

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Hypophosphatasia, HOPS, HPP, HP, Rathbun's disease, phosphoethanolaminuria.
1.2 OMIM# of the disease
146300, 241500, 241510.
1.3 Name of the analyzed genes or DNA/chromosome segments
ALPL (alkaline phosphatase liver-type), 1p36.1-p34.
1.4 OMIM# of the gene(s)
171760.
1.5 Mutational spectrum
Over 200 different disease-causing mutations have been reported in the ALPL gene mutations.
Database: http://www.sesep.uvsq.fr/03_hypo_mutations.php. The distribution is as follows: 79% missense mutations; 10% small deletions; 4% splicing mutations; 3% nonsense mutations; 2% small insertions; and ≤1%: complex insertion/deletions, large deletions and mutations in the regulatory sequence. The large proportion of missense mutations with various effects on the enzymatic activity of alkaline phosphatase has been correlated with the high clinical variability.1 A part of missense mutations exhibits a dominant-negative effect,2, 3, 4, 5 explaining dominant inheritance of mild forms of the disease.6, 7, 8
1.6 Analytical methods
The main strategy for mutation screening consists of sequencing of genomic exonic DNA, including flanking intronic sequences. This allows the detection of ∼95% of mutations in patients with hypophosphatasia (HP). The analysis may be completed by screening for large deletions by quantitative multiplex PCR of short fragments,9 but this does not significantly increase the detection rate, because large deletions seem rare in the ALPL gene. For prenatal diagnosis, a set of linked microsatellite sequences strongly linked to the ALPL gene may be used for indirect diagnosis and/or excluding maternal cell contamination.
1.7 Analytical validation
The existence of mutations is confirmed by testing the parental DNA or, when the parental DNA is not available, by sequencing a second and independant PCR product. Newly discovered missense mutations may be tested in vitro by site-directed mutagenesis in order to clarify their disease-causing role and estimate their degree of severity.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
According to a unique reported study performed in Toronto, Canada,10 the incidence of severe cases of HP (perinatal severe and infantile forms) is 1 in 100 000. Study of the prevalence of mild forms (prenatal benign, childhood, adult and odontohypophosphatasia) has not yet been reported, but it is expected to be higher because of low selective pressure and because heterozygotes for some mutations may express the disease.
1.9 If applicable, prevalence in the ethnic group of investigated person
Greenberg et al11 suggested that the disease prevalence in Canadian Mennonites could be up to 1 in 2500.
1.10 Diagnostic setting

Comment: See below points 3.1-3.4
2. TEST CHARACTERISTICS
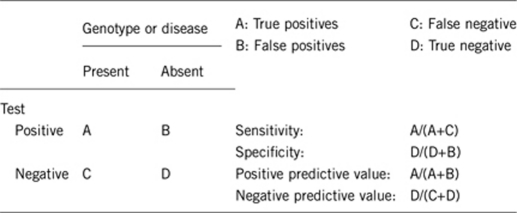
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
Nearly 95%.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
Nearly 100%.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
Nearly 95%.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
Nearly 100%.
2.5 Positive clinical predictive value (life-time risk of developing the disease if the test is positive)
100% in recessive inheritance.
Nearly 30–40% in mild forms with dominant inheritance.
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
100%.
Index case in that family had not been tested:
95%.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
(1) Diagnosis: Based on clinical courses and severity, HP has been divided into six major subtypes with different prognoses. The perinatal form is the most severe one. It results in stillbirth or death a few days after birth because of hypoplastic lungs, difficult-to-treat seizures, extensive hypomineralization, deformities of bone and disturbances of the calcium/phosphate metabolism. In the prenatal benign form, despite prenatal symptoms, there is a spontaneous improvement of skeletal defects. The patients manifest limb shortening and bowing and often dimples overlaying the long bone anomalies, and ultrasound scans reveal progressive improvement of the skeletal anomalies and mineralization during the third trimester of the pregnancy.12, 13 Clinical signs of the infantile form appear during the first 6 months of life, including rickets, premature craniosynostosis, irritability, seizures and nephrocalcinosis due to hypercalciuria. Later, premature shed of deciduous teeth are common. Death within the first year of life is common. The childhood form in most cases presents after the first year of life and is characterized by rickets causing short stature, delayed walking and a waddling gait due to bone deformities and pain of the lower extremities. Premature loss of teeth often leads to diagnosis. Adult HP presents with osteomalacia, chondrocalcinosis, osteoarthropathy and stress fractures during middle age in patients who had a history of mild rickets in childhood. Many patients present premature loss of permanent teeth. Odontohypophosphatasia is characterized by premature exfoliation of primary and/or permanent teeth and/or severe dental caries, often not associated with abnormalities of the skeletal system. However, it should be noticed that these six clinical subtypes can overlap significantly, for example, patients with adult HP often had musculoskeletal symptoms already in childhood. The infantile and the childhood form might be difficult to distinguish, because early symptoms might be present in the first months of life in both subtypes. In addition, dental abnormalities are frequent in other forms of HP. According to the severity of the disorder, some dental defects were infrequent, whereas others were always present.14 They consist of abnormal tooth shape (small bulbous crown, cervical constrictions, enlarged pulp spaces), abnormal tooth structure (enamel, dentin and cementum formation), tooth color, dental anomalies of tooth eruption/exfoliation with premature loss of predominantly the primary and also the permanent dentition. Delayed eruption of teeth and primary teeth impaction (ankylosis) are also recorded.
(2) Differential diagnosis: The differential diagnosis of HP depends on the age at which the diagnosis is considered.
In utero: Osteogenesis imperfecta type II, campomelic dysplasia and chondrodysplasias with bone mineralization defect.
At birth: Outwardly difficult to distinguish, radiographs readily distinguish osteogenesis imperfecta (type II), campomelic dysplasia and chondrodysplasias with bone mineralization defect, from HP.
Infancy and childhood: inborn errors of energy metabolism, organic acidemia, primary and secondary rickets, neglect and non-accidental trauma.
Nutritional and/or vitamin D deficiency, vitamin D resistance or renal osteodystrophy
Osteogenesis imperfecta (typically type III in infancy or type IV later on)
Cleidocranial dysostosis (OMIM 119600)
Cole–Carpenter syndrome (OMIM 112240).
Hadju–Cheney syndrome (OMIM 102500)
Idiopathic juvenile osteoporosis
Renal osteodystrophy Adult and odontohypophosphatasia
Osteoarthritis and pseudogout
Osteopenia/osteoporosis
Diseases with paraspinal ligament ossification (Forestier disease, arthropathy with calcium deposition)
Premature exfoliation of teeth can occur in the context of periodontal disease, as part of a connective tissue disorder such as Ehlers–Danlos syndrome (130050 type IV; 130080 type VIII), or associated with neutropenia, such as ELA2-related neutropenia (OMIM 202700), Papillon–Lefevre syndrome (OMIM 245000) and Haim–Munk syndrome (OMIM 245010), Chediak–Higashi syndrome (OMIM 214500) or as aggressive periodontitis (OMIM 170650). However, in HP, premature loss of primary and eventually permanent teeth occurs without root resorption (fully rooted teeth) and without inflammation of the gingivae and periodontium and X-rays show alveolar bone loss.
Dentin dysplasia type I (OMIM 125400).
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
Diagnosis of HP often causes considerable problems. On the one hand, it is a rare disease with a variety of differential diagnoses. On the other hand, different HP subtypes present a partly comparable spectrum of symptoms. Before genetic testing is applied, conventional alternative biochemical diagnosis is based on laboratory assays, genetic counseling and radiographic imaging. Total serum alkaline phosphatase (AP) activity or tissue-non-specific alkaline phosphatase (TNSAP) measured in leukocytes are below the age-related normal range. AP activity depends on age, sex and on laboratory procedures. However, a reduced AP activity is only a helpful diagnostic indicator, but it is not HP specific. Other conditions may also show reduced levels of AP, including early pregnancy, hypothyroidism, anemia, celiac disease or zinc deficiency. In general, residual serum AP activity has been directly linked to disease activity. Patients with perinatal forms often have a total serum AP <20% of the normal range, whereas a milder form (infantile or childhood form) has to be considered if AP values are around or slightly below the lower limit. In particular, patients with singular heterozygous mutations or autosomal dominant inheritance often exhibit a considerable residual AP activity. Separation of an autosomal dominant with a compound heterozygous disease state on the basis of the biochemical AP activity, however, is usually impossible.
Reduced enzyme activity results in accumulation of its substrates, including pyridoxal-5′-phosphate, inorganic pyrophosphate and phosphoethanolamine, which can be detected in serum, tissues and urine. Often, serum calcium and phosphate are normal or slightly increased. Urinary calcium excretion might be above the normal range. Careful surveillance including ophthalmological and neurological examination is recommended in patients with craniostenosis, eventually complemented by an invasive epidural monitoring of intracranial pressure.
Sequencing of the TNSAP gene is essential to confirm the diagnosis of HP when biochemical and clinical data are not clear enough or in the prenatal assessment of severe HP in couples with a previously affected child or pregnancy. Genetic consultation is recommended before genetic testing is done.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Biochemical testing does not exclude genetic testing and vice versa. Both diagnostic procedures add to the clinical picture and to aspects of prediction of severity and complications in the follow-up. Biochemical testing is cheap and may be measured in any routine laboratory. However, in order to avoid diagnosis pitfalls, appropriate collection tubes (serum separator tubes) and correct reference range matched for sex and age must be used.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
The age of onset is variable depending on the severity of the disease (see Whyte26 for review) and the penetrance is not complete in mild forms of HP. Thus, it makes sense to perform genetic testing in relatives of patients. This is interesting for genetic counseling and may help to prevent/delay the onset of clinical symptoms. However, owing to the clinical variability and reduced penetrance of some mutations, the predictive power may considerably vary between mutations. The test needs complete information of the tested person and about what to expect from the test, and it must be noticed that it may be proposed to relatives of probands only when the familial mutations have been previously characterized.
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
In case of positive test, medical management for prevention of clinical symptoms as described above.
If the test result is negative (please describe):
In case of negative result, the patient has to be reassured and considered as normal.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
In general, a person at-risk or with a high probability of being affected by the disease should follow the same recommendations for diagnosis and management of potential symptoms and complications, and should be referred to genetic counseling. In general, the clinical suspicion of the disease being present in addition to biochemical testing/confirmation makes the overall diagnosis quite likely.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
Severe forms of the disease (perinatal and infantile) are transmitted as an autosomal recessive trait, whereas both autosomal recessive and autosomal dominant transmission have been shown in clinically milder forms. Therefore, the risk of recurrence of severe forms is 25%. In moderate forms, it may be 25% (recessive transmission), 50% (dominant transmission) or still different (<50%), owing to incomplete penetrance in dominant forms. The mutations detected in dominant forms and responsible for moderate HP are also found in severe recessive HP, associated with other mutations. Thus, siblings of probands with severe HP may develop moderate symptoms, especially odontohypophosphatasia.
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
A negative test may resolve the genetic situation, as the disease is very rare and the tested person/couple have (almost) no risk for having affected children.
A positive test may lead to propose to test the partner, although the couple remain at low risk.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
In a family with phenotypically comparable affected siblings, a genetic test might not be essential in the yet untested individual.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
If a mutation of the index case has been shown to exhibit a dominant-negative effect, a relative carrying this mutation has a risk for developing mild HP (depending on the mutation). However, in this constellation, there is still a significant variability in the expectable clinical features.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
For both modes of inheritance, prenatal diagnosis for pregnancies at increased risk is possible if disease-causing mutation(s) of an affected family member is (are) known.
In the prenatal benign form of HP, despite prenatal symptoms, there is a spontaneous improvement of skeletal defects. Ultrasound scans reveal progressive improvement of the skeletal deformities and mineralization during the third trimester of the pregnancy.12, 13, 27 The distinction between severe and benign prenatal forms may be difficult and need to combine mutation characterization and careful ultrasound examination.28
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Genetic testing always has to be founded on genetic counseling. Genetic results with no particular immediate consequence, such as that in mild autosomal dominant forms of the disease, still do have significant impact on the long-term decisions in life such as partnership, parenthood and long-term surveillance of future medical symptoms. Thus, a test result can be quite useful in the long run, but it can also have stressful implications, especially for the development of a child, and, therefore, need to give a complete explanation for parents and psychological support.
Acknowledgments
This work was supported by EuroGentest, an EU-FP6 supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2).
The authors declare no conflict of interest
References
- Zurutuza L, Muller F, Gibrat JF, et al. Correlations of genotype and phenotype in hypophosphatasia. Hum Mol Genet. 1999;8:1039–1046. doi: 10.1093/hmg/8.6.1039. [DOI] [PubMed] [Google Scholar]
- Muller HL, Yamazaki M, Michigami T, et al. Asp361Val mutant of alkaline phosphatase found in patients with dominantly inherited hypophosphatasia inhibits the activity of the wild-type enzyme. J Clin Endocrinol Metab. 2000;85:743–747. doi: 10.1210/jcem.85.2.6373. [DOI] [PubMed] [Google Scholar]
- Lia-Baldini AS, Muller F, Taillandier A, et al. A molecular approach to dominance in hypophosphatasia. Hum Genet. 2001;109:99–108. doi: 10.1007/s004390100546. [DOI] [PubMed] [Google Scholar]
- Lia-Baldini AS, Brun-Heath I, Carrion C, et al. A new mechanism of dominance in hypophosphatasia: the mutated protein can disturb the cell localization of the wild-type protein. Hum Genet. 2008;123:429–432. doi: 10.1007/s00439-008-0480-1. [DOI] [PubMed] [Google Scholar]
- Fauvert D, Brun-Heath I, Lia-Baldini AS, et al. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet. 2009;10:51. doi: 10.1186/1471-2350-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MP, Teitelbaum SL, Murphy WA, Bergfeld MA, Avioli LV. Adult hypophosphatasia. Clinical, laboratory, and genetic investigation of a large kindred with review of the literature. Medicine (Baltimore) 1979;58:329–347. [PubMed] [Google Scholar]
- Eastman JR, Bixler D. Clinical, laboratory, and genetic investigations of hypophosphatasia: support for autosomal dominant inheritance with homozygous lethality. J Craniofac Genet Dev Biol. 1983;3:213–234. [PubMed] [Google Scholar]
- Eberle F, Hartenfels S, Pralle H, Kabisch A. Adult hypophosphatasia without apparent skeletal disease: ‘odontohypophosphatasia' in four heterozygote members of a family. Klin Wochenschr. 1984;62:371–376. doi: 10.1007/BF01716257. [DOI] [PubMed] [Google Scholar]
- Spentchian M, Brun-Heath I, Taillandier A, et al. Characterization of missense mutations and LARGE deletions in the ALPL gene by sequencing and quantitative multiplex PCR of short fragments. Genet Test. 2006;10:252–257. doi: 10.1089/gte.2006.10.252. [DOI] [PubMed] [Google Scholar]
- Fraser D. Hypophosphatasia. Am J Med. 1957;22:730–746. doi: 10.1016/0002-9343(57)90124-9. [DOI] [PubMed] [Google Scholar]
- Greenberg CR, Evans JA, McKendry-Smith S, et al. Infantile hypophosphatasia: localization within chromosome region 1p36.1-34 and prenatal diagnosis using linked DNA markers. Am J Hum Genet. 1990;46:286–292. [PMC free article] [PubMed] [Google Scholar]
- Pauli RM, Modaff P, Sipes SL, Whyte MP. Mild hypophosphatasia mimicking severe osteogenesis imperfecta in utero: bent but not broken. Am J Med Genet. 1999;86:434–438. doi: 10.1002/(sici)1096-8628(19991029)86:5<434::aid-ajmg8>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Moore CA, Curry CJ, Henthorn PS, et al. Mild autosomal dominant hypophosphatasia: in utero presentation in two families. Am J Med Genet. 1999;86:410–415. doi: 10.1002/(sici)1096-8628(19991029)86:5<410::aid-ajmg3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Reibel A, Maniere MC, Clauss F, et al. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J Rare Dis. 2009;4:6. doi: 10.1186/1750-1172-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb AA, Bruce SN, Morris AA, Cheetham TD. Infantile hypophosphatasia: disappointing results of treatment. Acta Paediatr. 2000;89:730–733. doi: 10.1080/080352500750044106. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Mumm S, Deal C. Adult hypophosphatasia treated with teriparatide. J Clin Endocrinol Metab. 2007;92:1203–1208. doi: 10.1210/jc.2006-1902. [DOI] [PubMed] [Google Scholar]
- Camacho PM, Painter S, Kadanoff R. Treatment of adult hypophosphatasia with teriparatide. Endocr Pract. 2008;14:204–208. doi: 10.4158/EP.14.2.204. [DOI] [PubMed] [Google Scholar]
- Gagnon C, Sims NA, Mumm S, et al. Lack of sustained response to teriparatide in a patient with adult hypophosphatasia. J Clin Endocrinol Metab. 2010;95:1007–1012. doi: 10.1210/jc.2009-1965. [DOI] [PubMed] [Google Scholar]
- Girschick HJ, Seyberth HW, Huppertz HI. Treatment of childhood hypophosphatasia with nonsteroidal antiinflammatory drugs. Bone. 1999;25:603–607. doi: 10.1016/s8756-3282(99)00203-3. [DOI] [PubMed] [Google Scholar]
- Girschick HJ, Schneider P, Haubitz I, et al. Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia. Orphanet J Rare Dis. 2006;1:24. doi: 10.1186/1750-1172-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan JL, Narisawa S, Lemire I, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777–787. doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girschick HJ, Haubitz I, Hiort O, Schneider P. Long-term follow-up of bone mineral density in childhood hypophosphatasia. Joint Bone Spine. 2007a;74:263–269. doi: 10.1016/j.jbspin.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Girschick HJ, Mornet E, Beer M, Warmuth-Metz M, Schneider P. Chronic multifocal non-bacterial osteomyelitis in hypophosphatasia mimicking malignancy. BMC Pediatr. 2007b;7:3. doi: 10.1186/1471-2431-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collmann H, Mornet E, Gattenlohner S, Beck C, Girschick H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst. 2009;25:217–223. doi: 10.1007/s00381-008-0708-3. [DOI] [PubMed] [Google Scholar]
- Lynch CD, Ziada HM, Buckley LA, O'Sullivan VR, Aherne T, Aherne S. Prosthodontic rehabilitation of hypophosphatasia using dental implants: a review of the literature andtwo case reports. J Oral Rehabil. 2009;36:462–468. doi: 10.1111/j.1365-2842.2009.01948.x. [DOI] [PubMed] [Google Scholar]
- Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994;15:439–461. doi: 10.1210/edrv-15-4-439. [DOI] [PubMed] [Google Scholar]
- Stevenson DA, Carey JC, Coburn SP, et al. Autosomal recessive hypophosphatasia manifesting in utero with long bone deformity but showing spontaneous postnatal improvement. J Clin Endocrinol Metab. 2008;93:3443–3448. doi: 10.1210/jc.2008-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinico M, Levaillant JM, Vergnaud A, et al. M Specific osseous spurs in a lethal form of hypophosphatasia correlated with 3D prenatal ultrasonographic images. Prenat Diagn. 2007;27:222–227. doi: 10.1002/pd.1648. [DOI] [PubMed] [Google Scholar]