

Abstract
A rare syndrome of rapid-onset obesity with hypothalamic dysfunction, hypoventilation and autonomic dysregulation (ROHHAD) has been recently described. We report the first patient with this syndrome in Southeast Asia and review reported cases to date. Our patient was good health with normal development until the age of 2. He then developed hyperphagic obesity, hypersomnolence, seizures, alveolar hypoventilation, central hypothyroidism, sodium and water dysregulation, gastrointestinal dysmotility, strabismus, disordered temperature and irregular heart rate, altered sweating, delayed puberty, mental retardation and recurrent respiratory tract infections. The cardiomyopathy with heart failure and abnormal cerebral spinal fluid (CSF) neurotransmitter analysis present in our patient have not been reported previously. Tumours of the sympathetic nervous system are known to be associated with this syndrome but had not been found in our patient at the time of reporting. We highlight the difficulty of achieving the diagnosis of ROHHAD syndrome and its overlap with other well-established disease entities. The mortality and morbidity resulting from the high incidence of cardiorespiratory arrest may be prevented by early ventilatory support.
Background
In 1965, Fishman applied the literary misnomer ‘Ondine's curse’ when describing a case of late-onset primary alveolar hypoventilation syndrome in the absence of primary lung, cardiac, brainstem and neuromuscular disease associated with hypothalamic dysfunction.1 This clinical entity consisted of symptoms and signs that overlapped those of two other disorders: congenital central hypoventilation syndrome (CCHS),2 which presents in the neonatal period, and late-onset central hypoventilation syndrome (LO-CHS), which is not associated with hypothalamic dysfunction. Studies attempting to delineate the origin of and relationship between CCHS and LO-CHS point to a genetic basis and both disorders can be inherited dominantly from the mutated paired-like homeobox 2B gene (PHOX2B) that encodes a transcription factor in the autonomic nervous system.3–6 However, recent reports have described another group of patients reminiscent of Fishman's case and not associated with PHOX2B mutation. This clinical spectrum has been renamed rapid-onset obesity with hypothalamic dysfunction, hypoventilation and autonomic dysregulation (ROHHAD). Molecular studies have also found it to be independent of the other genes involved in neuronal development, namely brain-derived neurotrophic factor (BDNF) and its receptor tyrosine kinase receptor b (TRKB),7 as well as ASCL1 (previously called HASH-1) and NECDIN.8 Clinically, the sequence of events in ROHHAD has been outlined as apparent normality in the first 2–4 years of life, followed by hyperphagic weight gain coupled with decrease in height velocity, then hypothalamic and autonomic dysregulation, change in behaviour and alveolar hypoventilation.7 9 We present the first case of ROHHAD in Malaysia and review the previously reported 52 cases, some diagnosed as late-onset central hypoventilation with hypothalamic dysfunction (LOCH/HD). In our patient the results of cerebrospinal fluid (CSF) neurotransmitter assay were consistent with mild inflammation.
Case presentation
This 11 ½-year-old Malaysian Chinese boy is the elder of two siblings born to non-consanguineous parents with a birth weight of 2.6 kg at full term via normal delivery. He had a normal perinatal period.
At 3 years of age, he presented with recurrent seizures and global developmental delay. On further interview, it was noted that he had experienced significant weight gain since the age of 2 and was always a ‘good’ and quiet child, sometimes even falling asleep in the bath. EEG showed evidence of absence seizures and cranial CT showed mild generalised cerebral atrophy.
Seizures were well controlled with anti-epileptics until the child was 5 years old when he presented with status epilepticus. He required prolonged ventilation in the paediatric intensive care unit with unsuccessful extubations on two occasions because of poor respiratory effort. Following discharge, he was dependent on continuous positive airway pressure (CPAP) ventilation via tracheostomy for 1 month. He was subsequently hypotonic with generalised weakness but was well enough to attend a school for disabled children. He remained overweight and was further admitted to hospital with intermittent but severe unexplained abdominal distension with apparent normal bowel habits. He was noted to have symptoms of obstructive sleep apnoea. Thyroid function tests revealed central hypothyroidism and thyroxine replacement was commenced.
At 6 ½ years of age, he presented with fever, drowsiness and shallow breathing. Investigations showed hypercapnia and respiratory acidosis accompanied by radiological evidence of pneumonia. He was reventilated via tracheostomy and has since remained dependent on BiPAP ventilation 24 h/day. During this admission, his weight was recorded as 35 kg (>97th percentile). A respiratory physiological study revealed obstructive sleep apnoea and alveolar hypoventilation during sleep. He also had bradycardia and transient Mobitz type 1 heart block. His echocardiogram was normal. Other investigations including creatine kinase, serum ammonia, serum amino acid, urine amino and organic acid, lactate (in both serum and CSF), CSF cultures, carnitine, random cortisol level, electromyography, nerve conduction studies, muscle biopsy, MRI and angiography of the brain, chromosome analysis, methylation study for Prader–Willi syndrome and MELAS (myeloencephalopathy, lactic acidosis, stroke-like episodes) 3243 mutation analysis were all normal. A glucose loading test showed mild elevation of postprandial lactate at 60 min. Formal assessments by the ophthalmologist revealed a right divergent squint, while a hearing test was reported as normal. Co-enzyme Q10 and a vitamin cocktail were administered for presumed mitochondrial cytopathy without any clear benefit.
Subsequently, the patient had eight further admissions for recurrent upper respiratory tract infections, pneumonia and encephalopathy secondary to CO2 retention. During one of these admissions, he had intermittent asymptomatic sinus bradycardia with a normal echocardiogram (ECHO). On another occasion, severe hypoxia resulted in cardiomyopathy (left ventricular ejection fraction of 27%), pleural and pericardial effusions, ascites and pre-renal impairment. These resolved with anti-failure medications and careful fluid management. Biochemical evidence of diabetes insipidus (hypernatraemia with highest Na+ recorded as 192 mmol/l, normal serum osmolality and low urine osmolality) with normal range urine output was documented during another admission. This resolved with desmopressin and fluid and electrolyte management. On another occasion, the patient experienced an episode of cardiorespiratory arrest after the administration of a sedative. Throughout this time, he demonstrated hyperphagia to the extent of stealing food in the middle of the night. There was also intermittent temperature dysregulation manifesting as both hyperthermia and hypothermia, as well as excessive sweating.
At the time of writing he was 11 of age and remained prepubertal. His BMI (body mass index) was 26. Follicle-stimulating hormone (FSH) was 0.3 U/l (normal range: 0.9–15), luteinising hormone (LH) <0.5 U/l (normal range: 1.3–12.9) and testosterone <0.5 nmol/l (normal range: 8.4–28.7), consistent with either a hypogonadotrophic hypogonadism or a prepubertal state. His bone age was 11 years. Prolactin was 465 U/l (normal range: 59–619), adrenocorticotropic hormone was <2 pmol/l (normal range: <49) and thyroid function test was normal on thyroxin replacement. Fasting lipid profile and blood sugars were normal. ECHO and 24-h Holter monitoring were normal. MRI of the brain and pituitary fossa showed normal pituitary and generalised cerebral atrophy (figure 1). CSF neurotransmitter analysis showed the following normal metabolites: homovanillic acid (HVA) 0.53 µmol/l, biopterin 15.3 nmol/l, 7-biopterin 1.3 nmol/l, neopterin 84.7 nmol/l and monapterin 9 nmol/l. However, 5-hydroxyindoleacetic acid (5-HIAA) was low at 0.09 nmol/l (normal range: 0.13–0.21) but was not specific for known neurotransmitter disorders. This may be attributed to low level central nervous system inflammation. Sequencing of PHOX2B gene was normal.
Figure 1.
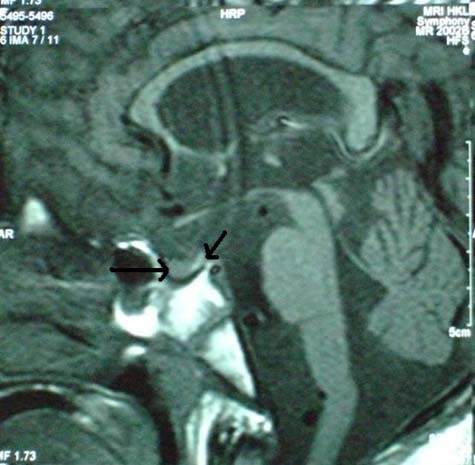
MRI of the pituitary fossa showing a normal anterior and posterior (arrows) pituitary.
Discussion
Almost all the previously reported patients with ROHHAD were of Caucasian and Arabic descent except for one case each of Japanese and Indian origin. We present here the first case of ROHHAD in the Southeast Asian population. The rarity of the condition, the lack of specific diagnostic tests and the likelihood of under-diagnosis may explain why no previous cases have been reported from this region.
Although many genes involved in neuronal development have been investigated for ROHHAD, no specific genetic marker has been established. Nevertheless, familial cases have been reported, suggesting that it may be a monogenic condition.8 The PHOX2B mutations implicated in CCHS are absent in children with ROHHAD, as was also the finding in our patient.
Several recent case series have examined the characteristics and natural progression of ROHHAD. The most consistent first symptom is hyperphagic obesity after the age of 2, followed by central hypoventilation months or years later.7–9 It is otherwise a heterogenous syndrome with some children exhibiting marked endocrine involvement,9 while others have neural crest tumours early on or marked behavioural disturbance.10 Our patient had a normal birth and development until the age of 2. However, he did not present with hyperphagic obesity or hypoventilation and medical attention was sought instead for seizures and developmental delay. Although these symptoms were previously noted common associations of ROHHAD (table 1), they were not the focus of description or presenting features. In addition, they were also the symptoms of many other neurodevelopmental disorders, thus further clouding the diagnosis. It was on retrospective review that this patient was noticed to have significant and rapid weight gain and hypersomnolence from the age of 2. Alveolar hypoventilation manifested more than 2 years after first presentation. Hypothalamic dysfunction manifested at the age of 5 and autonomic dysregulation at 6 ½. This long interval between the onset of rapid weight gain, hypoventilation, hypothalamic and autonomic dysfunction, adds to the difficulty of diagnosis and management.
Table 1.
Summary of reported phenotypes of patients with ROHHAD
| Present case | De Pontual et al8 | Bougnères et al9 | Ize-Ludlow et al7 | Aboushanab et al16 | Gothi et al17 | Kobayashi et al18 | Sirvent et al15 | Katz et al14 | Nunn et al10 | Del Carmen et al19 | North et al13 | Proulx et al20 | Gurewitz et al12 | duRivage et al11 | Frank et al21 | Dunger et al22 | Nattie et al23 | Fishman et al1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of cases | 1 | 13 | 6 | 15 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 |
| Gender, male:female | M | 7:6 | ? | 6:9 | F | F | M | F | F | M | F | M | F | M | 1:1 | F | M | F | M |
| Age at onset, years (range)* | (2) | (0; 9) | (1.5; 4.3) | (1.5; 4) | 3 | 8 | 2.25 | 1.5 | 2 | 3.5 | 2.5 | 2.3 | 4 | 4 | (4; 9) | 5 | 4.5 | 1.6 | 2.75 |
| Rapid onset ↑weight | + | 12/13 | 6/6 | 15/15 | + | + | + | + | + | + | + | + | + | + | 2/2 | + | + | + | + |
| Central ↓ventilation | + | 13/13 | 6/6 | 15/15 | + | + | + | + | + | + | + | + | + | + | 2/2 | + | + | + | + |
| ↑Na+ | + | 10/13 | 6/6 | 7/15 | + | + | + | + | + | + | 2/2 | + | |||||||
| ↓Na+ | + | 7/13 | 4/15 | + | + | SIADH | |||||||||||||
| DI | +† | 0/13 | 0/6 | 5/15 | + | + | + | ||||||||||||
| Hypodipsia/adipsia | + | 7/13 | + | + | + | + | + | 1/2 | + | + | |||||||||
| Central ↓T4 | + | 6/13 | ‡ | 5/15 | + | + | + | + | + | 2/2 | + | ||||||||
| GH def | 7/13 | 4/6 | 9/9 | + | + | + | + | + | + | + | 2/2 | + | |||||||
| ↑Prolactin | – | 5/13 | 4/6 | 7/15 | + | + | + | + | + | 2/2 | + | ||||||||
| Hypogonadism | Possible | 3/13 | 4/6 | 5/15 | 2/2 | + | |||||||||||||
| Precocious puberty | – | 1/13 | 0/6 | 2/15 | + | + | |||||||||||||
| Altered sweating | + | 8/15 | + | + | + | ||||||||||||||
| BP dys | – | 5/15 | + | ||||||||||||||||
| HR dys | + | + | + | ||||||||||||||||
| Temperature dys | + | 6/13 | 4/6 | 11/15 | + | + | + | + | + | + | |||||||||
| ↓Pain | – | 3/13 | 8/15 | + | + | + | + | + | + | ||||||||||
| Strabismus | + | 5/13 | 1/6 | 7/15 | + | + | + | + | |||||||||||
| Fixed dilated pupils | – | 6/13 | 2/6 | 8/15 | + | + | + | + | |||||||||||
| Neural crest tumours | – | GN [3/13] NB [2/13] |
GN [6/6] | GNB [3/15] GN [2/15] |
GN | NB | GN | Thoracic GNB | GNB | GN | GN | NB | |||||||
| Others | Seizures, mental retardation, recurrent respiratory tract infection, hypotonia, cardiomyopathy, cerebral atrophy | Behavioural disorders [6/13], mental retardation [6/13] | GI dysmotility [1/6], mental retardation [2/6], developmental regression [1/6], psychosis [1/6] | Ptosis [1/15], GI dysmotility [10/15], behavioural disorders [8/15], seizures [3/15], developmental delay [3/15] | Mood disorder | Mood disorder | ↓TSH and LH/FSH §, ptosis, abnormal ocular movement, personality change | Mood disorder | Equinovarus, scoliosis, mood disorder | Stuttering, behavioural disorders | Mood disorder | Mood disorder, developmental regression | Behavioural disorders, seizures, retardation | Ataxia/confusion [1/2] | Mood disorder, seizures | Mood disorder | Personality change | Stuttering, seizures | |
| PHOX2B | Normal | Normal [13/13] | ? | Normal [15/15] | Normal | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? |
| Outcome (age in years) | Alive (11), Tr/BiPAP full time | 2/13 died ¶ Tr [7/13] Mask PPV [6/13] 6/13 full time ventilation | 2/6 died (8.5 and 12), 2/6 full time ventilation | 1/15 died, 7/15 full time ventilation | Alive (8), Tr/BiPAP at night, full time O2 supplement | Alive (10) | Died (3.7) | Alive (3) | Alive (8.75), NIPPV at night | Died (4) CPAP for sleep | Alive (5.5) Tr/PPV | Died (4) Tr/PPV full time | Died (10.5) | Alive (20) | Alive (13.5 and 15.5) PNP [2/2] | Died (8) Tr/PPV | Alive (13) | Alive (2) Tr/PNP | Alive (4) Tr/PPV for sleep |
Age of onset of any symptoms of ROHHAD
Water deprivation test not done
Two patients died at 1 month and 5 ½ years after onset of disease respectively
4/6 patients exhibited TSH responses to TRH that indicated hypothalamic dysfunction but only 2 had low fT4 level
M, Male; F, Female; Wt, weight; ↓T4, hypothyroidism; GH def, growth hormone deficiency; DI, diabetes insipidus; SIADH, Syndrome of Inappropriate Antidiuretic Hormone; BP, blood pressure; HR, heart rate; T °, temperature; dys, dysregulation; Dev't, developmental; GNB, Ganglioneuroblastoma; GN, ganglioneuroma; NB, neuroblastoma; NIPPV, non-invasive positive pressure ventilation; Tr, tracheostomy; BiPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; PPV, positive pressure ventilation; PNP, phrenic nerve pacing; GI, gastrointestinal
The different symptoms of the constellation of symptoms in ROHHAD, appearing in stages, can be confused with those of many other disorders. Many differential diagnoses were considered in our patient even after neuroanatomical lesions of the central and peripheral nervous systems together with primary lung and cardiac diseases had been ruled out. Severe common obesity is often complicated with obstructive sleep apnoea, hypoventilation and hypersensitivity of the hypothalamic–pituitary axis resulting in hypercortisolism, depressed sex hormones, increased thryoid-stimulation hormone (TSH) and growth hormone unresponsiveness to stimulation tests. Nevertheless, in contrast to ROHHAD, obese children have elevated IGF-1 levels, and thus tall stature and related complications should improve with weight loss. Similar to previously reported cases, obesity syndromes were considered in our patient. Among them, the more common Prader–Willi syndrome shares many features with ROHHAD. However, the absence of dysmorphic facies and a negative DNA methylation test make Prader–Willi syndrome unlikely. Other obesity syndromes that may be considered include Bardet–Biedl and Alstrom syndromes. Again these can be distinguished by their distinctive phenotype. Congenital leptin deficiency presents with hyperphagic obesity and endocrine and autonomic manifestations that closely resemble those of ROHHAD. However, the onset of obesity in such patients is usually in early infancy and progresses more rapidly. Our review of reported ROHHAD cases revealed that the age of onset of disease ranges from 0 to 9 years old but the most common age of presentation is around 2 years old. Our patient has the clinical phenotype consistent with this; leptin level testing was not performed as this would have been an unnecessary expense for the family.
Consistent with the published literature, our patient had various degrees of autonomic dysfunction (table 1). This manifested as several episodes of symptomatic bradycardia, temperature dysregulation, increased sweating, right divergent strabismus and gastrointestinal dysmotility with severe abdominal distensions necessitating hospital admissions. These symptoms together with developmental delay and the multisystemic nature of the illness led to investigations for neurometabolic disorders, namely mitochondriapathy, cerebral organic acidaemia, fatty acid oxidation defects and neurotransmitter diseases. Mitochondrial cytopathy is a group of heterogeneous disorders with neuromuscular manifestations including psychomotor retardation or regression, hypotonia, seizures, ophthalmoplegia, autonomic disturbance, and pyramidal or extrapyrimidal signs, resulting in considerable overlap with the symptoms of ROHHAD. However, children with mitochondrial disease often fail to thrive and disease course is progressive, although it can be static for long periods of time. Investigations such as lactate, organic and amino acids, glucose challenge, neuroradiological studies and muscle biopsy findings may support the diagnosis. Disturbances of autonomic regulation may also be a prominent feature in neurotransmitter diseases. This together with ptosis, abnormal pupillary responses, mood or personality change, epilepsy, ataxia and other cerebellar symptoms and psychomotor retardation are features of both monogenic disorders of neurotransmission and ROHHAD. Indeed, it was speculated that a deficiency of serotonin, a neurotransmitter, could underlie the pathophysiology of ROHHAD11 and clomipramine has been used in treatment because of its function as a serotonin and norepinephrine reuptake inhibitor.12 Although manipulation of neurotransmitters appeared to have a beneficial effect on central apnoea, it was proved to be ineffective in treating the hypothalamic, autonomic or brainstem symptoms.10 12 In contrast to ROHHAD, neurotransmitter diseases are associated with severe encephalopathy, intractable seizures, and abnormal tone, posture and movements which when present are not easily confused with ROHHAD. CSF neurotransmitter examination was not performed in reported patients with ROHHAD except by Nunn et al who reported a normal result.10 We report for the first time the result of CSF neurotransmitter analysis that could be consistent with mild CNS inflammation in our patient. It remains to be seen if this is a consistent finding in other cases or important pathogenetically.
Searches for a neuroanatomical pathology that can explain the symptoms of ROHHAD have not yielded consistent findings. Reported cranial MRI pathologies associated with ROHHAD include bilateral basal ganglia hypodensities,13 Rathke's cleft cyst7 9 and hypointensities in the pons and midbrain.7 Hypothalamic inflammation with lymphocytic infiltrates was found in two cases on histopathological examination, but this was associated with tumours of the sympathetic nervous system (ganglioneuroma and ganglioneuroblastoma).10 13 CT and MRI of the cranium showed cerebral atrophy in our patient at ages 3 and 11, respectively. Both the anterior and posterior pituitary were reported to be normal on the MRI of the pituitary fossa (figure 1).
Patients with central hypoventilation syndrome usually have adequate ventilation while awake but a blunted response to hypercarbia results in hypoxaemia, necessitating artificial ventilation at night. More severely affected patients hypoventilate both when asleep and when awake. Our patient requires around the clock mechanical ventilation via tracheostomy. From the data available on the reported ROHHAD patients, 35 required artificial ventilation and 16 of these required full time ventilation. This may mean that patients with ROHHAD have the more severe type of alveolar hypoventilation. It is also evident from the literature that cardiorespiratory arrest and its related morbidity are common in this syndrome (table 1). Previous reports have suggested that early intervention with nocturnal artificial ventilation may improve daytime ventilation.14 To institute this, early recognition and diagnosis of ROHHAD is particularly important. Patients suspected of having this diagnosis need thorough respiratory physiological studies. Apart from aggressive early ventilation, it is also noted that sedation sometimes triggers respiratory arrest. This occurred in our patient as well as the case reported by Nunn et al.10
Careful investigations of the hypothalamic and autonomic systems are required as is follow-up on behavioural and developmental progress so that abnormalities can be addressed appropriately. ROHHAD has been postulated to be a paraneoplastic syndrome, especially when associated with tumours of the sympathetic nervous system.10 13 15 It is yet unclear whether neoplasm is a cause or effect of this syndrome. Twenty-six out of the 52 patients reviewed had neural crest tumours at the time of reporting. Although initial investigations in our patient did not reveal any tumours, regular surveillance for life is necessary. Reported cardiovascular involvement in ROHHAD included arrhythmias, blood pressure dysregulation and right ventricular hypertrophy secondary to cor pulmonale. Our patient had cardiomyopathy complicated by heart failure. This resolved with time and we postulate it to be due to a hypoxic event. Vigilant surveillance of cardiac function forms an important part of management.
Improved awareness and better understanding of the natural history and outcome of ROHHAD not only guide management of the physical abnormalities, but can also facilitate the counselling process which sometimes is central to providing optimal patient care.
Learning points.
-
▶
ROHHAD is a heterogenous syndrome with presenting features that mimic many other diseases.
-
▶
The cause of the disease is unknown.
-
▶
We present for the first time changes in CSF neurotransmitters and discuss their relevance to pathogenicity.
-
▶
The complication of cardiomyopathy with heart failure has not been previously reported.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Fishman LS, Samson JH, Sperling DR. Primary alveolar hypoventilation syndrome (Ondine's curse). Am J Dis Child 1965;110:155–61 [DOI] [PubMed] [Google Scholar]
- 2.Mellins RB, Balfour HH, Jr, Turino GM, et al. Failure of automatic control of ventilation (Ondine's curse). Report of an infant born with this syndrome and review of the literature. Medicine (Baltimore) 1970;49:487–504 [PubMed] [Google Scholar]
- 3.Trang H, Dehan M, Beaufils F, et al. ; French CCHS Working Group The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest 2005;127:72–9 [DOI] [PubMed] [Google Scholar]
- 4.Trochet D, de Pontual L, Straus C, et al. PHOX2B germline and somatic mutations in late-onset central hypoventilation syndrome. Am J Respir Crit Care Med 2008;177:906–11 [DOI] [PubMed] [Google Scholar]
- 5.Gallego J, Dauger S. PHOX2B mutations and ventilatory control. Respir Physiol Neurobiol 2008;164:49–54 [DOI] [PubMed] [Google Scholar]
- 6.Repetto GM, Corrales RJ, Abara SG, et al. Later-onset congenital central hypoventilation syndrome due to a heterozygous 24-polyalanine repeat expansion mutation in the PHOX2B gene. Acta Paediatr 2009;98:192–5 [DOI] [PubMed] [Google Scholar]
- 7.Ize-Ludlow D, Gray JA, Sperling MA, et al. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation presenting in childhood. Pediatrics 2007;120:e179–188 [DOI] [PubMed] [Google Scholar]
- 8.De Pontual L, Trochet D, Caillat-Zucman S, et al. Delineation of late onset hypoventilation associated with hypothalamic dysfunction syndrome. Pediatr Res 2008;64:689–94 [DOI] [PubMed] [Google Scholar]
- 9.Bougnères P, Pantalone L, Linglart A, et al. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab 2008;93:3971–80 [DOI] [PubMed] [Google Scholar]
- 10.Nunn K, Ouvrier R, Sprague T, et al. Idiopathic hypothalamic dysfunction: a paraneoplastic syndrome? J Child Neurol 1997;12:276–81 [DOI] [PubMed] [Google Scholar]
- 11.duRivage SK, Winter RJ, Brouillette RT, et al. Idiopathic hypothalamic dysfunction and impaired control of breathing. Pediatrics 1985;75:896–8 [PubMed] [Google Scholar]
- 12.Gurewitz R, Blum I, Lavie P, et al. Recurrent hypothermia, hypersomnolence, central sleep apnea, hypodipsia, hypernatremia, hypothyroidism, hyperprolactinemia and growth hormone deficiency in a boy–treatment with clomipramine. Acta Endocrinol Suppl (Copenh) 1986;279:468–72 [DOI] [PubMed] [Google Scholar]
- 13.North KN, Ouvrier RA, McLean CA, et al. Idiopathic hypothalamic dysfunction with dilated unresponsive pupils: report of two cases. J Child Neurol 1994;9:320–5 [DOI] [PubMed] [Google Scholar]
- 14.Katz ES, McGrath S, Marcus CL. Late-onset central hypoventilation with hypothalamic dysfunction: a distinct clinical syndrome. Pediatr Pulmonol 2000;29:62–8 [DOI] [PubMed] [Google Scholar]
- 15.Sirvent N, Bérard E, Chastagner P, et al. Hypothalamic dysfunction associated with neuroblastoma: evidence for a new Paraneoplastic syndrome? Med Pediatr Oncol 2003;40:326–8 [DOI] [PubMed] [Google Scholar]
- 16.Aboushanab OA, Alotaibi SA, Shaalan YA. Late-onset central hypoventilation syndrome with hypothalamic dysfunction in a Kuwaiti girl. Kuwait Medical J 2007;39:376–8 [Google Scholar]
- 17.Gothi D, Joshi JM. Late onset hypoventilation syndrome: is there a spectrum of idiopathic hypoventilation syndromes? Indian J Chest Dis Allied Sci 2005;47:293–7 [PubMed] [Google Scholar]
- 18.Kobayashi K, Miyamoto J, Hasegawa Y. A sudden death due to central hypoventilation in a 3 year old boy with idiopathic hypothalamic dysfunction. Clin Pediatr Endocrinol 2003;12:7–11 [Google Scholar]
- 19.Del Carmen Sanchez M, Lopez-Herce J, Carrillo A, et al. Late onset central hypoventilation syndrome. Pediatr Pulmonol 1996;21:189–91 [DOI] [PubMed] [Google Scholar]
- 20.Proulx F, Weber ML, Collu R, et al. Hypothalamic dysfunction in a child: a distinct syndrome? Report of a case and review of the literature. Eur J Pediatr 1993;152:526–9 [DOI] [PubMed] [Google Scholar]
- 21.Frank Y, Kravath RE, Inoue K, et al. Sleep apnea and hypoventilation syndrome associated with acquired nonprogressive dysautonomia: clinical and pathological studies in a child. Ann Neurol 1981;10:18–27 [DOI] [PubMed] [Google Scholar]
- 22.Dunger DB, Leonard JV, Wolff OH, et al. Effect of naloxone in a previously undescribed hypothalamic syndrome. A disorder of the endogenous opioid peptide system? Lancet 1980;1:1277–81 [DOI] [PubMed] [Google Scholar]
- 23.Nattie EE, Bartlett D, Rozycki AA. Central alveolar hypoventilation in a child: an evaluation using a whole body plethysmograph. Am Rev Respir Dis 1975;112:259–66 [DOI] [PubMed] [Google Scholar]