

Abstract
Although modern proteins consist of 20 different amino acids, it has been proposed that primordial proteins consisted of a small set of amino acids, and additional amino acids have gradually been recruited into the genetic code. This hypothesis has recently been supported by comparative genome sequence analysis, but no direct experimental approach has been reported. Here, we utilized a novel experimental approach to test a hypothesis that native-like globular proteins might be easily simplified by a set of putative primitive amino acids with retention of its structure and function than by a set of putative new amino acids. We performed in vitro selection of a functional SH3 domain as a model from partially randomized libraries with different sets of amino acids using mRNA display. Consequently, a library rich in putative primitive amino acids included a larger number of functional SH3 sequences than a library rich in putative new amino acids. Further, the functional SH3 sequences were enriched from the primitive library slightly earlier than from a randomized library with the full set of amino acids, while the function and structure of the selected SH3 proteins with the primitive alphabet were comparable with those from the 20 amino acid alphabet. Application of this approach to various combinations of codons in protein sequences may be useful not only for clarifying the precise order of the amino acid expansion in the early stages of protein evolution but also for efficiently creating novel functional proteins in the laboratory.
Introduction
Although modern proteins usually consist of 20 different amino acids, it has been proposed that amino acid members in primitive proteins varied during the early stage of protein evolution [1]–[6]. It has been inferred that the primordial genetic code was composed of a smaller set of amino acids because prebiotic synthesis on the primitive earth is thought to have been inadequate for 20 different amino acids [1]. In the coevolution hypothesis, it is proposed that the genetic code coevolved with the amino acid biosynthetic pathways, and additional amino acids were introduced after production through their synthetic pathways [4]. Comparative genome sequence analysis of orthologous proteins in the genomes of bacteria, archaea and eukaryota revealed that the frequencies of Gly, Ala, Glu and Pro in proteins consistently decrease (i.e., primitive amino acids that are assumed to have been the first incorporated into the genetic code), while the frequencies of Ser, His, Cys, Met and Phe increase (i.e., new amino acids that are assumed to have recently been added to the genetic code) over the course of protein evolution [5]. The trend of amino acid gain and loss is in agreement with the likely order of incorporation of amino acids into the genetic code, as deduced from other criteria [3].
Several protein design experiments have proved that the full set of 20 amino acids is not necessarily essential for protein structure and function [7]–[14]. For example, Riddle et al. generated simplified SH3 domains (a small β-sheet protein) from a combinatorial library that was composed of five different amino acids by using a phage display technique [8]. Further, Hecht’s group created four helix bundle proteins with 11 amino acids [7], [12], [13], and Jumawid et al. generated α3β3 de novo proteins with seven amino acids [14]. However, these experiments have attempted to generate simplified proteins with fewer amino acids than the natural proteins, and they have not focused on whether the accepted amino acids are primitive or not. Previously, Babajide et al. demonstrated in silico that native-like folded structures of several tested proteins are maintained with a restricted alphabet mainly containing primitive amino acids (Ala, Gly, Leu and Asp) but were not maintained with a set of nonprimitive amino acids (Gln, Leu and Arg) [15]. To test this hypothesis experimentally, we sought to compare the function and structure of tested proteins with different subsets of amino acids for the first time.
As a first attempt, we designed randomized src SH3 gene libraries in which approximately half the residues of the SH3 gene were replaced by randomized codons in the lower or upper half of the table of the genetic code (Fig. 1). The SH3 domain is one of the most common mediators in intracellular signaling pathways. Because the SH3 domain is a well-known protein and thus the conserved positions that play important roles in structure and function have already been examined [16], we can randomize only non-conserved regions. A subset of amino acids that are coded by the lower half of the genetic code are mainly putative primitive amino acids (e.g., Ala and Gly), whereas a subset of amino acids that are coded by the upper half contains many putative new amino acids (e.g., Cys, Phe, Tyr and Trp).
Figure 1. The universal genetic code.

The average rank represents the chronological order of amino acid addition to the genetic code. The values calculated from ∼60 criteria were Gly, 3.5; Ala, 4.0; Asp, 6.0; Val 6.3; Pro, 7.3; Ser, 7.6; Glu, 8.1; Thr, 9.4; Leu, 9.9; Arg, 11.0; Asn, 11.3; Ile, 11.4; Gln, 11.4; His, 13.0; Lys, 13.3; Cys, 13.8; Phe, 14.2; Tyr, 15.2; Met, 15.4; Trp, 16.5 (data from [3]). The upper half of the table of the genetic code corresponding to codon YNN (Y = T or C; N = T, C, A or G) contains a lot of newly added amino acids (e.g., Phe and Cys), while the lower half corresponding to codon RNN (R = A or G) contains the most primitive amino acids, Ala and Gly.
From these randomized libraries, functional SH3 sequences were selected using mRNA display [17], [18]. In mRNA display, each cell-free translated polypeptide in a library covalently binds to its corresponding mRNA through puromycin. After affinity selection via the protein portion of an mRNA-displayed protein library, selected proteins can be easily identified by amplification and sequencing of the mRNA portion. Moreover, mRNA display based on cell-free translation can handle larger number of molecules (approximately 1012–13) than the other cell-based display technique such as phage display, and it makes possible enrichment of active sequences with low abundance from a library with high diversity and complexity. Therefore, we used mRNA display to elucidate and compare the frequency of functional SH3 sequences in randomized SH3 libraries with different sets of amino acids.
Results
Design and construction of randomized SH3 libraries
First, we constructed partially (28 out of 57 amino acids) randomized SH3 gene libraries, SH3(RNN)28 and SH3(YNN)28, with randomized codons RNN (R = A or G; N = T, C, A or G) and YNN (Y = T or C), corresponding to the lower and upper half of the table of the genetic code, respectively (Fig. 1). We also prepared a randomized SH3 gene library SH3(NNN)28 with all 20 amino acids as a control. If particular amino acid residues are essential for a randomized position of the SH3 gene, the frequency of occurrence of functional proteins will be greatly affected. To exclude this possibility, the randomized codons were introduced into 28 out of 57 amino acid residues of the src SH3 domain and not in the highly conserved residues of the SH3 domain (Fig. 2), such as the ligand peptide binding region, the hydrophobic core region and the surface region, that prefers polar amino acids [16]. Furthermore, to match the biophysical properties (the proportion of hydrophobic residue and the tendency to form β-sheet) of randomized SH3 proteins among three libraries, the mixed-base compositions of random regions (N, R and Y) were designed to provide amino acid compositions resembling modern proteins.
Figure 2. The three-dimensional structure and sequence of the src SH3 domain.

(A) The three-dimensional structure of the complex of the SH3 domain with the VSL12 peptide. The randomized and conserved positions of the SH3 domain in this study are shown in blue and gray, respectively. The peptide ligand VSL12 is shown in red. Structure was visualized by Accelrys DiscoveryStudio 2.1 (PDBid: 1QWF, [21]). (B) The amino-acid sequence of the SH3 domain. The randomized amino acids (X) are shown in blue. In the highly conserved region (gray), red asterisks indicate residues contacting the peptide ligand and black dots indicate residues that are important in determing the domain structure [8], [16].
The random regions of the SH3(NNN)28 and SH3(YNN)28 libraries contain stop codons. In addition, when the randomized DNA was cloned and sequenced, more than 90% of sequences in each library contained unexpected frameshifts (data not shown), probably due to errors during chemical synthesis. Thus, we sought to eliminate sequences containing stop codons and frameshifts by preselection using mRNA display, as described previously [19], [20]. In mRNA display, the C-terminus of the in vitro translated polypeptide is covalently bound to the 3' terminus of the template mRNA without stop codons on the stalling ribosome. Thus, mRNA sequences with stop codons cannot form mRNA-displayed proteins. Further, the C-terminal FLAG tag encoded by mRNA sequences with frameshifts cannot be properly translated. Thus, these mRNA sequences with stop codons or frameshifts are principally removed from libraries by purification with anti-FLAG antibody-immobilized beads. Indeed, the percentages of sequences without stop codons or frameshifts in the SH3(RNN)28, SH3(YNN)28 and SH3(NNN)28 libraries increased from 8, 2 and 6% to 72, 25 and 33%, respectively, after one round of preselection.
Selection of functional sequences from the randomized SH3 libraries
In vitro selection of functional SH3 sequences that can bind to the ligand peptide VSL12 [21] from each library (containing 3×1013 molecules with up to 0.6–2.4×1011 diversity) was performed by mRNA display (Fig. 3). The procedure was the same as that of the preselection except for the following two points: (i) the mRNA portion of the mRNA-displayed protein was reverse-transcribed to form an RNA/DNA hybrid to prevent binding of RNA with a particular secondary structure, and (ii) the mRNA/DNA-displayed proteins were selected with VSL12-immobilized beads. After three rounds of selection, DNAs were amplified by polymerase chain reaction (PCR), translated en masse and analyzed by enzyme-linked immunosorbent assay (ELISA). Consequently, the fraction of functional SH3 sequences capable of binding to the VSL12 peptide increased in the SH3(RNN)28 library after 3 rounds of selection (Fig. 4), but not in the SH3(YNN)28 library after even 5 rounds of selection (Fig. S1).
Figure 3. Schematic representation of mRNA-display selection of functional SH3 sequences.

(1) A DNA library encoding a randomized SH3 domain with an N-terminal T7·tag and a C-terminal FLAG tag was transcribed and ligated with a PEG-puromycin spacer. (2) The modified mRNA library was translated in vitro, and (3) the resulting mRNA-protein conjugates were purified with anti-FLAG antibody-immobilized beads and reverse-transcribed. (4) The mRNA/DNA-protein conjugates were incubated with the ligand peptide-immobilized beads, washed and competitively eluted with the free ligand peptides. (5) The DNA portion of the eluted molecules was amplified by PCR to form a randomized DNA library for the next round.
Figure 4. Fraction of functional SH3 sequences at each round of selection.

The total amount of three kinds of libraries, SH3(NNN)28, SH3(RNN)28 and SH3(YNN)28, that bound to the peptide ligand before (0) and after 1–3 rounds of selection were quantified by ELISA (gray bars). Black bars show negative controls (no ligand peptide was immobilized). Error bars indicate s.d. of four samples.
Furthermore, the binding signal appeared in the 2nd round of the SH3(RNN)28 library but not in that of the SH3(NNN)28 library (Fig. 4). The selected DNAs from the 3rd round of SH3(RNN)28 and SH3(NNN)28 libraries were cloned, and over 90 randomly chosen clones from each library were sequenced. Because more than half of selected sequences contained frameshifts, we obtained 36 and 24 sequences without frameshift from SH3(RNN)28 and SH3(NNN)28 libraries, respectively (Fig. 5). All clones with no frameshift bound to VSL12 peptide by ELISA (see next section). The random regions of the selected amino acid sequences shared low-sequence similarity with the wild-type sequence (0% to 29% identical). Further, alignment of the selected sequences indicated that approximately half of the 3rd round of the SH3(RNN)28 library was dominated by the closely related sequences (R6, R8 and R12) and differed by only four residues (69–72 aa) (Fig. 5), suggesting that they were derived from a single ancestral sequence. On the other hand, the functional SH3 sequences from the 3rd round of the SH3(NNN)28 library have no such closely related sequences (Fig. 5). These results suggest that the functional SH3(RNN)28 sequences were enriched earlier than the functional SH3(NNN)28 sequences, and it presumably caused recombination between the formerly enriched sequences in the SH3(RNN)28 library during PCR.
Figure 5. Amino acid sequences of selected proteins.

Amino acid sequences of the full-length of wild-type (WT) SH3 domain and the randomized region (highlighted in gray) of selected proteins from the SH3(NNN)28 and SH3(RNN)28 libraries are shown. Each sequence contains the N-terminal T7·tag (1–11 aa) and the C-terminal FLAG tag (77–84 aa). Dashes indicate amino acids that are identical with WT.
Next, we characterized the function and structure of the selected proteins arbitrarily chosen from the 3rd round of the SH3(RNN)28 and SH3(NNN)28 proteins to test whether the biophysical properties of the SH3(RNN)28 and SH3(NNN)28 proteins were similar.
Binding properties of selected proteins
First, we characterized the binding specificity of 10 plurally-obtained clones (N8, N17, N44, N45, N47, R6, R7, R8, R12 and R66; Fig. 5) from the 3rd round of the SH3(RNN)28 and SH3(NNN)28 libraries by ELISA. Consequently, all selected proteins bound to the VSL12 peptide but did not bind to other nonrelated peptides (Fig. 6).
Figure 6. Binding specificity of selected proteins.

Relative binding of selected proteins from the SH3(NNN)28 and SH3(RNN)28 libraries to several kinds of peptide (black, none; dark gray, VSL12; light gray, p53371–380; white, VEGF84–91) was analyzed by ELISA. WT, wild-type SH3 domain. Error bars indicate s.d. of four samples.
Next, we characterized the binding affinity of purified proteins to VSL12 peptide by fluorescence perturbation assays. Eight SH3(RNN)28 and seven SH3(NNN)28 proteins were overexpressed in E. coli and purified by using the C-terminal His6-tag (Fig. S2) under denaturing condition because of their low solubility. Although we obtained two soluble proteins from the RNN library (data not shown), we did not use the soluble fraction for further characterization because of their low expression level. After refolding of denatured purified proteins, three SH3(RNN)28 proteins (R1, R12 and R13) and two SH3(NNN)28 proteins (N17 and N47) as well as the wild-type SH3 domain were obtained without aggregation. The affinities of the SH3(RNN)28 and SH3(NNN)28 proteins were similar to each other (0.44–0.74 µM) and 3- to 5-fold higher than the wild-type SH3 domain (Table 1).
Table 1. Kinetic and thermodynamic parameters for the wild-type and SH3 variants.
| Protein | K d (µM) | T m (K) | ΔH m (kJ/mol) |
| Wild-type | 2.30±0.47 | 345.2±0.5 | 195.9±13.5 |
| N17 | 0.55±0.07 | 312.9±0.7 | 122.8±9.0 |
| N47 | 0.53±0.09 | 306.6±1.8 | 88.7±10.6 |
| R1 | 0.70±0.11 | 304.5±1.6 | 92.6±9.4 |
| R12 | 0.74±0.13 | 309.7±6.2 | 73.4±23.9 |
| R13 | 0.44±0.07 | 330.8±0.5 | 123.8±6.0 |
Structural characterization of selected proteins
We analyzed the secondary structure of the purified proteins R1, R12, R13, N17 and N47 by means of circular dichroism (CD) spectroscopy. Although the CD spectra of β-sheet proteins usually have minima at ∼217 nm, native SH3 domain has unusual maxima at 220 nm that may be a result of the environment of aromatic residues or β-turn conformations [22]. Our results showed that all SH3 domain variants, especially R13 and N17, had the typical maxima at 220 nm for the wild-type SH3 domain (Fig. 7 and Fig. S3), though the peak intensities were varied, suggesting that they have similar secondary structure to the wild-type.
Figure 7. Circular dichroism spectra of selected proteins.

(A) The mean residue ellipticity values of wild-type SH3 domain (black solid line) and SH3 variants R1 (gray solid line), R12 (dashed line) and R13 (dotted line) selected from the SH3(RNN)28 library are shown. (B) The mean residue ellipticity values of wild-type SH3 domain (black solid line) and SH3 variants N17 (gray solid line) and N47 (dashed line) selected from the SH3(NNN)28 library are shown.
The thermal stabilities of the selected proteins were estimated from the thermal denaturation curves of the CD value at 220 nm. They exhibited two-state cooperative thermal unfolding (Fig. 8), and the denaturation processes were reversible. Though all variants were less stable than the wild-type SH3 domain, they showed a wide range (ΔH m values, 73.4 kJ/mol to 123.8 kJ/mol) of thermodynamic stabilities (Table 1).
Figure 8. Thermal unfolding curves of selected proteins.
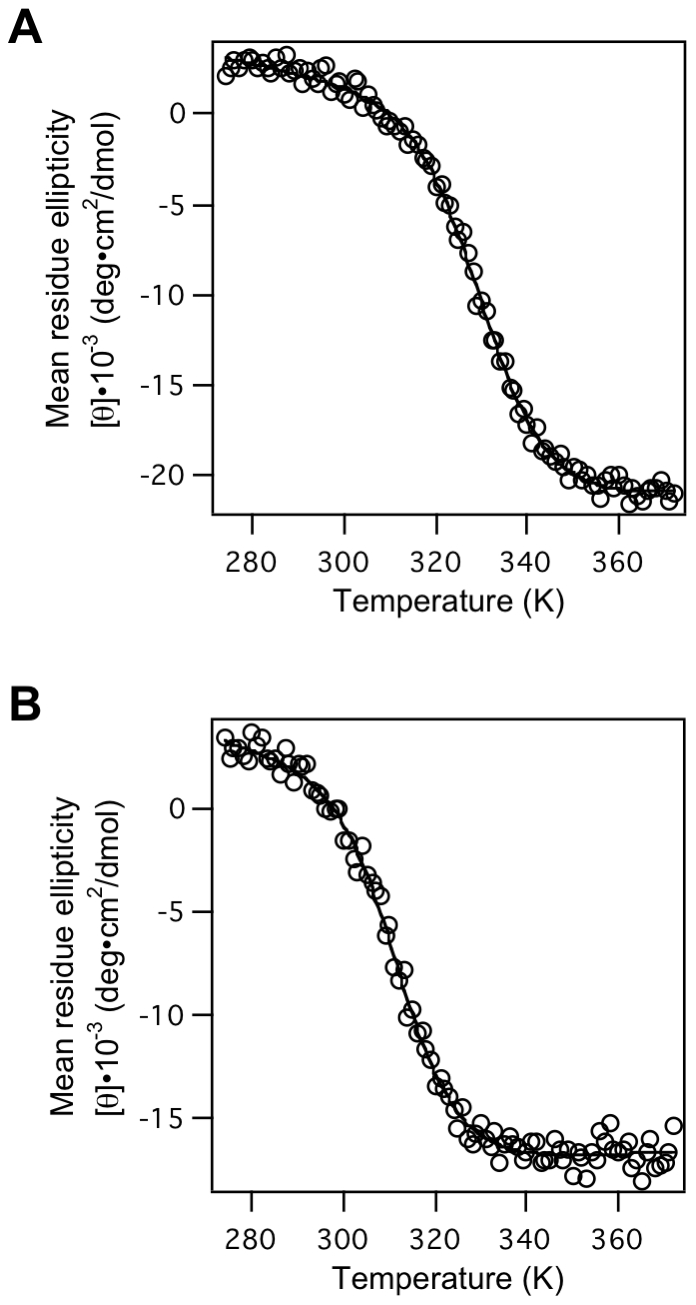
The CD mean residue ellipticity values at 220 nm for (A) R13 and (B) N17 were monitored as a function of temperature. The solid curves are the best fit of the whole set of data to eq. 1 (Materials and Methods).
These results indicated that the secondary structures and thermal stabilities of proteins selected from both the SH3(RNN)28 and SH3(NNN)28 libraries were similar to each other but lower than those of wild-type, in spite of their native-like specificity and higher affinity for the SH3 ligand peptide. These results are in contrast with those from a previous study in which SH3 variants with a simplified alphabet revealed a lower affinity and higher stability than the wild-type [8]. One of the reasons could be the difference in selection method, as we used mRNA display instead of phage display. Phage display is a multivalent display technique, and thus not only high-affinity binders but also low-affinity binders are captured by avidity effects, while mRNA display is a monovalent display technique. In addition, since phage-displayed proteins are expressed in E. coli, unstable SH3 variants might tend to be degraded by proteases or aggregated in E. coli, and thus only stable proteins may be selected in the previous study.
Discussion
In this study, functional SH3 sequences were enriched from a SH3(RNN)28 library but not from a SH3(YNN)28 library even after additional 2 rounds of selection. We roughly estimated that the SH3(RNN)28 library includes >10,000-fold larger number of functional SH3 sequences than the SH3(YNN)28 library because the enrichment efficiency was 200–3,000-fold per round calculated from the abundance in each round of two selected clones from each library by using real-time PCR (See Materials and Methods). We predicted that this would not be explained by the differences in typical biophysical properties (e.g., charge and hydrophobicity) of individual amino acids coded by RNN with those by YNN because we reconstructed the randomized SH3 domain in which highly conserved positions were fixed. If negatively charged amino acids (Glu and Asp) in a position of the randomized region of SH3 domain are essential for the SH3 activity, no functional sequence will be obtained from the SH3(YNN)28 library because YNN codes no negatively charged amino acids. However, negatively charged amino acids in the randomized region would not be essential, because the number of Glu and Asp in the region was zero in a selected active clone N36 (Fig. 4). Further, the percentage of hydrophobic residues (Ala, Val, Leu, Ile, Met, Phe, Trp and Tyr) in the initial SH3(YNN)28 library (43%) is almost equal to that of the 3rd round of the SH3(RNN)28 library (42%) as well as the SH3(NNN)28 library (44%) (Table S1).
Our result experimentally supports the Babajide's hypothesis [15], for the first time, that modern proteins might be able to be simplified by a set of putative primitive amino acids more easily than by a set of putative new amino acids. The reason is still unknown but may reflect an evolutionary constraint that primordial proteins consisted of a small set of primitive amino acids and gradually acquired new amino acids in the course of neutral evolution. To strengthen this hypothesis, not only a β-sheet protein used in this study but also an α-helical protein and other protein folds should be tested. Moreover, application of this approach to various combinations of codons in a protein sequence may be useful for clarifying the precise order of the amino acid expansion in the early stages of protein evolution.
Further, interestingly, the functional SH3 sequences were enriched from the SH3(RNN)28 library slightly earlier than from the SH3(NNN)28 library, while the function and structure of selected SH3(RNN)28 proteins with the primitive alphabet were comparable with those of SH3 domains with the 20 alphabet. The results imply that the protein sequence variety with a limited set of primitive amino acids includes a larger number of functional sequences than that with the current 20 amino acid alphabet. Previously, it has been reported that such reduced alphabets are effective for functional selection from randomized libraries [23], [24]. However, in these studies, only a few amino acids in the active sites were randomized. In this study, we showed that a limited set of primitive amino acids are also effective for wide frame regions, excluding the active sites.
In future work, it would be extremely interesting to randomize both active sites and frame regions and to examine whether the resulting wholly random-sequence library with limited alphabets is suited for in vitro selection of functional sequences, as the occurrence rate of functional sequences in a random-sequence library with a natural 20 alphabet has been shown to be quite low [25]. In our previous study, the random-sequence proteins with primitive alphabets tended to be more soluble as compared to random-sequence proteins with the natural alphabet [20], [26]. Similarly, in this study, 2 of 8 functional proteins from the SH3(RNN)28 library were expressed in the soluble fraction in E. coli, while none of the 7 functional proteins from the SH3(NNN)28 library were expressed in the soluble fraction (data not shown). Thus, the design of proteins with a higher content of primitive amino acids may improve the solubility as well as the rate of folded and functional proteins. Again, various subsets of amino acids including putative primitive amino acids should be tested for functional selection depending on the target function, because some putative new amino acids may be essential for some function. For example, His and Cys are essential residues for binding to zinc ions in the zinc-finger motif, and Cys is required for stabilization of extracellular domains by disulfide bonds. Combining of putative primitive amino acids and some particular new amino acids depending on the target protein would provide attractive resource for design and evolution of novel proteins in the laboratory.
Materials and Methods
Construction of randomized DNA libraries
All oligonucleotides used in this study were synthesized by Sigma-Aldrich, Japan (Table S2). Each of three randomized libraries, SH3(RNN)28, SH3(YNN)28 and SH3(NNN)28, was constructed by overlap-extension PCR from an equimolar mixture (8 pmol each) of four DNAs (Fragment 1–4; Tables S2 and S3) containing a random sequence region flanked by constant sequences using the primers SPO7tagF-mut2 and FLAG1A-mut2 (Table S2). The PCR products were purified with a QIAquick PCR purification kit (Qiagen).
Preselection of randomized DNA libraries using mRNA display
Preselection by mRNA display was performed as previously described [19], [20]. Briefly, the purified DNA (∼5 pmol, 3×1012 molecules) was transcribed with a RiboMax large-scale RNA production system-SP6 (Promega). The resulting RNA was purified with an RNeasy mini kit (Qiagen) and ligated with polyethylene glycol (PEG)-puromycin spacer [p(dCp)2-T(Fluor)p-PEGp-(dCp)2-puromycin] using T4 RNA ligase (Takara). The ligated RNA was purified with the RNeasy mini kit and translated with wheat germ extract plus (Promega) for 1 h at 25°C. The reaction mixture containing mRNA-displayed proteins (6×1013 molecules with 3×1012 potentially different sequences) was added to anti-FLAG M2 antibody-immobilized agarose beads (Sigma-Aldrich) and mixed on a rotator for 1 h at 4°C. The beads were washed with 500 µl of TBST (Tris-buffered saline with 0.2% Tween 20, pH 7.4) four times. The mRNA-displayed proteins were eluted with TBST containing 1 mg/ml FLAG M2 peptide (Sigma-Aldrich) at 4°C for 1 h. The mRNA portion of the eluted mRNA-displayed proteins was amplified by reverse-transcription (RT)-PCR with a OneStep RT-PCR kit (Qiagen) using the primers SPO7tagF-mut2 and FLAG1A-mut2. The RT-PCR products were purified with the QIAquick PCR purification kit and were served as randomized DNA libraries for further functional selection.
In vitro selection of functional SH3 sequences using mRNA display
From the above DNA libraries, the mRNA-displayed protein libraries were generated as described above, mixed with the anti-FLAG M2 antibody-immobilized agarose beads again, and washed with 300 µl of TBST three times. Then the RT reaction mixture with Superscript II (Invitrogen) and FLAG M2 peptide were added and incubated for 1 h at 37°C to form the RNA/DNA hybrid. The resulted mRNA/DNA-displayed protein libraries were exchanged into TBST on Bio-gel P-30 (BioRad) gel filtration columns and then incubated for 1 min at 4°C with Streptavidin coating Magnotex-SA beads (Takara) preblocked with DIG blocking Buffer (Roche), salmon sperm DNA (Stratagene) and yeast RNA (Sigma-Aldrich) to avoid non-specific binders. The supernatants were incubated with biotinylated SH3 peptide ligand VSL12 (Invitrogen; Biotin-XXXVSLARRPLPPLP, X = Aminohexanoic acid) for 1 h at 4°C, and the complexes of mRNA/DNA-displayed proteins and the biotinylated peptides were captured on Magnotex-SA beads for 1 min at 4°C. After washing with 300 µl of TBST three times, the bound mRNA/DNA-displayed proteins were eluted with TBST containing 1 mM free VSL12 peptide (Invitrogen; VSLARRPLPPLP) for 5 min at 4°C. The eluate of each library was used for PCR amplification with the primers SPO7tagF-mut2 and FLAG1A-mut2. The resulted DNA was purified, and served as template DNA for next round of selection or cloned using a TOPO TA cloning kit (Invitrogen) followed by sequencing with an ABI PRISM 3100 genetic analyzer (Applied Biosystems).
Real-time PCR analysis
Real-time PCR was performed with SYBR Premix Ex Taq II (Takara) using protein-specific primer sets, N8-F and N8-R, N16-F and N16-R, R2-F and R2-R, R6-F and R6-R (Table S2) on the LightCycler (Roche).
Enzyme-linked immunosorbent assay (ELISA)
Streptavidin transparent C8 plates (Nunc) were incubated with 1 µM biotinylated peptide [VSL12, p53371–380 (Invitrogen; Biotin-SKKGQSYSRH), and VEGF84–91 (Invitrogen; Biotin-XXPHQGQHIG, X = Aminohexanoic acid)] for 1 h at 25°C, and washed with TBST. The RNA libraries from each round of the selection or the RNA of selected clones were translated using wheat germ extract (Promega) for 2 h at 25°C. The translated product was transferred into wells of the above plate with or without immobilized peptide and then incubated for 1 h at 25°C. After washing with TBST, the plate was incubated with HRP-conjugated anti-FLAG M2 antibody (Sigma-Aldrich) for 1 h at 25°C. After washing, the amounts of bound molecules of each library or the selected clones were detected by using TMB substrate kit (Nacalai Tesque). The absorbance at 450 nm (reference wavelength at 655 nm) was measured with a microplate reader (Safire, Tecan).
Cloning, overexpression and purification of selected proteins
The random regions of the clones were digested with BglII and XhoI and subcloned into the pET20 vector (Novagen) containing the N-terminal T7·tag sequence and the C-terminal His6 tag sequence. The individual plasmids were transformed into Escherichia coli BL21(DE3)-CodonPlus cells (Stratagene). The bacteria were grown in LB broth containing 100 µg/ml ampicillin and 34 µg/ml chloramphenicol at 37°C, and protein expression was induced by adding 0.5 mM isopropyl-β-D-thiogalactopyranoside. After an additional 5 h of growth, the bacteria were harvested by centrifugation and lysed in a BugBuster (Novagen) containing a protease inhibitor cocktail (Sigma-Aldrich). The centrifuged supernatants were used as soluble fractions. The pellets were resuspended in a buffer containing 8 M urea, and the supernatants after centrifugation were used as insoluble fractions. The proteins of selected clones were purified by affinity chromatography under denaturing condition using Ni-NTA Superflow resin (Qiagen), from which they were eluted with a pH gradient under denaturing condition. The purified denatured proteins were dialyzed against 50 mM phosphate buffer (pH 7.4) for refolding. The soluble and insoluble fractions and purified proteins were separated by 16.5% Tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and detected by Coomassie brilliant blue staining. The protein concentrations were determined using a BCA protein assay kit (Pierce).
Circular dichroism (CD) measurements
CD measurements were performed with a J-820 spectropolarimeter (Jasco). CD spectra of purified proteins (10 µM) were measured from 190 to 250 nm at 20°C using a 2 mm path-length cell. The results were expressed as mean residue molar ellipticity [θ]. Thermal denaturation was monitored by following the change in ellipticity at 220 nm using a 10 mm path-length cell. The temperature was increased at 2°C/min. The reversibility of thermal denaturation was tested by stepwise cooling of the protein solution back to 20°C. Thermal denaturation data were fit to standard equations by nonlinear least-squares regression using the Igor Pro (Wave Metrics, Inc.) assuming a two-state transition. All denaturation curves were fit to following equation:
| (1) |
where y represents the observed ellipticity; y n and m n, y d and m d are the y-intercept and slope of the pre- and posttransitional baselines respectively; T is the temperature (in degrees Kelvin); T m is the midpoint transition temperature; and ΔH m is the enthalpy change for unfolding at T m [27].
Fluorescence perturbation assays
The affinities of SH3 domain variants for peptide VSL12 were measured by fluorescence perturbation assays, as described previously [28]. Aliquots of peptide solution were added to solutions of SH3 domain (0.5 µM) in PBS (Phosphate-buffered saline, pH 7.4). The mixture was incubated for 10 min at 20°C and then analyzed by a FP-777 fluorescence spectrophotometer (Jasco). The excitation wavelength was 278 nm (10 nm slit), and the emission wavelength was 350 nm (5 nm slit) for all experiments.
Supporting Information
ELISA of SH3(YNN)28 libraries at each round of selection. The total amount of SH3(YNN)28 library that bound to the peptide immobilized (gray bars) and non-immobilized well (black bars) before (0) and after 1–5 rounds of mRNA-display selection were quantified by ELISA. Consequently, after 5 rounds of selection, the translated products of SH3(YNN)28 library non-specifically bound to ELISA plates, and no ligand-specific binder was enriched. Because the sequences of the non-specific binders contain a partial frameshift in the fixed region in the SH3 gene (data not shown), the non-specific binders might have no SH3-like structure. Further, their sequences contain a lot of basic amino acids (data not shown), suggesting that they would probably bind to carboxylic acid group on the surface of the affinity beads and the ELISA plates. Such non-specific binders might also be included in the initial SH3(RNN)28 and SH3(NNN)28 libraries, but not be observed after selection probably due to the competition with a lot of specific-binders in the libraries.
(TIFF)
Purification of proteins selected from the SH3(RNN)28 and SH3(NNN)28 libraries. The selected proteins with His6 tag, were overexpressed in E. coli. The insoluble fractions of the crude lysate of selected proteins were purified on Ni-NTA resins. The samples before (N, non purified) and after purification (P, purified) were resolved by 16.5% Tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis and stained with Coomassie brilliant blue. The purified proteins (∼9 kDa) showed single bands.
(TIFF)
Circular dichroism spectra of SH3 domains in folded and unfolded states. The folded and unfolded samples were measured at 20°C and 99°C, respectively. (A) wild-type; (B) R13; (C) N17. Although the CD spectra of β-sheet proteins usually have minima at ∼217 nm, the folded SH3 domains have unusual maxima at 220 nm (solid line) that are thought to be caused by the environment of the aromatic residues or β-turn conformations [22]. Further, the CD spectra of the unfolded SH3 domains have unusual minima at 220 nm (broken line), probably due to the presence of non-native hydrophobic clusters organized by Trp rings within disordered states [29].
(TIFF)
Percentage of each amino acid in the randomized region of the initial and third rounds of libraries.
(DOC)
Oligonucleotide sequences used in this study.
(DOC)
Percentage of each nucleotide at each position of the designed codons.
(DOC)
Acknowledgments
We wish to thank members of our lab for helpful comments and discussions: Hideaki Takashima, Kenichi Horisawa, Seiji Tateyama and Etsuko Miyamoto-Sato for help with mRNA display, and Toru Tsuji for help with denaturation curve fitting.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by a Grant-in-Aid for Scientific Research (19657073) from the JSPS (Japan Society for the Promotion of Science) and a Grant-in-Aid for the Keio University Global Center of Excellence (G-COE) Program entitled 'Center of Human Metabolomic Systems Biology' from the MEXT (Ministry of Education, Culture, Sports, Science and Technology) of Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Miller SL. Which organic compounds could have occurred on the prebiotic earth? Cold Spring Harb Symp Quant Biol. 1987;52:17–27. doi: 10.1101/sqb.1987.052.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Cronin JR, Pizzarello S. Amino acids in meteorites. Adv Space Res. 1983;3:5–18. doi: 10.1016/0273-1177(83)90036-4. [DOI] [PubMed] [Google Scholar]
- 3.Trifonov EN. The triplet code from first principles. J Biomol Struct Dyn. 2004;22:1–11. doi: 10.1080/07391102.2004.10506975. [DOI] [PubMed] [Google Scholar]
- 4.Wong JT. Coevolution theory of the genetic code at age thirty. BioEssays. 2005;27:416–425. doi: 10.1002/bies.20208. [DOI] [PubMed] [Google Scholar]
- 5.Jordan IK, Kondrashov FA, Adzhubei IA, Wolf YI, Koonin EV, et al. A universal trend of amino acid gain and loss in protein evolution. Nature. 2005;433:633–638. doi: 10.1038/nature03306. [DOI] [PubMed] [Google Scholar]
- 6.Koonin EV, Novozhilov AS. Origin and evolution of the genetic code: the universal enigma. IUBMB Life. 2009;61:99–111. doi: 10.1002/iub.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Protein design by binary patterning of polar and nonpolar amino acids. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 8.Riddle DS, Santiago JV, Bray-Hall ST, Doshi N, Grantcharova VP, et al. Functional rapidly folding proteins from simplified amino acid sequences. Nat Struct Biol. 1997;4:805–809. doi: 10.1038/nsb1097-805. [DOI] [PubMed] [Google Scholar]
- 9.Silverman JA, Balakrishnan R, Harbury PB. Reverse engineering the (α/β)8 barrel fold. Proc Natl Acad Sci U S A. 2001;98:3092–3097. doi: 10.1073/pnas.041613598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akanuma S, Kigawa T, Yokoyama S. Combinatorial mutagenesis to restrict amino acid usage in an enzyme to a reduced set. Proc Natl Acad Sci U S A. 2002;99:13549–13553. doi: 10.1073/pnas.222243999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walter KU, Vamvaca K, Hilvert D. An active enzyme constructed from a 9-amino acid alphabet. J Biol Chem. 2005;280:37742–37746. doi: 10.1074/jbc.M507210200. [DOI] [PubMed] [Google Scholar]
- 12.Go A, Kim S, Baum J, Hecht MH. Structure and dynamics of de novo proteins from a designed superfamily of 4-helix bundles. Protein Sci. 2008;17:821–832. doi: 10.1110/ps.073377908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel SC, Bradley LH, Jinadasa SP, Hecht MH. Cofactor binding and enzymatic activity in an unevolved superfamily of de novo designed 4-helix bundle proteins. Protein Sci. 2009;18:1388–1400. doi: 10.1002/pro.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jumawid MT, Takahashi T, Yamazaki T, Ashigai H, Mihara H. Selection and structural analysis of de novo proteins from an α3β3 genetic library. Protein Sci. 2009;18:384–398. doi: 10.1002/pro.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babajide A, Hofacker IL, Sippl MJ, Stadler PF. Neutral networks in protein space: a computational study based on knowledge-based potentials of mean force. Fold Des. 1997;2:261–269. doi: 10.1016/S1359-0278(97)00037-0. [DOI] [PubMed] [Google Scholar]
- 16.Larson SM, Davidson AR. The identification of conserved interactions within the SH3 domain by alignment of sequences and structures. Protein Sci. 2000;9:2170–2180. doi: 10.1110/ps.9.11.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemoto N, Miyamoto-Sato E, Husimi Y, Yanagawa H. In vitro virus: Bonding of mRNA bearing puromycin at the 3'-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997;414:405–408. doi: 10.1016/s0014-5793(97)01026-0. [DOI] [PubMed] [Google Scholar]
- 18.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho G, Keefe AD, Liu R, Wilson DS, Szostak JW. Constructing high complexity synthetic libraries of long ORFs using in vitro selection. J Mol Biol. 2000;297:309–319. doi: 10.1006/jmbi.2000.3571. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka J, Doi N, Takashima H, Yanagawa H. Comparative characterization of random-sequence proteins consisting of 5, 12, and 20 kinds of amino acids. Protein Sci. 2010;19:786–795. doi: 10.1002/pro.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng S, Kasahara C, Rickles RJ, Schreiber SL. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc Natl Acad Sci U S A. 1995;92:12408–12415. doi: 10.1073/pnas.92.26.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maxwell KL, Davidson AR. Mutagenesis of a buried polar interaction in an SH3 domain: sequence conservation provides the best prediction of stability effects. Biochemistry. 1998;37:16172–16182. doi: 10.1021/bi981788p. [DOI] [PubMed] [Google Scholar]
- 23.Reetz MT, Kahakeaw D, Lohmer R. Addressing the numbers problem in directed evolution. Chembiochem. 2008;9:1797–1804. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- 24.Wu S, Acevedo JP, Reetz MT. Induced allostery in the directed evolution of an enantioselective Baeyer-Villiger monooxygenase. Proc Natl Acad Sci U S A. 2010;107:2775–2780. doi: 10.1073/pnas.0911656107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keefe AD, Szostak JW. Functional proteins from a random-sequence library. Nature. 2001;410:715–718. doi: 10.1038/35070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doi N, Kakukawa K, Oishi Y, Yanagawa H. High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Protein Eng Des Sel. 2005;18:279–284. doi: 10.1093/protein/gzi034. [DOI] [PubMed] [Google Scholar]
- 27.Knapp S, Mattson PT, Christova P, Berndt KD, Karshikoff A, et al. Thermal unfolding of small proteins with SH3 domain folding pattern. Proteins. 1998;31:309–319. doi: 10.1002/(sici)1097-0134(19980515)31:3<309::aid-prot7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 28.Chen JK, Lane WS, Brauer AW, Tanaka A, Schreiber SL. Biased combinatorial libraries: novel ligands for the SH3 domain of phosphatidylinositol 3-kinase. J Am Chem Soc. 1993;115:12591–12592. [Google Scholar]
- 29.Crowhurst KA, Tollinger M, Forman-Kay JD. Cooperative interactions and a non-native buried Trp in the unfolded state of an SH3 domain. J Mol Biol. 2002;322:163–178. doi: 10.1016/s0022-2836(02)00741-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ELISA of SH3(YNN)28 libraries at each round of selection. The total amount of SH3(YNN)28 library that bound to the peptide immobilized (gray bars) and non-immobilized well (black bars) before (0) and after 1–5 rounds of mRNA-display selection were quantified by ELISA. Consequently, after 5 rounds of selection, the translated products of SH3(YNN)28 library non-specifically bound to ELISA plates, and no ligand-specific binder was enriched. Because the sequences of the non-specific binders contain a partial frameshift in the fixed region in the SH3 gene (data not shown), the non-specific binders might have no SH3-like structure. Further, their sequences contain a lot of basic amino acids (data not shown), suggesting that they would probably bind to carboxylic acid group on the surface of the affinity beads and the ELISA plates. Such non-specific binders might also be included in the initial SH3(RNN)28 and SH3(NNN)28 libraries, but not be observed after selection probably due to the competition with a lot of specific-binders in the libraries.
(TIFF)
Purification of proteins selected from the SH3(RNN)28 and SH3(NNN)28 libraries. The selected proteins with His6 tag, were overexpressed in E. coli. The insoluble fractions of the crude lysate of selected proteins were purified on Ni-NTA resins. The samples before (N, non purified) and after purification (P, purified) were resolved by 16.5% Tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis and stained with Coomassie brilliant blue. The purified proteins (∼9 kDa) showed single bands.
(TIFF)
Circular dichroism spectra of SH3 domains in folded and unfolded states. The folded and unfolded samples were measured at 20°C and 99°C, respectively. (A) wild-type; (B) R13; (C) N17. Although the CD spectra of β-sheet proteins usually have minima at ∼217 nm, the folded SH3 domains have unusual maxima at 220 nm (solid line) that are thought to be caused by the environment of the aromatic residues or β-turn conformations [22]. Further, the CD spectra of the unfolded SH3 domains have unusual minima at 220 nm (broken line), probably due to the presence of non-native hydrophobic clusters organized by Trp rings within disordered states [29].
(TIFF)
Percentage of each amino acid in the randomized region of the initial and third rounds of libraries.
(DOC)
Oligonucleotide sequences used in this study.
(DOC)
Percentage of each nucleotide at each position of the designed codons.
(DOC)