

Abstract
The potential of wild-type and mutant glycosyltransferases to produce glycoconjugates carrying sugar moieties with chemical handles has made it possible to conjugate biomolecules with orthogonal reacting groups at specific sites. The synthesis of UDP-2-(2-ketopropyl)galactose has been previously carried out, albeit with difficulty and low efficiency. A modified approach has been developed for the synthesis of UDP-2-(2-ketopropyl)glucose and UDP-2-(2-ketopropyl)galactose, allowing better access to the desired test compounds, the UDP-2-(2-ketopropyl)glucose and UDP-2-(2-ketopropyl)galactose analogs were synthesized in 8 steps and 4.8% and 5.3% overall yield respectively, an improvement over the 1st generation synthesis involving 8 steps and an overall yield of 0.7%.
Keywords: Carbohydrate chemistry, Organic synthesis, Modified sugars, Glycosyltransferases
1. Introduction
Structural information on glycosyltransferases has revealed that the specificity of these enzymes can be broadened to include modified sugars with chemical handles that can then be utilized for site-specific conjugation chemistry. As such, this approach represents a powerful general method for the site-specific modification of glycoproteins. Substitution of Tyr289 to Leu in the catalytic pocket of bovine β-1,4-galactosyltransferase generates a novel glycosyltransferase which can transfer not only galactose, but also modified galactosyl moieties such as GalNAc1 and C2-modified galactose containing various chemical handles, from the corresponding uridine diphosphate (UDP) derivatives.2,3 Similarly, the wild-type N-acetylgalactosaminyltransferase, which naturally transfers GalNAc from UDP-GalNAc, can also transfer C2-modified galactose units from their UDP-derivative to the Ser/Thr residue of a polypeptide acceptor substrate tagged as a fusion peptide to a non-glycoprotein.4 The potential of wild-type and mutant glycosyltransferases to produce glycoconjugates carrying sugar moieties with chemical handles has made it possible to conjugate biomolecules with orthogonal reacting groups at specific sites.5 This methodology has enabled the assembly of novel bio-nanoparticles useful for targeted drug-delivery and as MRI contrast agents.6,7
The synthesis of UDP-2-(2-ketopropyl)glucose (Figure 1. 8-Glc) has not been previously reported, while the synthesis of UDP-2-(2-ketopropyl)galactose (Figure 1. 8-Gal) was reported in low yields. The lack of efficient syntheses of these substrates represents a roadblock to the continuing development of this promising technology, as both are required for the site-specific conjugation of biomolecules via glycan residues using glycosyltransferases. The linking technique enables conjugation of the glycoprotein-appended 2-(2-ketopropyl)sugar to aminooxy derivatives such as aminooxybiotin and has wide potential for future conjugation of cytotoxic drugs, cytokines, radionuclides for imaging and radioimmunotherapy, and lipids for the assembly of liposomes for targeted drug delivery.6,7
Figure 1.
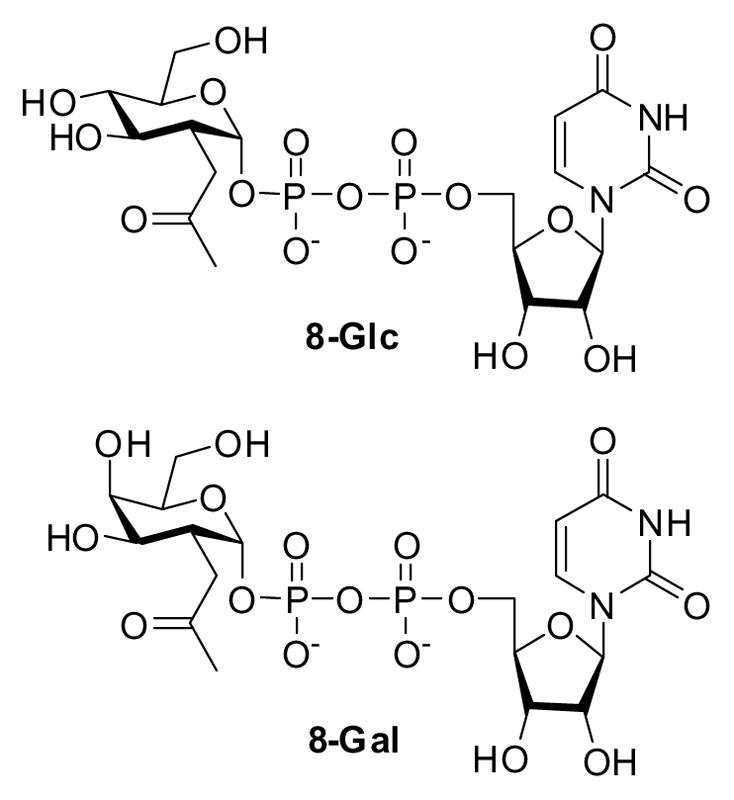
UDP-2-(2-ketopropyl)glucose 8-Glc and UDP-2-(2-ketopropyl)galactose 8-Gal
2. Results and discussion
“2-(2-Ketopropyl)sugars”, which are C2-carbon isosteres of 2-N-acetamidosugars, were first introduced by Bertozzi8 as novel analogues possessing a ketone group for chemoselective reaction with aminooxy- or hydrazido-reagents. The 1st generation synthesis of UDP-2-(2-ketopropyl)galactose (8-Gal) was carried out by Hsieh-Wilson’s group2, albeit with difficulty and low efficiency. The modified approach for the synthesis of UDP-2-(2-ketopropyl)glucose (8-Glc) and UDP-2-(2-ketopropyl)galactose (8-Gal), developed at the Imaging Probe Development Center is displayed in Scheme 1.
Scheme 1.

Reagents and conditions. (a) N-Iodosuccinimide (1.5 eq.), AcOH (2 eq.) CH2Cl2, R T, 45 min. (b) Methallyltributylstannane (2 eq.), AIBN (0.15 eq), Benzene, Reflux, 3h. (c) Hydrazine acetate (1.2 eq.), DMF, RT, 2hr. (d) 1-H -tetrazole (4 eq.), dibenzyl N,N -diisopropylphosphoramidite (2.5 eq.), CH2Cl2, −30 °C to RT 3 hr. (e) i) Ozone, CH2Cl2-CH3OH, −78 °C, 40 min. Then ii) Me2S (excess), −78 °C to RT overnight. (f) 1) 10% Pd/C (10 wt.%), trioctylamine (0.6 eq.), CH3OH, H2, 24hr. 2) UMP-morpholidate 4-morpholine-N,N′-dicyclohexylcarboxamide salt (1.5 eq.), 1-H -tetrazole (4 eq.), pyridine, 4A MS, 3 days. (g) CH3OH, H2O, Et3N (4:2:1), 24hr.
Thus, from commercially available tri-O-acetyl-D-glucal (1-Glc) or tri-O-acetyl-D-galactal (1-Gal), equatorial iodination was effected with N-iodosuccinimide (NIS) and acetic acid in dry dichloromethane to afford equatorial iodosugars 2-Glc and 2-Gal in 25% and 35% yield respectively, with an α:β ratio of ~ 1:10 for the Glc series and ~ 1:2 for the Gal series. Keck radical allylation9 with methallyltributylstannane and catalytic AIBN (2,2′-Azobis(2-methylpropionitrile)) afforded the desired equatorial methallyl compound 3-Glc and 3-Gal in 70% and 68% yield respectively, along with <5% of the axial epimer, separable chromatographically.
Anomeric deprotection was effected with hydrazine acetate in DMF, obtaining lactol 4-Glc and 4-Gal, which was coupled with dibenzyl N,N-diisopropylphosphoramidite at −30 °C. Compounds 5-Glc and 5-Gal were not isolated. After a quick workup, 5-Glc and 5-Gal were subjected to ozonolysis at −78 °C followed by addition of dimethyl sulfide to obtain α-D-glucopyranoside phosphate 6-Glc in 68% yield from 4-Glc, and α-D-glucopyranoside phosphate 6-Glc in 60% yield from 4-Gal.
Hydrogenolysis of the benzyl groups in 6-Glc and 6-Gal, followed by condensation with UMP-morpholidate and deprotection of the acetyl groups afforded the target UDP-2-(2-ketopropyl)glucose (8-Glc) in 49% yield from 6-Glc, and UDP-2-(2-ketopropyl)galactose (8-Gal) in 53% yield from 6-Gal.
A major obstacle was presented by the sequence illustrated in Scheme 2, employed in the 1st generation synthesis.2 The sequence was troubled by low-yielding transformations and unwanted byproduct formation due to the instability of the intermediates, along with a need to perform two oxidation steps.
Scheme 2.

Problematic step and unwanted side-product generation via Aldol condensation.
The yield reported in the 1st generation synthesis from intermediate 3-Gal to intermediate 6-Gal was of only 26%. These shortcomings would prove detrimental if the synthesis was to be scaled up eventually, as requested. The new approach shown in Scheme 1 applies redox economy in organic synthesis,10 and illustrates how unwanted byproduct formation (Aldol condensation) was avoided by performing just one combined oxidation step11 after the phosphoramidite coupling had taken place (4 to 5 to 6 in Scheme 1). This aided in scale up and afforded increased yields for key intermediates 6-Gal and 6-Glc with the yields from intermediate 3 to intermediate 6 now 42% in the galactose series and 54% in the glucose series (Scheme 1).
3. Conclusion
The modified approach developed for the synthesis of UDP-2-(2-ketopropyl)glucose and UDP-2-(2-ketopropyl)galactose, herein described in full experimental detail, has allowed much improved access to the desired test substrates. The UDP-2-(2-ketopropyl)glucose analog 8-Glc was synthesized in 8 steps and 4.8% overall yield, and the UDP-2-(2-ketopropyl)galactose analog 8-Gal was obtained in the same number of steps with and overall yield of 5.3%. With the latter previously described compound a 7.5-fold yield improvement was obtained over the 1st generation synthesis,2 which also involved 8 steps but resulted in an overall yield of only 0.7%.
4. Experimental section
4.1 General
All commercially available organic precursors and dry solvents were obtained from Sigma-Aldrich, and used as received unless otherwise noted. Reactions were magnetically stirred under an argon atmosphere and monitored by thin layer chromatography (TLC) with 0.25 mm Sigma-Aldrich pre-coated aluminum-backed silica gel plates with fluorescent indicator. TLC visualization was achieved using 254 nm or 360 nm UV lamp detection and/or staining with cerium molybdate (Hannesian’s stain), phosphomolybdic acid (PMA), or potassium permanganate. Flash column chromatography was performed on an AnaLogix IntelliFlash 280 system, using Biotage® SNAP Cartridges and SNAP Samplet Cartridges with KP-Silica 60 μm. Analytical HPLC analyses were performed on an Agilent 1200 Series instrument equipped with multi-wavelength detectors using a Zorbax Stable Bond C-18 column (4.6 × 50 mm, 3.5 μm) with a flow rate of 0.5 mL/min or 1.0 mL/min. Solvent A was 0.05% trifluoroacetic acid (TFA) in water (H2O), solvent B was 0.05% TFA in acetonitrile (ACN), and a linear gradient of 5% B to 95% B over 10 minutes was used. ESI or APCI mass spectrometry (MS) were performed on an LC/MSD TrapXCl Agilent Technologies instrument or on a 6130 Quadrupole LC/MS Agilent Technologies instrument equipped with a diode array detector. High resolution mass spectrometry (HRMS) was performed by The Scripps Research Institute Center for Metabolomics and Mass Spectrometry (La Jolla, CA). 1H, 13C and 31P NMR spectra were recorded on a Varian spectrometer operating at 400 MHz, 100 MHz and 162 MHz, respectively. Chemical shifts are reported relative to either chloroform (δ 7.26), dimethyl sulfoxide (δ 2.50), acetone (δ 2.05) or deuterium oxide (δ 4.79) for 1H NMR and chloroform (δ 77.0), dimethyl sulfoxide (δ 39.5) or acetone (δ 29.8) for 13C NMR.
4.1.1. 1,3,4,6-Tetra-O-acetyl-2-deoxy-2-iodo-D-glucopyranose (2-Glc)
N-Iodosuccinimide (NIS) (2.04 g, 9.07 mmol) was added at room temperature to a solution of tri-O-acetyl-D-galactal (1.51 g, 5.55 mmol) and acetic acid (AcOH) (635 μL, 11.1 mmol) in dry dichloromethane (CH2Cl2) (20 mL) in an aluminum foil-wrapped flask. The resulting reaction mixture was allowed to stir in the dark for 45 min, after which TLC [2:1 hexane (Hex): ethyl acetate (EtOAc)] showed completion. The reaction was quenched by addition of a saturated aqueous solution of sodium thiosulfate (Na2S2O3) and allowed to stir for 15 min. The reaction mixture was diluted with CH2Cl2 and washed with Na2S2O3 (1 time), H2O (1 time) and brine, dried over magnesium sulfate (MgSO4) and concentrated. The crude mixture was purified by flash column chromatography on silica gel eluted with a gradient of 3:1→2:1 Hex:EtOAc to afford the unwanted 2-iodomannose isomer (lower Rf) as the major product, and the desired 2-iodoglucose derivative 2-Glc (higher Rf) as the minor product (634 mg, 25% yield) as a clear syrup with an α:β anomeric ratio of 1:10. β anomer: 1H NMR (400 MHz, CDCl3) δ 5.85 (d, J = 9.6 Hz, 1H), 5.32 (t, J = 10.0 Hz, 1H), 4.99 (t, J = 10.0 Hz, 1H), 4.30 (dd, J = 12.4, 4.4 Hz, 1H), 4.07 (dd, J = 12.4, 2.0 Hz, 1H), 3.97 (t, J = 11.2 Hz, 1H), 3.86 (ddd, J = 10.0, 4.4, 2.0 Hz, 1H), 2.15 (s, 3H), 2.08 (s, 3H), 2.06 (s, 3H), 2.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 169.7, 169.4, 168.4, 93.8, 75.1, 72.9, 68.4, 61.4, 25.6, 20.6, 20.5; MS (m/z) = 459.7 (M+1)+; HRMS (ESI) calcd for C14H19IO9Na+ [M+Na]+ 480.9966, found 480.9966.
4.1.2. 1,3,4,6-Tetra-O-acetyl-2-deoxy-2-iodo-D-galactopyranose (2-Gal)
Tri-O-acetyl-D-Galactal (2.30 g, 8.45 mmol), NIS (2.28 g, 10.1 mmol), AcOH (1 mL, 16.9 mmol), CH2Cl2 (25 mL) to obtain 2-Gal (1.34 g, 34% yield) as syrup, minor product with higher Rf. β anomer: 1H NMR (400 MHz, CDCl3) δ 5.88 (d, J = 9.2 Hz, 1H), 5.32 (m, 1H), 5.22 (d, J = 4 Hz, 1H), 5.12 (dd, J = 12.0, 3.6 Hz, 1H), 4.33 (t, J = 6.8 Hz, 1H), 4.11 (m, 1H), 4.04 (m, 1H), 2.15 (s, 3H), 2.11 (s, 3H), 2.04 (s, 3H), 2.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 169.8, 169.5, 168.4, 93.3, 73.0, 72.3, 68.8, 61.1, 24.7, 20.5, 19.5; MS (m/z) = 459.1 (M+1)+; HRMS (ESI) calcd for C14H19IO9Na+ [M+Na]+ 480.9966, found 480.9973..
4.1.3. 1,3,4,6-Tetra-O-acetyl-2-deoxy-2-(methallyl)-D-glucopyranose (3-Glc)
A solution of 2-Glc (1.30 g, 2.84 mmol) and methallyltributylstannane9 (1.96 g, 5.67 mmol) in benzene (15 mL) was thoroughly degassed by bubbling argon through it for 20 min. 2,2′-Azobis(2-methylpropionitrile) (AIBN) (70.0 mg, 0.43 mmol) was then added in one portion and degassing continued for another 5 min. The resulting reaction mixture was refluxed for 3 h, after which TLC (2:1 Hex:EtOAc) showed completion. The reaction mixture was allowed to cool to room temperature and concentrated under reduced pressure. The residue was taken up in acetonitrile and washed with hexanes (3 times). The acetonitrile layer was concentrated and the residue purified by flash column chromatography on silica gel using 2:1 Hex:EtOAc to afford 3-Glc as a clear oil (768 mg, 70% yield). β anomer: 1H NMR (400 MHz, CDCl3) δ 5.54 (d, J = 8.8 Hz, 1H), 5.01 (t, J = 6.8 Hz, 1H), 4.67 (s, 1H), 4.62 (s, 1H), 4.31 (dd, J = 12.4, 4.4 Hz, 1H), 4.06 (dd, J = 12.4, 2.4 Hz, 1H), 3.79-3.74 (m, 1H), 2.10-2.07 (m, 10H), 2.00 (s, 3H), 1.98 (s, 3H), 1.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.2, 169.8, 168.9, 142.3, 112.2, 94.3, 72.3, 69.1, 61.9, 42.1, 37.1, 21.6, 20.8, 20.7, 20.6; MS (m/z) = 387.9 (M+1)+; HRMS (ESI) calcd for C18H26O9Na+ [M+Na]+ 409.1469, found 409.1467.
4.1.4. 1,3,4,6-Tetra-O-acetyl-2-deoxy-2-(methallyl)-D-galactopyranose (3-Gal)
2-Gal (2.20 g, 4.80 mmol), methallyltributylstannane8 (4.14 g, 12.0 mmol), AIBN (118 mg, 0.72 mmol), benzene (30 mL) afforded 3-Gal (1.26 g, 68% yield) as a clear oil. β anomer: 1H NMR (400 MHz, CDCl3) δ 5.54 (d, J = 9.2 Hz, 1H), 5.36-5.33 (m, 1H), 5.26 (d, J = 2.4 Hz, 1H), 4.82 (dd, J = 11.6, 3.2 Hz, 1H), 4.66 (s, 1H), 4.59 (s, 1H), 4.16-4.03 (m, 3H), 3.94 (t, J = 6.8 Hz, 1H), 2.12-2.09 (m, 4H), 2.05 (s, 3H), 2.01 (s, 3H), 1.95 (s, 3H), 1.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 169.9, 168.8, 142.6, 113.6, 113.0, 112.1, 93.7, 71.4, 68.9, 67.0, 66.8, 37.9, 36.8, 35.7, 21.8, 20.9, 20.7, 20.3, 19.7; MS (m/z) = 387.5 (M+1)+; HRMS (ESI) calcd for C18H26O9Na+ [M+Na]+ 409.1469, found 409.1475.
4.1.5. 3,4,6-Tri-O-acetyl-2-deoxy-2-(methallyl)-D-glucopyranose (4-Glc)
Hydrazine acetate (102 mg, 1.11 mmol) was added in one portion to a solution of 3-Glc (357 mg, 0.92 mmol) in N,N-dimethylformamide (DMF) (5 mL). The reaction was allowed to stir at room temperature for 2 h. TLC (2:1 Hex:EtOAc) showed completion. The reaction mixture was partitioned between H2O and EtOAc, and the organic layer washed with H2O (3 times), followed by brine, dried over MgSO4 and concentrated under reduced pressure. Product was purified by flash column chromatography on silica gel with 2:1 Hex:EtOAc to afford the lactol 4-Glc as a syrup (265 mg, 83% yield). α anomer: 1H NMR (400 MHz, CDCl3) δ 5.27 (t, J = 10.0 Hz, 1H), 5.15 (d, J = 3.2 Hz, 1H), 4.96 (t, J = 9.6 Hz, 1H), 4.78-4.68 (m, 3H), 4.26-4.21 (m, 1H), 4.17-4.12 (m, 1H), 4.09-4.02 (m, 1H), 3.33 (d, J = 3.6 Hz, 1H), 2.07-2.04 (m, 5H), 2.01 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H), 1.96 (s, 3H), 1.69 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.9, 170.6, 170.1, 142.4, 114.0, 112.5, 93.6, 72.7, 71.3, 70.5, 69.3, 68.5, 67.7, 65.7, 62.8, 42.2, 40.0, 35.9, 23.1, 21.6, 20.6, 19.7; MS (m/z) = 345.8 (M+1)+; HRMS (ESI) calcd for C16H24O8Na+ [M+Na]+ 367.1363, found 367.1368.
4.1.6.3,4,6-Tri-O-acetyl-2-deoxy-2-(methallyl)-D-galactopyranose (4-Gal)
Hydrazine acetate (145 mg, 1.56 mmol), 3-Gal (550 mg, 1.42 mmol), DMF (5 mL) to obtain 4-Gal (343 mg, 70% yield). α anomer: 1H NMR (400 MHz, CDCl3) δ 5.31 (d, J = 2.8 Hz, 1H), 5.23 (t, J = 3.2 Hz, 1H), 5.14 (dd, J = 11.6, 3.2 Hz, 1H), 4.77 (d, J = 10.0 Hz, 2H), 4.41 (t, J = 6.4 Hz, 1H), 4.13-4.11 (m, 1H), 4.07 (t, J = 5.6 Hz, 1H), 2.98 (d, J = 3.6 Hz, 1H), 2.14-2.10 (m, 5H), 2.03 (s, 3H), 1.97 (s, 3H), 1.75-1.69 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.4, 142.2, 112.5, 93.2, 69.5, 67.1, 66.5, 62.4, 36.4, 35.3, 22.6, 22.3, 20.7 (x2); MS (m/z) = 345.6 (M+1)+; HRMS (ESI) calcd for C16H24O8Na+ [M+Na]+ 367.1363, found 367.1369.
4.1.7. Dibenzyl (2-acetonyl-2-deoxy-3,4,5-tri-O-acetyl-α-D-glucopyranosyl) phosphate (6-Glc)
A solution of lactol 4-Glc (250 mg, 0.73 mmol) and 1 H-tetrazole (200 mg, 2.90 mmol) in dry CH2Cl2 (5 mL) was cooled to −30 °C and treated with dibenzyl N,N-diisopropylphosphoramidite (627 mg, 1.81 mmol). The resulting reaction mixture was allowed to slowly warm from −30 °C to room temperature over 3 h. TLC (2:1 Hex:EtOAc) showed complete disappearance of lactol 4-Glc. The reaction mixture was diluted with Et2O and washed with cold brine (2 times), dried over MgSO4 and concentrated under reduced pressure. The residue was then taken up in dry CH2Cl2 (6 mL) and cooled to −78 °C. A stream of ozone was bubbled through the reaction mixture until a light blue color became evident (~30–40 min). Argon was then bubbled through the reaction mixture until the blue color disappeared and Me2S (1 mL) added. The resulting reaction mixture was allowed to stir overnight as the temperature rose to ambient. The solvent was removed under reduced pressure and the residue purified by flash column chromatography: silica gel, 1:1 Hex:EtOAc to afford 6-Glc as a syrup (298 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.37-7.32 (m, 10H), 5.81 (dd, J = 6.4, 3.2 Hz, 1H), 5.37 (t, J = 3.2 Hz, 1H), 5.23 (t, J = 10.2 Hz, 1H), 5.09-4.97 (m, 4H), 4.21 (t, J = 4.4 Hz, 1H), 4.14-4.06 (m, 2H), 4.04-3.99 (m, 1H), 3.88 (dd, J = 12.4, 2.0 Hz, 1H), 3.31 (d, J = 2.4 Hz, 1H), 2.13 (s, 3H), 2.03 (s, 3H), 2.00 (s, 3H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 205.5, 170.5, 170.4, 169.5, 135.3, 128.7 (x2), 128.0, 97.1, 70.5, 69.7 (x2), 69.6, 68.6, 61.6, 39.7, 29.7, 20.6, 20.5; 31P NMR (162 MHz, CDCl3) δ −2.31; MS (m/z) = 607.9 (M+1)+; HRMS (ESI) calcd for C29H35O12P+ [M+H]+ 607.1939, found 607.1930.
4.1.8. Dibenzyl (2-acetonyl-2-deoxy-3,4,5-tri-O-acetyl-α-D-galactopyranosyl) phosphate (6-Gal)
4-Gal (60 mg, 0.17 mmol), 1 H-tetrazole (49 mg, 0.70 mmol), dibenzyl N,N-diisopropylphosphoramidite (150 mg, 0.44 mmol), ozone, Me2S (1 mL), to afford 6-Gal as a syrup (63 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 7.35-7.30 (m, 10H), 5.86 (dd, J = 6.0, 2.8 Hz, 1H), 5.29 (s, 1H), 5.09-5.00 (m, 4H), 4.92 (dd, J = 12.4, 3.2 Hz, 1H), 4.25 (t, J = 6.4 Hz, 1H), 4.08-4.03 (m, 2H), 3.95-3.91 (m, 1H), 2.36-2.34 (m, 2H), 2.10 (s, 3H), 1.97 (s, 3H), 1.92 (s, 3H), 1.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 205.9, 170.2 (x2), 170.0, 135.3, 128.7 (x2), 128.6, 128.0, 97.8, 92.9, 69.6, 69.5 (x2), 68.9, 66.7, 66.1, 65.9, 61.7, 40.8, 39.7, 34.4, 30.3, 29.8, 20.6, 20.4; 31P NMR (162 MHz, CDCl3) δ −2.24; MS (m/z) = 607.3 (M+1)+; HRMS calcd for C29H35O12P+ [M+H]+ 607.1939, found 607.1935.
4.1.9. Uridine 5′-diphospho-2-acetonyl-2-deoxy-α-D-glucopyranose diammonium slat (8-Glc)
10% Pd/C (30 mg) was added to a solution of 6-Glc (290 mg, 0.48 mmol) and trioctylamine (101 mg, 0.29 mmol) in dry CH3OH (4 mL). The resulting reaction mixture was subjected to an H2 atmosphere via a balloon and allowed to stir for 20 h. TLC (1:1 Hex:EtOAc) showed complete disappearance of 6-Glc starting material. The reaction mixture was filtered through Celite™ and concentrated to dryness. The residue was taken up in dry pyridine (3 mL) and UMP-morpholidate 4-morpholine-N,N′-dicyclohexylcarboxamide salt (492 mg, 0.72 mmol) was added along with activated 4Å molecular sieves (~300 mg), followed by addition of 1 H-tetrazole (134 mg, 1.91 mmol) in pyridine (2 mL). The resulting reaction mixture was allowed to stir at room temperature for 3 days and then filtered through Celite™. The filtrate was concentrated under reduced pressure and the residue taken up in CH3OH (4 mL). H2O (2 mL) was added, followed by Et3N (1 mL). The resulting reaction mixture was allowed to stir at room temperature for 24 h then diluted with H2O and extracted with CH2Cl2 (2 times). The aqueous layer was lyophilized and the crude mixture purified by HPLC using a preparative Agilent Zorbax XDB C-18 column, 21.4 × 250 mm, 5 μm, eluted with 100 mM ammonium bicarbonate buffer (NH4HCO3, pH = 7.9) at a rate of 20 mL/min. The desired product eluted at a retention time of 4.1 – 6.2 min to afford 8-Glc (150 mg, 49% yield) as a white powder after lyophilization. 1H NMR (400 MHz, D2O) δ 7.93 (d, J = 8.0 Hz, 1H), 6.00-5.95 (m, 2H), 5.57 (dd, J = 7.2, 3.2 Hz, 1H), 4.40-4.34 (m, 2H), 4.28-4.26 (m, 1H), 4.24-4.16 (m, 2H), 3.91-3.78 (m, 3H), 3.63 (t, J = 9.2 Hz, 1H), 3.46 (t, J = 9.2 Hz, 1H), 2.86 (dd, J = 18.0, 5.2 Hz, 1H), 2.79-2.72 (m, 2H), 2.62 (s, 3H); 13C NMR (100 MHz, D2O) δ 215.5, 167.2, 151.0, 141.9, 103.5, 96.2, 88.5, 84.6, 74.3, 72.1, 69.6, 68.2, 65.2, 63.9, 63.2, 44.5, 40.6, 30.1; 31P NMR (162 MHz, D2O) δ −11.24 (d, J = 18.0 Hz), −12.33 (d, J = 19.2 Hz); MS (m/z) = 605.0 (M+1)+; HRMS (ESI) calcd for C18H26N2O17P2+ [M-H]− 605.0785, found 605.0801.
4.1.10. Uridine 5′-diphospho-2-acetonyl-2-deoxy-α-D-galactopyranose diammonium salt (8-Gal)
6-Gal (60 mg, 99.0 μmol), trioctylamine (21 mg, 59.4 μmol), 10% Pd/C (8 mg), CH3OH (3 mL). Then UMP-morpholidate 4-morpholine-N,N′-dicyclohexylcarboxamide salt (102 mg, 0.15 mmol), 1 H-tetrazole (28 mg, 0.40 mmol), pyridine (3 mL), activated 4Å molecular sieves (~300 mg). Then CH3OH (2 mL), H2O (1 mL), Et3N (0.5 mL). Preparative HPLC conditions same as for 8-Glc, afforded 8-Gal as white powder (34 mg, 53% yield) after lyophilization. 1H NMR (400 MHz, D2O) δ 7.95 (d, J = 8.1 Hz, 1H), 5.97-5.95 (m, 2H), 5.57 (dd, J = 7.5, 3.4 Hz, 1H), 4.35-4.30 (m, 2H), 4.25-4.23 (m, 1H), 4.21-4.16 (m, 2H), 4.11 (t, J = 5.3 Hz, 1H), 3.91-3.88 (m, 1H), 3.78-3.70 (m, 3H), 2.80-2.75 (m, 1H), 2.56 (m, 1H), 2.60 (s, 3H); 13C NMR (100 MHz, D2O) δ 214.5, 167.9, 152.0, 142.1, 104.1, 96.9, 88.4, 84.1, 74.3, 72.1, 69.8, 68.3, 65.2, 64.0, 63.3, 44.6, 40.1, 30.5; 31P NMR (162 MHz, D2O) δ −10.24 (d, J = 19.0 Hz), -12.43 (d, J = 19.9 Hz); MS (m/z) = 605.4 (M+1)+; HRMS (ESI) calcd for C18H26N2O17P2+ [M-H]− 605.0785, found 605.0798.
Supplementary Material
Acknowledgments
This work was supported by the NIH Roadmap for Medical Research Initiative through its establishment of the Imaging Probe Development Center, administered by the National Heart, Lung, and Blood Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Ramakrishnan B, Qasba PK. J Biol Chem. 2002;277:20833–20839. doi: 10.1074/jbc.M111183200. [DOI] [PubMed] [Google Scholar]
- 2.Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. J Am Chem Soc. 2003;125:16162–16163. doi: 10.1021/ja038545r. [DOI] [PubMed] [Google Scholar]
- 3.Boeggeman E, Ramakrishnan B, Kilgore C, Khidekel N, Hsieh-Wilson LC, Simpson JT, Qasba PK. Bioconjugate Chem. 2007;18:806–814. doi: 10.1021/bc060341n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramakrishnan B, Boeggeman E, Qasba PK. Bioconjugate Chem. 2007;18:1912–1918. doi: 10.1021/bc7002346. [DOI] [PubMed] [Google Scholar]
- 5.Pradman KQ, Elizabeth B, Boopathy R. Biotechnol Progr. 2008;24:520–526. [Google Scholar]
- 6.Qasba PK, Ramakrishnan B, Boeggeman E. Am Ass Pharm Soc J. 2006;8:190–195. [Google Scholar]
- 7.Ramakrishnan B, Boeggeman E, Qasba PK. Expert Opin Drug Deliv. 2008;5:149–153. doi: 10.1517/17425247.5.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hang HC, Bertozzi CR. J Am Chem Soc. 2001;123:1242–1243. doi: 10.1021/ja002962b. [DOI] [PubMed] [Google Scholar]
- 9.Keck GE, Enholm EJ, Yates JB, Wiley MR. Tetrahedron. 1985;41:4079–4094. [Google Scholar]
- 10.Burns NZ, Baran PS, Hoffmann RW. Angew Chem Int Ed. 2009;48:2–16. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 11.Campbell RE, Tanner ME. J Org Chem. 1999;64:9487–9492. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.