

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Laing distal myopathy (MPD1, early-onset distal myopathy).
1.2 OMIM# of the disease
160500.
1.3 Name of the analysed genes or DNA/chromosome segments
Myosin heavy chain 7, cardiac muscle, beta: MYH7.
1.4 OMIM# of the gene(s)
160760.
1.5 Mutational spectrum
10 mutations in MYH7 have been published in full papers or abstracts as causing Laing distal myopathy.1, 2, 3 All mutations to date are dominant. There is a relatively high new mutation rate.1, 2 The p.Lys1617del mutation is a recurrent mutation.4 The mutations published have been missense mutations to proline, charge reversing mutations (Glu>Lys) or deletions of amino acids in short repeat sequences.1, 2, 3
1.6 Analytical methods
Mutation screening in MYH7 is carried out by direct sequencing analysis of exons 30–40. A mutation of the head of MYH7 has been reported in one case where the patient had a distal myopathy similar to Laing distal myopathy and an associated cardiomyopathy.5 Therefore, in cases where no mutations are identified, where the clinical picture points convincingly to an MYH7 mutation, the remaining MYH7 exons may also be screened. A mutation in Kelch-like homologue 9 (KLHL9) has recently been identified in a family with a similar phenotype to Laing distal myopathy,6 and therefore KLHL9 should be screened in any patient with a Laing distal myopathy phenotype in which no MYH7 mutation is identified. Recessive missense mutations in nebulin (NEB) can cause a similar phenotype to Laing distal myopathy and NEB may be screened in patients with a Laing distal myopathy clinical phenotype and a family structure compatible with recessive inheritance.7
1.7 Analytical validation
When a mutation is identified by bi-directional direct sequencing, the test is repeated from a fresh dilution of genomic DNA.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
Unknown.
1.9 If applicable, prevalence in the ethnic group of investigated person
Not applicable.
1.10 Diagnostic setting

Comment:
Although Laing distal myopathy (MPD1) is a dominant disease, the high new mutation rate means that isolated cases with a compatible clinical phenotype should be analysed for mutations. Although the disease is relatively mild, except in certain cases with associated cardiomyopathy, prenatal diagnosis may be requested by family members. Other mutations in MYH7 may cause pure cardiomyopathy, myosin storage myopathy or scapuloperoneal myopathy.8, 9
2. TEST CHARACTERISTICS
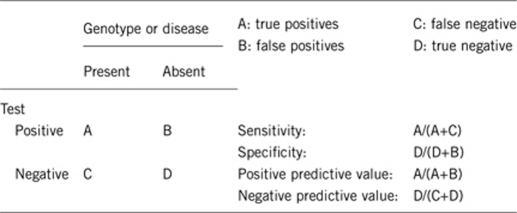
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
Approximately 100%.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
Approximately 100%.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Approximately 50% of cases of early-onset distal myopathy with anterior tibial predominance have MYH7 mutations.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Approximately 100%.
2.5 Positive clinical predictive value (lifetime risk of not developing the disease if the test is positive)
Approximately 100%: incomplete penetrance has not been described.
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Approximately 100%.
Index case in that family had not been tested:
Approximately 50%.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
Not applicable.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Not applicable.
3.1.4 Will disease management be influenced by the result of a genetic test?

Therapy (please describe): Physiotherapy and splinting are helpful for footdrop. Medical therapy may be applied for cardiac involvement, although this is not common.
Prognosis (please describe): Good. Patients in their seventh decade are most often still ambulant. Cardiac involvement is not usual. In the largest series of patients published to date, the disease was not associated with reduced life expectancy.10
Management (please describe): Supportive.
3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
In the event of a positive gene result, all patients must have a cardiac evaluation. Also, early referral to a physiotherapist can prevent the secondary tightening of the tendo Achilles, which occurs in all patients with time. A physiotherapist can also devise an appropriate exercise programme, which is also beneficial for prevention of back pain and preservation of strength in minimally affected muscles.
If the test result is negative (please describe):
If a genetic test is negative, tests of other disease-causing genes causing similar phenotypes should be pursued.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
All at-risk family members must have a cardiac evaluation.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 2.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnostic?
Yes.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
A positive genetic test result, in a patient with the recognisable clinical phenotype, provides a molecular diagnosis for the patient's symptoms, and removes the need for further expensive and possibly invasive investigations, such as electromyography, muscle or nerve biopsy. It also allows accurate genetic counselling both for the patient and for his/her relatives. However, since the largest patient cohort published to date confirms that the clinical, EMG and muscle pathology phenotypes may be highly variable,10 in the index patient of a new family or in a ‘sporadic' patient with possible de novo mutation, the orientation towards molecular genetic diagnosis may need both EMG and muscle biopsy of an affected muscle.
Acknowledgments
This work was supported by EuroGentest, an EU-FP6-supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2) and the Australian National Health and Medical Research Council (NH&MRC) Fellowship 403904 to NGL.
The authors declare no conflict of interest.
References
- Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1) Am J Hum Genet. 2004;75:703–708. doi: 10.1086/424760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd B. 165th ENMC international workshop: distal myopathies 6–8th February 2009 Naarden, The Netherlands. Neuromusc Disord. 2009;19:429–438. doi: 10.1016/j.nmd.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Lamont PJ, Wallefeld W, Junckerstorff R, Laing NG. Multiminicore myopathy caused by a mutation in MYH7. Neuromusc Disord. 2009;19:590–591. [Google Scholar]
- Lamont PJ, Udd B, Mastaglia FL, et al. Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psych. 2006;77:208–215. doi: 10.1136/jnnp.2005.073825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darin N, Tajsharghi H, Ostman-Smith I, Gilljam T, Oldfors A. New skeletal myopathy and cardiomyopathy associated with a missense mutation in MYH7. Neurology. 2007;68:2041–2042. doi: 10.1212/01.wnl.0000264430.55233.72. [DOI] [PubMed] [Google Scholar]
- Cirak S, von Deimling F, Sachdev S, et al. Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain. 2010;133:2123–2135. doi: 10.1093/brain/awq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Lehtokari VL, Kalimo H, et al. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain. 2007;130:1465–1476. doi: 10.1093/brain/awm094. [DOI] [PubMed] [Google Scholar]
- Oldfors A. Hereditary myosin myopathies. Neuromusc Disord. 2007;17:355–367. doi: 10.1016/j.nmd.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Pegoraro E, Gavassini BF, Borsato C, et al. MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromusc Disord. 2007;17:321–329. doi: 10.1016/j.nmd.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Muelas N, Hackman P, Luque H, et al. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology. 2010;75:732–741. doi: 10.1212/WNL.0b013e3181eee4d5. [DOI] [PubMed] [Google Scholar]