVHD animals were studied. Total spleen cells from 5–10 animals per group were pooled and magnetically enriched for T cells using negative selection resulting in a >90% purity. Lysates were prepared as described and immunoblotted with pSTAT1, pSTAT3, p-p65, p-MEK1,2, p-pERK1.2, p-p38 and GAPDH as housekeeping control. Data are representative of one out of two independent experiments.
B. Expansion of pSTAT1/3 positive CD4 and CD8 T cells. Splenocytes were harvested on day +3 and +6 post-BMT. Phosphorylation status of STAT1 (Tyr701) and STAT3 (Tyr705) in CD4+ and CD8+ T cells was assessed using phosphotyrosine-specific antibodies using flow cytometry. The mean number of p-STAT1 and p-STAT3 expressing cells (+/− SEM) is depicted for animals with GVHD and syngeneic controls (n= 3). Statistical significance was calculated using the student t-test. There was no significant different between syngeneic controls and untreated normal recipients (data not shown).
3.3 MAPK activation in the spleen
It is conceivable that the multitude of cytokine and growth factors induced during GVHD could also lead to activation of other inflammatory signaling pathways including the MAPK pathway. We therefore studied the activation of p38, ERK1,2 and JNK in total spleen cells. To our surprise, phosphorylated-p38 was detected in total splenocytes and splenic T cells of both control and GVHD animals alike (Figures 2a, 3) suggesting that p38 activation is more a reflection of non-specific conditioning-dependent toxicity than of GVHD. Activation of ERK1,2 occurred in whole splenocytes transiently in a GVHD-specific pattern on day +1. Of interest, analysis of purified splenic T cells by western blot demonstrated absence of ERK1,2 activation on day +3. (Figure 2a). Interestingly, on day +3 activation of MEK1,2 occurred only in splenic T cells from GVHD animals, but not from syngeneic recipients. We were not able to detect a significant JNK activation in spleen samples.
Figure 3. Activation of MAPK pathway during GVHD in spleen cells.
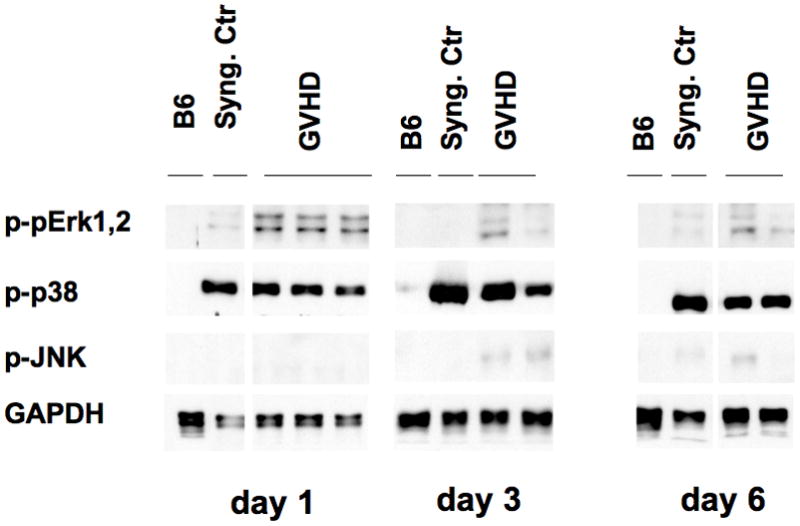
As described above total spleen cells were harvested and protein lysates prepared from normal B6, syngeneic controls, and animals with GVHD on day +1, +3, and +6 post-BMT. Data are representative of one out of three independent experiments.
3.4 NF-kB activation in the spleen
Given the crucial role, which has been attributed to TNF and LPS in the course of GVHD, we focused next on NF-kB signaling within the spleen during GVHD. NF-kB activation as determined by IκB and p-65 phosphorylation was detected in total splenocytes only on day +6 (Figure 1b) and was no longer detectable at later time points. Thus, NF-kB activation occurred within a very narrow window in the spleen during the course of GVHD and was lost after day +6.
3.5 STAT1 and STAT3 are progressively activated in parenchymal GVHD target organs
We next studied STAT1 and STAT3 activation in the GVHD target organs colon and small bowel during the early post-BMT course in animals with GVHD or syngeneic controls. Induction of STAT1 and STAT3 protein expression became readily detectable in the colon of transplanted animals as early as day +1 (syngeneic controls and GVHD animals, Figure 4a). Phosphorylation of STAT1 and STAT3 became detecable on day +3 in control and GVHD animals. In syngeneic controls STAT1 phosphorylation was only transient and disappeared by day +6. In contrast, STAT1 and STAT3 were progressively phosphorylated in GVHD animals peaking on day +6. To address the possibility that STAT1 expression in western blot was due to the presence of infiltrating hematopoietic cells or contaminating lymphocytes from secondary lymphoid organs of the gut, we performed immunohistochemistry. Expression of STAT1 and phosphorylated STAT1 (Tyr701) was completely absent in normal untreated colon samples (data not shown). In line with the western blot data, maximum expression of STAT1 and p-STAT1 was observed on day +6 in GVHD samples (Figure 4b). Bright staining of STAT1 and p-STAT1 was observed in colonic epithelial cells and correlated with typical morphological signs of GVHD-induced tissue damage (luminal sloughing, crypt cell apoptosis) on day +6 post-BMT indicating that STAT1 may be required for gut organ pathology. Similar results were obtained for the small bowel (Figure 4c). In syngeneic controls phosphorylated and unphosphorylated STAT1 expression peaked on day +3 (Figure 4c) with some p-STAT1 staining still observable on day + 6. Since we observed a biphasic pattern of GVHD development, we were interested in determining the expression and activation status of STAT1 and STAT3 also during the late phase (i.e. > day+10 post-BMT) in GVHD target tissues. For this purpose liver tissue samples harvested on days +13 and +26 post-BMT were evaluated. pSTAT1 expression was not or only weakly detectable on day +13 and was completely undetectable on day +26 in the GVHD group (Suppl. Figure 3). However, we did see an increased and persistent expression of unphosphorylated STAT1 on day+13 (and to a lesser extent on day+26). In contrast, expression of phosphorylated STAT3 was significantly upgregulated in the GVHD group compared to the syngeneic controls (Suppl. Figure 3) on days +13 and +26 post-BMT.
Figure 4. STAT1 activation in the gut.
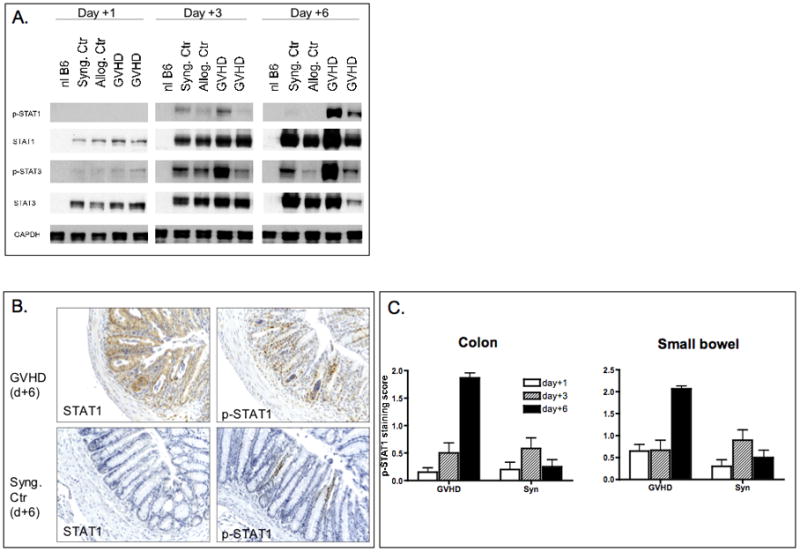
A. Colon samples were harvested on days +1, +3 and +6 post-BMT following lethal irradiation and BMT using the BALB/c to B6 strain combination. Protein was extracted and analyzed by western blotting for STAT1, p-STAT1 (Tyr701), STAT3 and pSTAT3 (Tyr705) expression. Data are representative of one out of two independent experiments.
B. Immunohistochemical analysis of STAT1 and p-STAT1 in colon tissues. 100x magnification of representative colon tissue from syngeneic controls or GVHD animals. Slides were stained with STAT1 and p-STAT1 specific antibodies. STAT1 and p-STAT1 expression are significantly increased in GVHD animals compared to syngeneic controls on day +6. This coincided with signs of tissue damage (crypt apoptosis and luminal sloughing).
C. Semiquantitative scoring of staining intensity of STAT1 and pSTAT1 in colon and small bowel samples. A total of four animals per group were studied.
3.6 NF-kB activation in GVHD target epithelial organs
Next, we assessed the role of NF-κB pathway in the development of GVHD in host parenchymal tissues. In an initial experiment we utilized a Luminex transcription factor (TF) multiplex assay to screen for the activation of various transcription factors during GVHD in target organs. DNA binding activity of various TF was assayed using nuclear extracts from liver tissue obtained from animals with GVHD, syngeneic controls or normal B6 mice. DNA binding activity of NF-κB, AP2 and EGR was identified to be significantly upregulated in liver samples of animals with GVHD compared to syngeneic controls and normal B6 (Figure 5a). To confirm these results and further characterize the NF-κB binding activity, EMSA studies were conducted. We demonstrated that in comparison to syngeneic controls, NF-κB DNA binding activity was increased progressively in the liver and gut of GVHD animals between day +3 and +6 post-BMT (Figure 5b, d). Supershifting studies in the liver on day +6 demonstrated that both p50 and p65 were contributing to the NF-κB complex in GVHD animals (Figure 5c). In the colon (Figure 5d) we observed an increase in both p50 homodimers and p50/p65 heterodimers. In liver tissues we noted that p50 homodimers were the major NF-κB complex on day +3 and p50/p65 heterodimers were dominant on day +6. As shown in Figure 5e, analysis of p65-phosphorylation in liver samples confirmed prolonged activation of NF-κB on day +9.
Figure 5. NF-kB activation in GVHD target organs.
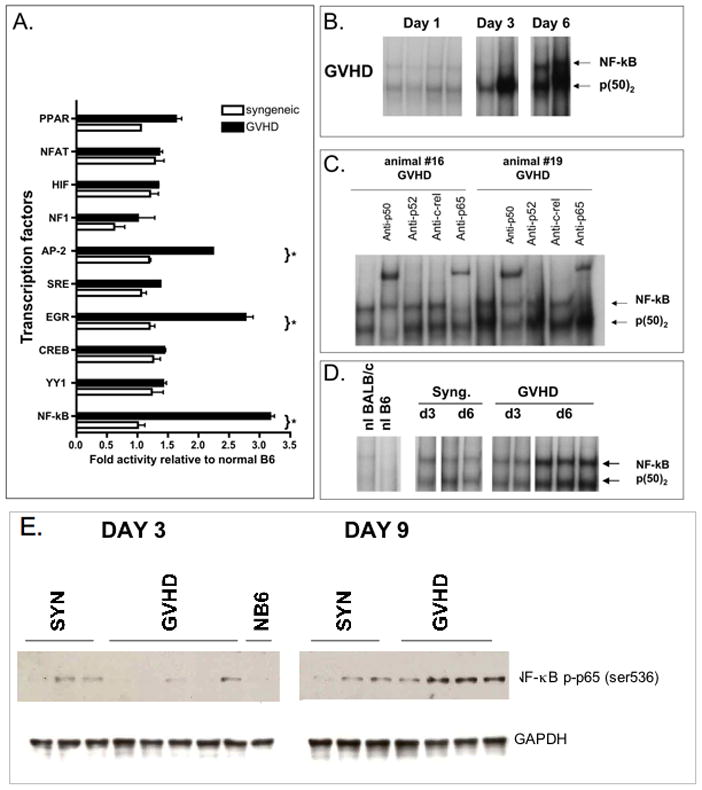
A. Transcription factor profiling of liver tissue. Nuclear extracts were prepared as described and subjected to transcription multiplex analysis. DNA binding activity was normalized to unstimulated normal liver.
B. Electrophoretic mobility shift assays were performed on nuclear extracts from liver tissue samples obtained from GVHD animals.
C. Supershift assays were conducted to identify the NF-kB subunit composition in liver samples. Antibodies against c-rel, anti-p65, anti-p50 and anti-p52 were added to the nuclear extracts. p50 homodimers and p50/p65 heterodimers are the predominant NF-kB complexes during induction of GVHD.
D. Electrophoretic mobility shift assays were performed on nuclear extracts from colon tissue samples obtained from GVHD animals or syngeneic controls. Data shown in A–D are representative of three independent experiments performed.
E. Total cell lysates were prepared from livers of animals with GVHD (n=4) or from syngeneic controls (n=3) on days +3 and +9 post-BMT. Blots were probed with antibodies against phospho-p65 and protein loading was controlled using a GAPDH-specific antibody. One of two similar experiments is shown.
3.7 Evidence for STAT1-dependent gene activation in the liver
To further substantiate the relevance of the STAT1 pathway in GVHD target organs, we performed gene expression profiling studies in livers from animals with GVHD or syngeneic controls on day +3 post-BMT. Pooled RNA from three mice per group was studied. As shown in Figure 6a, GVHD resulted in a significant upregulation of a number of IFN-γ-dependent transcripts. Most prominently, STAT1 and STAT1-dependent transcripts were upregulated (IP-10 [CXCL10], Mig [CXCL9], IRF-1, SOCS1 etc). Confirming our previously published results, STAT1 was the most significantly upregulated gene. The data from our microarray were confirmed by quantitative Real Time-PCR for IRF-1 and SOCS1 (Figure 6b).
Figure 6. STAT1 and STAT3-dependent gene activation.
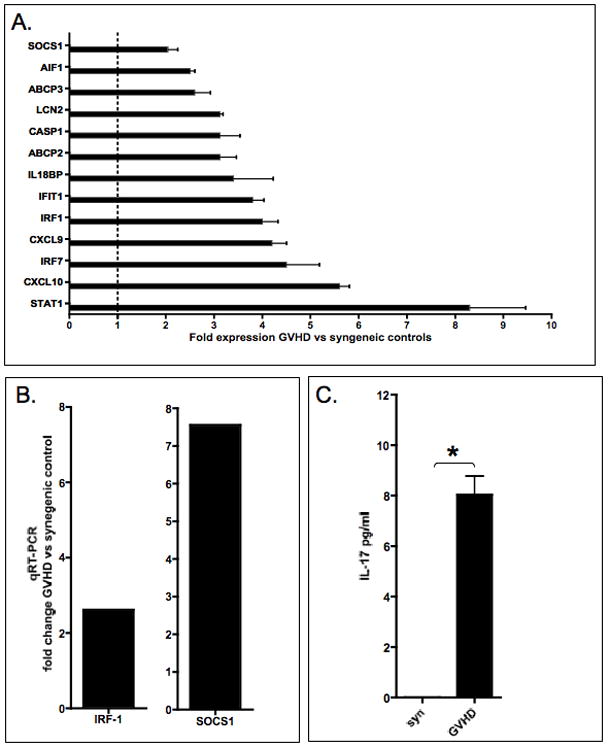
A. IFN-gamma-induced signaling profile in liver samples
To validate activation of STAT1 gene expression program in GVHD target organs, total RNA was extracted from livers of GVHD animals or syngeneic controls (three animals per group) on day +3 post-BMT, pooled and analyzed by gene expression profiling. Gene expression profiles from GVHD animals were compared to syngeneic controls. Two-fold change in expression was considered significant. Each gene was spotted 4 times on the array. SD were calculated based on the mean hybridization (= expression level) signal of the four spots.
B. Expression of STAT1 dependent genes IRF1 and SOCS1 were studied by q-RT-PCR in liver samples from animals with GVHD or syngeneic controls. Significant GVHD-induced upregulation of IRF1 and SOCS1 was detectable.
C. Activation of STAT3 dependent gene expression in vivo was studied by correlating STAT3 activation with IL-17 secretion during induction of GVHD. On day +3 post-BMT animals were sacrificed, serum were collected. Sera from animals with GVHD (n=6) and syngeneic controls (n=3) were studied with an IL-17-specific ELISA. Bars represent the mean ± Standard Error of Mean (SEM). Statistical significance was determined by Mann Whitney test. (p<0.001)
3.8 STAT3–dependent Th17 activation during GVHD
In addition to ROR-γt, STAT3 is one of the key transcriptional regulators of the Th17 pathway. Given the significant expression of STAT3 in splenic T cells of GVHD animals (Figure 1b) and to support the notion that activation of STAT3 is important for the induction of Th17 cells during GVHD, we studied expression of IL-17 in the serum of animals with GVHD or syngeneic controls. IL-17, which was below the detection limit in syngeneic controls, was upregulated in the serum of GVHD animals on day +3 and +9 (Figure 6c). These results support the concept of STAT3 inflammatory pathway activation during the early course of GVHD and thus leading to Th17 pathways differentiation.
4. DISCUSSION
The goal of this study was to gain a better understanding of STAT1 and STAT3 activation, and their temporal and spatial sequence during the initiation phase of GVHD in target organs and in secondary lymphoid organs. In addition we focused on the MAPK and NF-kB pathway. Apart from a transient activation of ERK1,2 in spleen cells on day+1, activation of the MAPK pathway did not appear to be GVHD specific. We did not detect activation of JNK. Activation of p38 was observed in a non-GVHD specific manner in total spleen cells and splenic T cells (also in GVHD target organs, data not shown). The relevance of the GVHD-specific transient ERK1,2 activation observed on day+1 in the spleen, but not in parenchymal target organs requires further evaluation. There is currently no functional data addressing the role of ERK1,2 in GVHD. Furthermore, detailed studies are warranted to determine splenic cell type activated during GVHD. As we did not detect ERK1,2 activation in our splenic T cell lysates, it is possible that other accessory cells monocytes or dendritic cells may demonstrate ERK1,2 activation during GVHD. Furthermore, it is possible that sensitivity of our western blot studies was not sufficient to detect low levels of ERK1,2 activation in the splenic T cell lysates as recent studies from Lu et al show evidence for ERK1,2 activation in CD4 T cells using phosphoprotein array studies [19].
Activation of NF-kB was detectable by different methods in both spleen cells and also GVHD target organs. Splenic NF-kB activation was very short-lived following GVHD induction. In contrast, progressive activation of NF-kB was detectable in the GVHD target organs liver and colon. Furthermore, a tissue specific subset composition was detectable. These results have several implications: it was recently reported that proteasome inhibitors which affect the NF-kB pathway can either abrogate or accelerate GVHD depending on the timing of administration [11, 12]. Based on our results it could be speculated that early inhibition of NF-kB may reduce GVHD by affecting primarily the hematopoietic compartment with subsequent inhibition of donor T cell expansion or host APC maturation. Conversely, delayed inhibition of NF-kB may interfere with tissue regeneration or promotion of inflammation leading to worsening of GVHD. Indeed, it has been shown that intestinal epithelial-cell-specific inhibition of NF-kB caused severe chronic intestinal inflammation in mice. NF-kB deficiency led to apoptosis of colonic epithelial cells, impaired expression of antimicrobial peptides and enhanced translocation of bacteria into the mucosa[20]. It is therefore not surprising that NF-kB inhibitors may have opposing effects on GVHD. Furthermore, NF-kB subset composition may determine its function in different GVHD target tissues. Thus, it has been shown that p50 homodimers may have anti-inflammatory properties by inducing IL-10 expression or by inhibiting production of pro-inflammatory cytokines [21].
In line with our previously reported findings, STAT1 activation was an early and highly specific event during the induction of GVHD in total spleen cells, splenic T cells and GVHD epithelial target organs and correlated with expansion of pSTAT1 expressing CD4+ and CD8+ T cells. Based on our results, activation of STAT1 in the spleen preceded its activation in the GVHD target organs liver and colon. Whereas conditioning-induced STAT1 activation was only transient in syngeneic controls, GVHD animals showed progressive activation of STAT1 of the gut and in the liver as already demonstrated [7]. Since pSTAT1 expression was dramatically downregulated at the late phases of GVHD in target tissues, this implies that expression of pSTAT1 is a GVHD-specific event, which correlates primarily with the early cytokine storm. The relevance of the persistently increased expression of unphosphorylated STAT1 is currently unclear. However, unphosphorylated STAT1 has been demonstrated to be transcriptionally active [22, 23] and it is therefore conceivable that the persistently elevated levels of unphosphorylated STAT1 could further contribute to GVHD-associated tissue damage even in the absence of continuing IFN-γ signalling.
In agreement with these results we were able to detect STAT1 dependent gene-expression programs in the GVHD target organ at early stage as determined by gene array studies and confirmed by quantitative RT-PCR. This data corroborates our previous findings on systemic and local expression of CXCR3 Ligands IP-10 (CXCL10), Mig(CXCL9), ITAC (CXCL11) - major STAT1-dependent chemokine [24] and underscore the notion that GVHD in the gut is a Th1-driven process [25]. This concept is further supported by recent results demonstrating that absence of host IFN-γ-R leads to reduced GI damage [26]. Based on the strong activation of STAT1 in GVHD target organs (liver and GI) and donor T cells, blocking STAT1 may provide a novel attractive approach for inhibition of GVHD. Further functional studies will be necessary to address how host versus donor STAT1 affects GVHD development.
STAT3 was also identified as another GVHD-specific signaling pathway in GVHD target organs and also secondary lymphoid tissue. STAT3 activation in the spleen correlated with high levels of the STAT3-activating cytokines IL-6 and also IL-10. The marked change in IL-6/IL-10 ratio (Figure 1A) during the development of GVHD could suggest that STAT3 may act as a promoter of inflammation during the early priming and induction phase of GVHD (high IL-6/IL-10 ratio)and may mediate anti-inflammatory signals at later time points (low IL-6/IL-10 ratio). Studies are ongoing for a more detailed characterization of cell-type specific STAT3 activation during GVHD in spleen cells. Recent results from Lu et al. also provide evidence for alloactivation-induced STAT3 phosphorylation in CD4 T cells [19]. Furthermore, ex vivo inhibition of STAT3 in donor T cells prior to transplantation using small molecule inhibitors appeared to attenuate GVHD. However, given the fact that the inhibitors utilized in that study (e.g. Cucurbitacin I) have significant off-target effects which can also be observed in the absence of STAT3 ([27] and H.H.Ma and M.Y. Mapara manuscript in preparation), it is still not quite clear which functional role donor STAT3 plays during acute GVHD. In contrast, using conditional STAT3 knockout mice as donors Radojcic et al were able to demonstrate that STAT3 is critical for CD4-driven chronic GVHD [28]. In summary these studies and our data suggest that blocking STAT3 may provide a novel approach for inhibition of GVHD. In contrast to STAT1, we observed persistent expression of pSTAT3 in GVHD liver samples even at the late phase, suggesting ongoing transcriptional STAT3 activity in the GVHD target tissues. Further studies are warranted to determine the cellular source of this persistent STAT3 activation observed by western blot (e.g. presence of infiltrating inflammatory donor cells vs. STAT3 activation in host hepatocytes or Kupffer cells).
The notion that early STAT3 activation may reflect a proinflammatory response is further supported by the finding that early activation of STAT3 in splenic T cells was accompanied by systemic secretion of IL-17 only in GVHD animals, but not in syngeneic controls. Th17 cells and CD4 memory T cells are the primary source of IL-17 [29, 30]. Th17 cells have been recently recognized as mediators of autoimmunity, inflammation, acute and chronic GVHD [31–33] [34]. However, the precise role of Th17 in GVHD is still to be clarified [31, 34, 35]. Differentiation of Th17 cells requires functional STAT3 and the orphan nuclear receptor ROR-γt[36–39]. We postulate that STAT1 and STAT3 may have a critical role in controlling the host tissue inflammatory response, shaping the donor T cell response and mediating host tissue damage. Further studies will be necessary to functionally dissect the role of STAT1 and STAT3 and their crosstalk in development of GVHD.
Supplementary Material
Recipients were lethally irradiated with 10.75 Gy and reconstituted within 4 hours with a single intravenous inoculum of 5×06 allogeneic or syngeneic bone marrow cells. GVHD was induced by co-injection of allogeneic spleen cells (4 × 107). A. Mice in the GVHD group had a median survival time [MST] of 8 days (log rank test p=0.018) compared to the syngeneic control animals which mremained healthy. Clinical GVHD scores (B) and weight change (C) were assessed everyday following BMT. Clinical GVHD scores increased significantly in the GVHD group after day+2, reaching a first peak on day+7 (4.6±1.4). After a brief recovery period GVHD scores started to increase again from 2.1±0.4 on day+14 to 6.8±1.7 on day+26 (*p<0.001 at indicated time points). Weight loss was also significantly increased in the GVHD group compared to syngeneic control (#p<0.05 and * p<0.01 at indicated time points). Representative results of one out of 5 independent experiments are shown (n=4/group)
{kind=link}
Splenocytes GVHD or control animals were harvested on day +3 and +6 post-BMT. Phosphorylation status of STAT1 (Tyr701) and STAT3 (Tyr705) in donor CD4+ and CD8+ T cells was assessed using phosphotyrosine-specific antibodies using flow cytometry. Results are representative of 3 similar experiments.
{kind=link}
Liver samples were harvested on day+13 and day+26 post-BMT following lethal irradiation and BMT using the BALB/c to B6 strain combination. Protein was extracted and analyzed by western blotting for STAT1, p-STAT1 (Tyr701), STAT3 and pSTAT3 (Tyr705) expression, GAPDH or β-actin was used as loading control. Data are representative of one out of two independent experiments.
{kind=link}
Acknowledgments
M.Y.M. is a Hillman Fellow for Innovative Cancer Research. This work was supported by NHLBI RO1HL093716 and the Pittsburgh Foundation (M.Y.M). We thank Betsy Ryce-Platt for secretarial assistance and Drs. Richard Steinman, Michael Lotze and Elizabeth Stenger for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, Baker EM, Cao YA, Contag CH, Negrin RS. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. 2005;106:1113–1122. doi: 10.1182/blood-2005-02-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS. Leukocyte migration and graft-versus-host disease. Blood. 2005;105:4191–4199. doi: 10.1182/blood-2004-12-4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mapara MY, Kim YM, Marx J, Sykes M. Donor lymphocyte infusion-mediated graft-versus-leukemia effects in mixed chimeras established with a nonmyeloablative conditioning regimen: extinction of graft-versus-leukemia effects after conversion to full donor chimerism. Transplantation. 2003;76:297–305. doi: 10.1097/01.TP.0000072014.83469.2D. [DOI] [PubMed] [Google Scholar]
- 4.Mapara MY, Kim YM, Wang SP, Bronson R, Sachs DH, Sykes M. Donor lymphocyte infusions mediate superior graft-versus-leukemia effects in mixed compared to fully allogeneic chimeras: a critical role for host antigen-presenting cells. Blood. 2002;100:1903–1909. doi: 10.1182/blood-2002-01-0023. [DOI] [PubMed] [Google Scholar]
- 5.Billiau AD, Fevery S, Rutgeerts O, Landuyt W, Waer M. Crucial role of timing of donor lymphocyte infusion in generating dissociated graft-versus-host and graft-versus-leukemia responses in mice receiving allogeneic bone marrow transplants. Blood. 2002;100:1894–1902. doi: 10.1182/blood-2002-02-0419. [DOI] [PubMed] [Google Scholar]
- 6.Xia G, Truitt RL, Johnson BD. Graft-versus-Leukemia and Graft-versus-Host Reactions after Donor Lymphocyte Infusion Are Initiated by Host-Type Antigen-Presenting Cells and Regulated by Regulatory T Cells in Early and Long-Term Chimeras. Biol Blood Marrow Transplant. 2006;12:397–407. doi: 10.1016/j.bbmt.2005.11.519. [DOI] [PubMed] [Google Scholar]
- 7.Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S, Mapara MY. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006;34:776–787. doi: 10.1016/j.exphem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, Ferrara JL. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101:3921–3926. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, Maeda Y, Tawara I, Krijanovski O, Gatza E, Liu C, Malter C, Mascagni P, Dinarello CA, Ferrara JL. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008;118:2562–2573. doi: 10.1172/JCI34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Y, Chin YE, Weisiger E, Malter C, Tawara I, Toubai T, Gatza E, Mascagni P, Dinarello CA, Reddy P. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. 2009;182:5899–5903. doi: 10.4049/jimmunol.0804388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun K, Wilkins DE, Anver MR, Sayers TJ, Panoskaltsis-Mortari A, Blazar BR, Welniak LA, Murphy WJ. Differential effects of proteasome inhibition by bortezomib on murine acute graft-versus-host disease (GVHD): delayed administration of bortezomib results in increased GVHD-dependent gastrointestinal toxicity. Blood. 2005;106:3293–3299. doi: 10.1182/blood-2004-11-4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun K, Welniak LA, Panoskaltsis-Mortari A, O’Shaughnessy MJ, Liu H, Barao I, Riordan W, Sitcheran R, Wysocki C, Serody JS, Blazar BR, Sayers TJ, Murphy WJ. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci U S A. 2004;101:8120–8125. doi: 10.1073/pnas.0401563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vodanovic-Jankovic S, Hari P, Jacobs P, Komorowski R, Drobyski WR. NF-kappaB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood. 2006;107:827–834. doi: 10.1182/blood-2005-05-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugerman PB, Faber SB, Willis LM, Petrovic A, Murphy GF, Pappo J, Silberstein D, van den Brink MR. Kinetics of gene expression in murine cutaneous graft-versus-host disease. Am J Pathol. 2004;164:2189–2202. doi: 10.1016/S0002-9440(10)63776-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichiba T, Teshima T, Kuick R, Misek DE, Liu C, Takada Y, Maeda Y, Reddy P, Williams DL, Hanash SM, Ferrara JL. Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood. 2003;102:763–771. doi: 10.1182/blood-2002-09-2748. [DOI] [PubMed] [Google Scholar]
- 16.Lentzsch S, Gries M, Janz M, Bargou R, Dorken B, Mapara MY. Macrophage inflammatory protein 1-alpha (MIP-1 alpha ) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood. 2003;101:3568–3573. doi: 10.1182/blood-2002-08-2383. [DOI] [PubMed] [Google Scholar]
- 17.Hill GR, Cooke KR, Teshima T, Crawford JM, Keith JC, Jr, Brinson YS, Bungard D, Ferrara JL. Interleukin-11 promotes T cell polarization and prevents acute graft-versus-host disease after allogeneic bone marrow transplantation. J Clin Invest. 1998;102:115–123. doi: 10.1172/JCI3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathas S, Rickers A, Bommert K, Dorken B, Mapara MY. Anti-CD20- and B-cell receptor-mediated apoptosis: evidence for shared intracellular signaling pathways. Cancer Res. 2000;60:7170–7176. [PubMed] [Google Scholar]
- 19.Lu SX, Alpdogan O, Lin J, Balderas R, Campos-Gonzalez R, Wang X, Gao GJ, Suh D, King C, Chow M, Smith OM, Hubbard VM, Bautista JL, Cabrera-Perez J, Zakrzewski JL, Kochman AA, Chow A, Altan-Bonnet G, van den Brink MR. STAT-3 and ERK 1/2 phosphorylation are critical for T-cell alloactivation and graft-versus-host disease. Blood. 2008;112:5254–5258. doi: 10.1182/blood-2008-03-147322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, Gumucio D, Neurath MF, Pasparakis M. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 21.Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–26050. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111–4122. doi: 10.1093/emboj/19.15.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer T, Gavenis K, Vinkemeier U. Cell type-specific and tyrosine phosphorylation-independent nuclear presence of STAT1 and STAT3. Exp Cell Res. 2002;272:45–55. doi: 10.1006/excr.2001.5405. [DOI] [PubMed] [Google Scholar]
- 24.Mapara MY, Leng C, Kim YM, Bronson R, Lokshin A, Luster A, Sykes M. Expression of chemokines in GVHD target organs is influenced by conditioning and genetic factors and amplified by GVHR. Biol Blood Marrow Transplant. 2006;12:623–634. doi: 10.1016/j.bbmt.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 cells both contribute to acute GVHD, and each subset has distinct end-organ targets. J Clin Invest. 2000;105:1289–1293. doi: 10.1172/JCI7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burman AC, Banovic T, Kuns RD, Clouston AD, Stanley AC, Morris ES, Rowe V, Bofinger H, Skoczylas R, Raffelt N, Fahy O, McColl SR, Engwerda CR, McDonald KP, Hill GR. IFNgamma differentially controls the development of idiopathic pneumonia syndrome and GVHD of the gastrointestinal tract. Blood. 2007;110:1064–1072. doi: 10.1182/blood-2006-12-063982. [DOI] [PubMed] [Google Scholar]
- 27.Graness A, Poli V, Goppelt-Struebe M. STAT3-independent inhibition of lysophosphatidic acid-mediated upregulation of connective tissue growth factor (CTGF) by cucurbitacin I. Biochem Pharmacol. 2006;72:32–41. doi: 10.1016/j.bcp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Radojcic V, Pletneva MA, Yen HR, Ivcevic S, Panoskaltsis-Mortari A, Gilliam AC, Drake CG, Blazar BR, Luznik L. STAT3 signaling in CD4+ T cells is critical for the pathogenesis of chronic sclerodermatous graft-versus-host disease in a murine model. J Immunol. 2010;184:764–774. doi: 10.4049/jimmunol.0903006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Lohr J, Knoechel B, Wang JJ, Villarino AV, Abbas AK. Role of IL-17 and regulatory T lymphocytes in a systemic autoimmune disease. J Exp Med. 2006;203:2785–2791. doi: 10.1084/jem.20061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. 2009;113:1365–1374. doi: 10.1182/blood-2008-06-162420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM, Ligh C, Holland AM, Grubin J, Mark NM, Liu C, Iwakura Y, Heller G, van den Brink MR. IL-17 contributes to CD4-mediated graft-versus-host disease. Blood. 2009;113:945–952. doi: 10.1182/blood-2008-08-172155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yi T, Zhao D, Lin CL, Zhang C, Chen Y, Todorov I, LeBon T, Kandeel F, Forman S, Zeng D. Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood. 2008;112:2101–2110. doi: 10.1182/blood-2007-12-126987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Vodanovic-Jankovic S, Johnson B, Keller M, Komorowski R, Drobyski WR. Absence of regulatory T-cell control of TH1 and TH17 cells is responsible for the autoimmune-mediated pathology in chronic graft-versus-host disease. Blood. 2007;110:3804–3813. doi: 10.1182/blood-2007-05-091074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iclozan C, Yu Y, Liu C, Liang Y, Yi T, Anasetti C, Yu XZ. T helper17 cells are sufficient but not necessary to induce acute graft-versus-host disease. Biol Blood Marrow Transplant. 2010;16:170–178. doi: 10.1016/j.bbmt.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 37.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG. Cutting Edge: An In Vivo Requirement for STAT3 Signaling in TH17 Development and TH17-Dependent Autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 38.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007 doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ivanov, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Recipients were lethally irradiated with 10.75 Gy and reconstituted within 4 hours with a single intravenous inoculum of 5×06 allogeneic or syngeneic bone marrow cells. GVHD was induced by co-injection of allogeneic spleen cells (4 × 107). A. Mice in the GVHD group had a median survival time [MST] of 8 days (log rank test p=0.018) compared to the syngeneic control animals which mremained healthy. Clinical GVHD scores (B) and weight change (C) were assessed everyday following BMT. Clinical GVHD scores increased significantly in the GVHD group after day+2, reaching a first peak on day+7 (4.6±1.4). After a brief recovery period GVHD scores started to increase again from 2.1±0.4 on day+14 to 6.8±1.7 on day+26 (*p<0.001 at indicated time points). Weight loss was also significantly increased in the GVHD group compared to syngeneic control (#p<0.05 and * p<0.01 at indicated time points). Representative results of one out of 5 independent experiments are shown (n=4/group)
Splenocytes GVHD or control animals were harvested on day +3 and +6 post-BMT. Phosphorylation status of STAT1 (Tyr701) and STAT3 (Tyr705) in donor CD4+ and CD8+ T cells was assessed using phosphotyrosine-specific antibodies using flow cytometry. Results are representative of 3 similar experiments.
Liver samples were harvested on day+13 and day+26 post-BMT following lethal irradiation and BMT using the BALB/c to B6 strain combination. Protein was extracted and analyzed by western blotting for STAT1, p-STAT1 (Tyr701), STAT3 and pSTAT3 (Tyr705) expression, GAPDH or β-actin was used as loading control. Data are representative of one out of two independent experiments.