

Abstract
ADAR2, a member of the adenosine deaminase family of proteins, is the enzyme that edits the Q/R site in the GluR-B transcript, an important physiological A-to-I editing event. ADAR2 pre-mRNA undergoes a number of known alternative splicing events, affecting its function. Here we describe a novel alternatively spliced exon, located within intron 7 of the human gene, which we term “exon 7a”. This alternatively spliced exon is highly conserved in the mammalian ADAR2 gene. It has stop codons in all three frames and is down regulated by NMD. We show that the level of exon 7a inclusion differs between different human tissues, with the highest levels of inclusion in skeletal muscle, heart and testis. In the brain, where the level of editing is known to be high, the level of exon 7a inclusion is low. The new alternative form was also found in supraspliceosomes, which constitute the nuclear pre-mRNA processing machine. The high conservation of the novel ADAR2 alternative exon in mammals indicates a physiological importance for this exon.
Keywords: ADAR2, alternative splicing, tissue specificity, supraspliceosome
Introduction
A to I RNA editing is the process in which adenosines (A) are deaminated into inosines (I). Because inosines base pair with cytosines they are recognized as guanosines by the translation and splicing machineries, and therefore can lead to changes in the splicing patterns and coding capacities of the edited RNA (reviewed in refs. 1–3). The deamination is performed by the ADAR (adenosine deaminase that acts on RNA) family of proteins, which appear in several isoforms in both vertebrates and invertebrates. In mammals there are 3 known ADAR proteins—ADAR1-3. The mammalian ADAR2 is homologous to the drosophila sole ADAR protein.4 Other vertebrates show sequence homology with the 3 mammalian ADARs. Because ADARs act on double stranded RNA, they contain double stranded RNA binding motifs (dsRBMs) in addition to the deaminase domain.
Although the amount of inosines in rat RNAs implies massive editing,5 the number of known transcripts that contain specifically edited adenosines within coding regions is small, though recent analyses extended the list of specific editing events.6,7 On the other hand, bioinformatics analyses have predicted many editing events in non-coding regions, mostly in Alu sequences.8–10 The implications of these editing events are not yet fully understood. Yet, A-to-I changes in non-coding regions can have a pleiotropic effect: generate, hinder, or otherwise affect cis-acting splicing elements, influence RNAi, and may cause RNA instability or nuclear retention.3, 11, 12
ADAR1 and ADAR2 are expressed in several tissues at different levels, suggesting their general importance. Nonetheless, most of the editing found in coding regions occurs in mRNAs expressed in the central nervous system. A vital editing site of ADAR2 is in the GluR-B subunit of the AMPA receptor.13,14 This editing event changes a glutamine codon to an arginine one (the Q/R site) and thereby affects the efficiency of the splicing of the transcript, the assembly of the AMPA receptor, and the calcium permeability of the channel.15 It has also been reported that A-to-I editing is associated with epilepsy, cancer, amyotrophic lateral sclerosis and depression.6,16
The human ADAR2 transcript is comprised of 15 known exons and can undergo a number of alternative splicing events (ref. 17; see also Fig. 1A). A number of splice options occur at the 3′ end – some changing the C-terminus of the protein and some the 3′ UTR. Additional alternative options are: an alternative Alu exon (exon 5a) can be included; exon 2, which contains the two dsRBMs of ADAR2, can be skipped; and there are two alternative exons upstream of exon 1, which extend the coding region of the alternatively spliced ADAR2 mRNA.17–23 Also, ADAR2 can edit its own transcript and create an alternative 3′ splice site close to the AUG, whose use causes the insertion of 47 nt and introduces a very early premature termination codon (PTC).24 Some of the above-mentioned splicing events (such as the latter one) appear to be conserved; whereas in other cases similar, but not identical, splicing events have been observed.17 Although not yet well understood, ADAR2 alternative splicing affects its function.
Figure 1.

A new alternative exon in ADAR2 is conserved in mammals. (A) A diagram of the ADAR2 gene spanning exon 7 through exon 8 with the alternative exon 7a in the intron in-between [the lengths (in nucleotides) of exon 7a and flanking introns are marked]. (B) RT-PCR analysis of RNA from human HeLa, rat PC12 and mouse NIH3T3 cells using primers from exons 7 (sense) and 8 (antisense). The upper and lower bands represent inclusion or skipping of exon 7a, respectively, as indicated on the left. (C) RT-PCR analysis of RNAs as in B, using primer pairs from the splice junction of exon 7-exon 7a (sense) and exon 7a-exon 8 (antisense), specific for each species. The bands represent inclusion of exon 7a. (D) Sequence alignment of a part of intron 7 of the human ADAR2 with other mammals (the sequences were taken from the UCSC genome browser, the alignment from ClustalW). The boundaries of exon 7a are marked by a blue vertical line. The stop codons in the exon are marked with different colors for the 3 frames. Percentages of homology to the human exon 7a of the listed sequences are: mouse, 93; rat, 95; rabbit, 91; dog, 64; armadillo, 80; elephant, 88. Percentages of homology to the entire human sequence shown are: mouse, 81; rat, 75; rabbit, 75; dog, 65; armadillo, 61; elephant, 72.
Alternative splicing has been associated with the control of mRNA levels through the introduction of PTCs, which direct the alternatively spliced transcripts to the nonsense mediated mRNA decay (NMD) RNA surveillance pathway (reviewed in ref. 25). The scope of this phenomenon has not yet been clearly assessed, because bioinformatics analyses have predicted many PTC-containing transcripts, whereas microarray data suggest that most of these events are not part of a control mechanism mediated through NMD.26–28 Nevertheless, it seems likely that NMD does play a role in certain families of splicing factors, as well as in other RNA-associated genes.25 For example, all members of the SR proteins family and hnRNP genes, are expressed as alternatively spliced mRNAs having PTCs in highly conserved regions.29–32 It has also been shown that abrogation of NMD resulted in elevated levels of selected PTC-containing mRNAs in these gene families. Some of these genes even show autoregulation; namely, when over expressed, the relevant protein causes a shift in the splicing of its pre-mRNA in favor of the PTC-containing mRNA.29,32
Here we report on a newly discovered alternatively spliced isoform of the human ADAR2 gene, in which a 93-nt sequence located within intron 7 is included as exon 7a. This alternative exon is conserved in mammals and its expression appears to be tissue-specific. It is found at very low levels in many tissues, amongst them the brain, but is found at higher levels in muscle, heart and testis. Exon 7a contains at least one PTC in each reading frame, which makes it a substrate for NMD. Thus, abrogation of NMD up regulated the level of the alternatively spliced mRNA, and a similar effect was observed when ADAR2 was over expressed in HeLa cells. This effect is independent of ADAR2 editing, because over expression of an ADAR2 inactive mutant also up regulates the level of the alternatively spliced isoform. Finally, we show that the new alternative splicing event is associated with supraspliceosomes - the nuclear ribonucleoprotein complexes that have been proposed to constitute the machine where RNA splicing occurs in living cells.33 Taken together, our results suggest a role for exon 7a in controlling the expression levels of the ADAR2 gene.
Results
A conserved new exon is located within intron 7 of the human ADAR2 transcript
Fig. 1B (lane 1) shows an RT-PCR analysis of ADAR2 RNA expressed in HeLa cells, using PCR primers that flank intron 7. Gel electrophoresis of the PCR products revealed the expected product joining exons 7 and 8, and a longer additional band. DNA sequencing showed that the longer product contained an additional 93-nt long exon, located 924 nt downstream of the 5′ end of the ~16 kb intron 7 (Fig. 1A). This newly discovered exon is termed hereafter as exon 7a. Alignment of the human, mouse, rat, rabbit, elephant, dog and armadillo gene sequences from the UCSC genome browser showed that the sequence of exon 7a and the flanking intronic sequences, including the 5′ and 3′ splice sites, are highly conserved in mammals (Fig. 1D). Namely, exon 7a sequences of the above species (excluding the dog) contain extensive homology (80–95%) to the human sequence, whereas the homology is lower (61–81%) when intronic sequences flanking exon 7a are included (Fig. 1D). Interestingly, in all mammalian species compared above, the new alternative ADAR2 exon contains a substantial number of stop codons in all three reading frames (Fig. 1D). RT-PCR analysis of ADAR2 RNA expressed in rat (PC12) and mouse (NIH3T3) cells, using PCR primers that flank the new exon, revealed the expression of this alternative exon (Fig. 1B, lanes 2 and 3, respectively). We confirmed the expression of exon 7a in all three species by RT-PCR using specific splice-junction primer pairs for each of the three different species: a sense primer that flanks the exon 7-exon 7a splice junction, and the respective antisense primer that flanks the exon 7a-exon 8 spice junction (Fig. 1C). The inclusion of exon 7a in mouse and rat was further confirmed by DNA sequencing. Examination of the vertebrate Multiz alignment and conservation of 17 species from the UCSC genome browser revealed that exon 7a has 2 main conserved regions, but it is not conserved in other vertebrates besides mammals (Fig. 2). A third conserved region is found in the intron of exon 7a. Its conservation suggests the presence of a control element(s) that might affect the alternative splicing of exon 7a.
Figure 2.

The conservation of ADAR2 between 17 species (human, chimp, macaque, mouse, rat, rabbit, dog, cow, armadillo, elephant, tenrec, opossum, chicken, frog, zebrafish, tetraodon and fugu). The upper panel shows the entire ADAR2 gene, and the lower panel shows exon 7a and its neighborhood (taken from the UCSC genome browser).
Inhibition of NMD up regulates the alternatively spliced exon 7a-containing ADAR2 mRNA
The alternatively spliced ADAR2 isoform was predicted to be a substrate for the NMD RNA surveillance mechanism,34 because exon 7a has stop codons in all three protein reading frames, and most of them are located more than 55 nt upstream of the exon 7a-exon 8 splice junction (Fig. 1D). To check this prediction, we analyzed the expression of ADAR2 in HeLa cells treated with the protein synthesis inhibitor cycloheximide (CHX). Because NMD is known to be dependent on protein translation,35,36 we expected that the level of the 7a-containing isoform of ADAR2 mRNA would increase upon treatment with CHX. This indeed was the case as can be seen in the qualitative PCR analysis using primers from exon 7 and exon 8 (Fig. 3A). Increase in the expression level of exon 7a after treatment with CHX was also observed in rat and mouse (Fig. 3A, PC12 and NIH3T3). A quantitative analysis of this effect in HeLa cells was carried out by real-time RT-PCR (Fig. 3C). For this aim we designed primer pairs that each should specifically amplify only one of the expected splicing products (Fig. 3B). The sense primer for the constitutive exon 7-exon 8 mRNA flanks the exon 7-exon 8 splice junction, and the anti-sense primer is from exon 8. The sense primer for the alternative exon 7-exon 7a-exon 8 mRNA flanks the exon 7-exon 7a splice junction, and the respective antisense primer flanks the exon 7a-exon 8 spice junction. The results of four independent experiments are shown in the bar diagram of Figure 3C. As can be seen, the increase in the level of the alternative ADAR2 isoform in the CHX treated cells was about 3-fold relative to that in the untreated cells, indicating that the inclusion of exon 7a rendered the alternative isoform a substrate for NMD. As a control we show that under the same conditions of CHX treatment, the mRNA level expressed from a known substrate of NMD – the β-globin Ter39 construct37 – is up regulated, while that of the wild type is unaffected (Fig. 3D). We next used RNAi to abrogate NMD by down regulating the expression of the hUpf1 essential NMD gene.38,39 As can be seen in Figure 3E, quantitative real-time RT-PCR analysis, using the primer pairs depicted in Figure 3B, revealed a similar increase in the level of exon 7a after down regulation of hUpf1. These results confirmed that the exon 7a isoform was subject to NMD. Figure 3F shows that indeed the level of hUpf1 was down regulated by the siRNA treatment.
Figure 3.

The level of ADAR2 exon 7a is down regulated by NMD. (A) RT-PCR of RNA expressed in HeLa, PC12 and NIH3T3 cells, untreated (−) or treated (+) with CHX. The primers are from exons 7 (sense) and 8 (antisense). Diagrams, on the left, show the two different products. (B) A diagram showing the two primer pairs used for the real-time RT-PCR analyses on RNA extracted from HeLa cells either untreated (control), or treated with CHX (CHX) (C), or treated with siRNA against hUpf1 (E). (C) A graph showing the real-time RT-PCR results on HeLa cells either untreated (control) or treated with CHX (CHX). The ratio between the levels of the product from the upper primer pair (containing exon 7a) and the product from lower primer pair (lacking exon 7a) was taken from 4 different experiments. The bars represent the average of this ratio normalized to that of the control samples (error bars represent standard deviation). Similar results were obtain when GAPDH was used for normalization. (D) HeLa cells were transfected with either β-globin wt (left panel) or β-globin Ter 39 (right panel) and with GFP as a reference for transfection efficiency. Twenty-four hours later, CHX was either added or not to the medium of the different cells. RNA was then extracted and RT-PCRs with primers for β-globin and GFP were performed. (E) Quantitative real-time RT-PCR analysis of the effect of RNAi of hUpf1 on the inclusion of exon 7a, using the primer pairs depicted in (B). (F) Western blot analyses of total proteins extracted from the above treated cells were performed with antibodies against hUpf1 (upper panel) and anti-Sm antibodies as a loading control (lower panel).
The expression of exon 7a-containing ADAR2 mRNA differs between tissues
In order to gain further insight into the role of ADAR2 exon 7a we examined how it is expressed in different tissues. We analyzed RNA from 18 human tissues by RT-PCR, using the primers in exons 7 and 8 and the results are presented in Figure 4. Whereas the exon 7a-containing ADAR2 isoform was expressed in the majority of tissues tested, its level of expression varied significantly. Particularly high levels of expression were observed in skeletal muscle, heart and testis (35%, 27% and 27%, respectively). Notably, in brain, where the function of ADAR2 is crucial,14 the level of exon 7a ADAR2 was low (3%).
Figure 4.

Expression of exon 7a is tissue specific. RT-PCR analysis of RNA expressed from different human tissues using primers from exons 7 (sense) and 8 (antisense). The amplified products were analyzed on polyacrylamide urea denaturing gels. Diagrams, on the left, show the two different products. The percentage of exon 7a inclusion is presented. RT-PCR for GAPDH is presented as loading controls.
Does the inclusion of exon 7a affect editing activity?
To address this question, we chose the skeletal muscle and heart as representing tissues with high exon 7a levels, and the brain and spinal cord as representing tissues with low exon 7a levels. We analyzed by RT-PCR and sequencing the A-to-I editing of the Q/R site of the GluR-6 gene transcript. This gene transcript is expressed in all four tissues and knockout of ADAR2 revealed it as the principal RNA-editing enzyme of GluR-6 in the Q/R, I/V and especially the Y/C sites.14 We found that in the brain, where the inclusion of exon 7a was low (3%; Fig. 4), the editing level of the Q/R site in GluR-6 was very high (83%; Fig. 5). On the other hand, in skeletal muscle and in the heart, where the inclusion of exon 7a was high (35 and 27%, respectively; Fig. 4), the editing of the Q/R site in GluR-6 was low (below 15%; Fig. 5). Similar results were obtained for the editing levels of two additional (I/V and Y/C ) editing sites of GluR-6. Namely, the editing levels found for the I/V and Y/C sites, respectively, were: brain 52 and 88%; skeletal muscle 0 and 26%; and heart 0% for both sites. This correlation was not observed in the spinal cord, where the inclusion of exon 7a was low (<5%; Fig. 4) while the editing level of GluR-6 was 19% for the Q/R site and 20 and 26% for the I/V and Y/C sites, respectively. Although there appears to be a negative correlation between the editing levels of the three editing sites of GluR-6 and exon 7a inclusion in heart, skeletal muscle and brain, such correlation is not found for the spinal cord. Nevertheless, our results suggest that the inclusion of exon 7a might be one of the factors that affect the level of editing, at least in the case of the GluR-6 transcript. In this context, it should be noted that a recent study has shown that regulation of ADAR2 editing activity is not only dependent on ADAR2 concentration; rather, it is involved in a network of interactions that affect its editing activity.40
Figure 5.
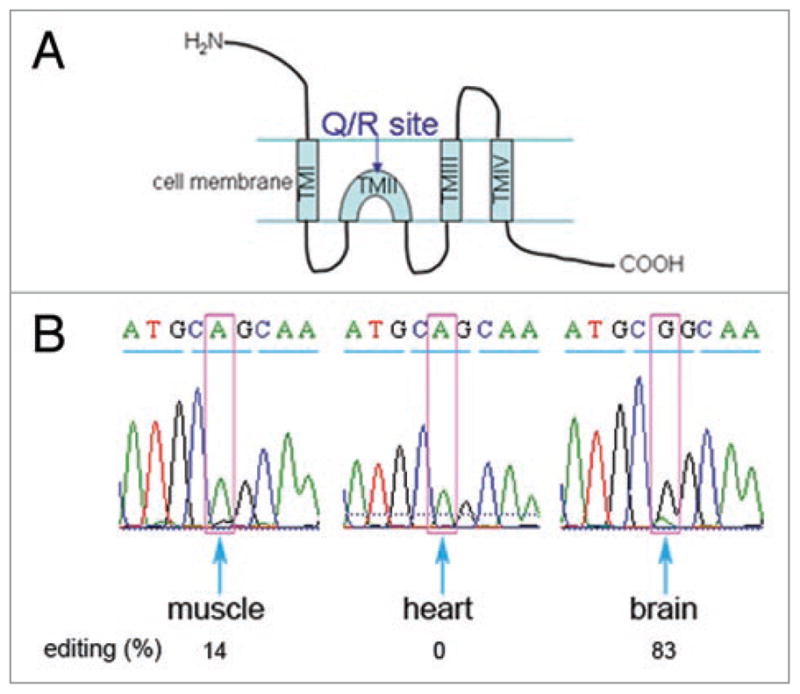
The editing levels of the Q/R site in GluR-6 in muscle, heart and brain. (A) A schematic diagram of GluR-6. The horizontal lines represent the cell membrane. TM is “trans-membrane”. The Q/R site position (in TMII) is indicated. (B) RT-PCR with primers flanking the Q/R editing site in the GluR-6 gene was performed on RNA from brain, heart and skeletal muscle. The products were extracted and sequenced. The results shown are for the codon including the Q/R site (marked in pink) and its two neighboring codons. The triplets are underlined by blue lines. The percentage of editing is indicated below.
Over expression of ADAR2 increases the inclusion of exon 7a
Over expression of SR proteins, as well as other splicing factors that have PTC-containing splice variants, was shown to upregulate the splicing levels of their respective PTC-containing isoforms.29,32 Here we asked whether the level of ADAR2 also affects the inclusion of the PTC-containing exon 7a. To this end we first show that HeLa cells transfected with human ADAR2 cDNA over express the ADAR2 protein (Fig. 6A). Next we compared the expression level of the exon 7a-inlcuded isoform to that in untreated cells by quantitative real-time RT-PCR (Fig. 6). To avoid the inclusion of transcripts expressed from the ADAR2 cDNA construct, we determined the total level of the endogenously expressed ADAR2 mRNAs using a primer pair from the 5′ UTR, which is absent from the ADAR2 cDNA construct. The sense primer flanked the exon (−2)- exon (−1) splice junction, and the anti-sense primer was from exon (−1) (Fig. 6B). This region of the ADAR2 gene is not known to undergo alternative splicing and would therefore appear in all transcripts. The bars in Figure 6C represent the ratio between the levels of the 7a-included transcript to that of the total endogenous ADAR2 transcripts, normalized to the control. As can be seen (Fig. 6C), over expression of ADAR2 increased the inclusion of exon 7a by about 1.8-fold, indicating that increased levels of the ADAR2 protein up regulated the alternative splicing event that led to the inclusion of exon 7a in ADAR2 mRNA. We also tested the effect of an ADAR2 mutant (ADAR2 E397A), which is inactive in editing.41 Quantitative real-time RT-PCR analysis revealed that over expression of this inactive mutant also up regulated the expression of exon 7a (Fig. 6E), indicating that the effect of ADAR2 over expression on exon 7a inclusion is not dependent on editing. Over expression of the ADAR2 E397A mutant is presented in Figure 6D.
Figure 6.

Over expression of ADAR2 increases exon 7a inclusion. HeLa cells were transfected with the human ADAR2 cDNA construct (A and C), or with the inactive ADAR2 mutant E397A construct41 (D and E), or mock transfected (control) for 48 h. RNA and proteins were extracted. The RNA was analyzed by quantitative real-time RT-PCR and the proteins by Western blots. (A, D) Western blots with antibodies against ADAR2 (upper panel) and Sm as a loading control (lower panel). (B) A diagram showing the primer pairs used for the real-time RT-PCR analyses. (C) A graph showing the real-time RT-PCR results for over expression of ADAR2. The ratio between the levels of the product from the primer pair amplifying exon 7a and the primer pair amplifying the endogenous ADAR2 was taken from 3 different experiments. The bars represent the average of this ratio normalized to that of the control samples (error bars represent standard deviation). (E) Real-time RT-PCR analysis of ADAR2 E397A over expression.
The alternatively spliced isoform of ADAR2 exon 7a is found in supraspliceosomes
Supraspliceosomes are ribonucleoprotein complexes whose major components are the five spliceosomal U small nuclear ribonucleoproteins (snRNPs), as well as regulatory splicing factors such as the SR protein family, RBM4 and hnRNP G.33,42,43 Supraspliceosomes have been shown to have splicing activity,44 thus alternative splicing is expected to take place in them. Accordingly, it was recently shown that hnRNP G affects the alterantive splicing of the Tau gene in supraspliceosomes.43 We therefore looked for the presence of the alternative spliced form of ADAR2 containg exon 7a in supraspliceosomes. For this aim, HeLa cells nuclear supernatants enriched for supraspliceosomes was fractionated in a glycerol gradient as previously described.45,46 RNA was then extracted from each of the fractions and RT-PCR was performed using primers from the flanking exons of exon 7a. As shown in Figure 7 there is a major peak of the two PCR products at the 200S region of the gradient, where supraspliceosomes sediment, and a minor peak at the 70S region, where native spliceosomes sediment.44 Thus the alternatively spliced form of ADAR2 containing exon 7a is found in supraspliceosomes.
Figure 7.

The new ADAR2 alternatively spliced exon is found in supraspliceosomes. HeLa nuclear supernatant enriched in supraspliceosomes was fractionated on a glycerol gradient and RNA was extracted. RT-PCR with primers from exons 7 (sense) and 8 (antisense) was then performed. The upper panel shows 30 cycles of PCR, and the lower panel shows 35 cycles of PCR. Diagrams on the left show the 2 different products. Supraspliceosomes sediment at 200S and native spliceosomes at 60–70S.
Discussion
In the present study we describe a previously unreported alternative exon located within intron 7 of the human ADAR2 RNA transcript. This 93-nt exon, named exon 7a, is highly conserved in mammals (Fig. 1D), indicating that it might have a physiological regulatory role in the ADAR2 gene expression. Bioinformatics analyses have predicted a frequent occurrence of alternative splicing events that lead to PTC-containing mRNAs, in conserved as well as nonconserved gene regions.26,27,47 It has thus been suggested that such alternative splicing events could be part of a control pathway on mRNA levels through the degradation of the PTC-containing molecules by the NMD RNA surveillance pathway.25 It seems likely that the NMD RNA surveillance pathway is involved also in the regulation of ADAR2, because exon 7a harbors stop codons in all 3 protein reading frames (Fig. 1D), thus making it an appropriate NMD-substrate. Indeed, inhibition of NMD by the addition of the translation inhibitor CHX, or by down regulation of the essential NMD factor hUpf1, up regulated the alternative exon 7a-containing isoform (Fig. 3). Our findings add ADAR2 to the list of splicing factors and RNA related genes in which the introduction of PTCs through a splicing isoform seems to be regulatory.29–32 As ADAR2 proteins lacking only a small fragment of the carboxyl terminus have little deaminase activity,18 it is likely that if a small fraction of mRNAs with exon 7a escapes NMD, the protein will not have a catalytic activity.
One of the factors that may affect tissue-dependent variations in the expression levels of PTC-containing mRNAs, as observed here for the exon 7a-included ADAR2, is the efficiency of NMD in the respective tissue. This issue has been addressed lately by Zetoune et al.48 who reported differences of up to 2-fold in the efficiency of NMD on a mutated Men1 gene, although in all tested tissues the Men1 RNA level was reduced to 40% or less of the control. Interestingly, the same authors48 have shown that in the brain, where the level of the inclusion of exon 7a in our case was very low, the efficiency of NMD was high. This could perhaps partially account for the low level of exon 7a-containing ADAR2 mRNA in the brain, since abrogation of NMD by CHX in human medulloblastoma TE-671 cells did not show a significant elevation in the level of exon 7a inclusion compared to the elevation viewed in HeLa cells (data not shown). Because the editing by ADAR2 in the brain appears to be extremely important, it is plausible that the alternative splicing event leading to the inclusion of exon 7a is somehow suppressed or not activated and NMD plays a role of a fail-safe mechanism.
Consistent with this idea are reports showing that in heart and testis,48 where the inclusion levels of exon 7a were relatively high (27%), NMD was highly efficient. Also, in human skeletal muscle, Upf1 and Upf2, components of the NMD pathway, are abundant and NMD is functioning.49 It should be noted that the relative differences of exon 7a inclusion between the different tissues were larger than 5-fold (Fig. 4), while the reported differences in NMD were about 2-fold.48 It seems therefore likely, that the differences in NMD efficiencies may not be the major source for the tissue dependent differences in exon 7a inclusion. It is possible that combinatorial interplay of tissue specific alternative splicing regulatory factor/s, ADAR2 over expression and NMD, is affecting the levels of inclusion of ADAR2 exon 7a, to account for the different requirements of ADAR2 editing in the different tissues.
Previous studies have demonstrated that ADAR2 can auto-regulate its activity by editing its own transcript, thereby creating a new 3′ splice site whose usage for splicing led to a PTC in the spliced mRNA.24 Our report adds another example of the auto-regulation of the ADAR2 enzyme through alternative splicing, by showing that when ADAR2 is over expressed the inclusion of the PTC-containing exon 7a is enhanced. This effect is not dependent of the editing activity of ADAR2, because we showed that over expression of an inactive ADAR2 41 also increased the expression of exon 7a. It is likely that the effect of ADAR2 over expression on exon 7a inclusion is correlated to its RNA binding capacity. This observation is consistent with the findings by Heale et al.,41,50 that ADAR2 can affect RNA processing independent of its enzymatic activity, presumably by acting as RNA-binding proteins. In line with these observations is our finding that the alternative splicing of exon 7a is associated with supraspliceosomes, which have been proposed to constitute the machine where RNA splicing occurs in living cells.33,44 These large multi-component ribonucleoprotein complexes have been shown to harbor different alternative splicing regulatory proteins,43,51 as well as ADAR2 protein and activity.52 It is therefore plausible that ADAR2 affects its own splicing through interactions with other splicing factors within the supraspliceosome.
What might be the significance of the fact that the inclusion of exon 7a is tissue-specific? The ADAR2 enzyme is responsible for editing the Q/R site in the GluR-B of the AMPA receptor in the brain.13 It has also been reported that ADAR2 knockout mice died very early, but could be rescued when the A moiety at the edited site of the GluR-B gene had been mutated to a G.14 It seems therefore that the presence of a functional ADAR2 in the brain is essential. Consistent with this finding, we show here that the inclusion of exon 7a in ADAR2 expressed in the brain could hardly be detected, which might be significant in light of the abovementioned observations. It is also noteworthy that in skeletal muscle, heart and testis, the level of inclusion of exon 7a is relatively high (35, 27 and 27%, respectively). We therefore tested the editing levels of the GluR-6 ion channel, which is expressed in the brain, heart and skeletal muscle and, like GluR-B, contains a Q/R editing site. Our results show (Fig. 5) that in the brain, where there is hardly any inclusion of exon 7a, almost all Q/R sites in GluR-6 appeared to be edited, whereas in the skeletal muscle and heart tissues, where the inclusion of exon 7a was relatively high, the editing level of the Q/R site in GluR-6 was very low. However, in spinal cord, where the inclusion of exon 7a is low, the editing level of the Q/R site of GluR-6 is low (20%). It seems therefore likely that the inclusion of exon 7a may be one of the factors affecting the editing activity of ADAR2.
In conclusion, defects in A-to-I RNA editing have been shown to be associated with neuronal and muscular abnormalities. Thus, elucidating the control of A-to-I editing could be important for the understanding of these abnormalities. Although RNA editing exhibits tissue specificity and changes in development, very little is known about the regulation of these processes. Yet the large number of alternatively spliced isoforms of the ADAR enzymes indicates that alternative splicing may play a role in this regulation. In this study we describe a new tissue-specific alternative exon of ADAR2. The tissue specific expression of this alternative exon is likely the result of combinatorial interplay of tissue specific regulatory splicing factors, ADAR2 level and NMD. The high conservation in mammals of this exon and its characteristics described above indicate its potential physiological importance.
Materials and Methods
Reverse transcription RT-PCR
Total RNA from cell lines was prepared and treated with RNase-free DNase I as previously described.53 RNA from human tissues was from Clontech. cDNA primer by using was synthesized from 0.5–5 μg of RNA with dT15 Moloney murine leukemia virus reverse transcriptase (Promega). PCRs (25 μl) contained 10 pmol of each of the indicated primer pairs (see below) and 1X taq master mix, purple (λ biotech). The amplified products were run on either a 2% agarose gel or a 10% polyacrylamide/7M urea denaturing gel.
Quantitative real-time RT-PCR
Sets of specific primers were designed, enabling the amplification of a single specific product at a time. Total RNA and cDNA were prepared as described above. cDNA was mixed with 10 μl of 2x AbSolute blue Syber Rox MIX (ABgene), the appropriate forward and reverse primers were added to 150 nM in a final volume of 20 μl. Amplification was carried out using an ABI PRISM 7700 sequence detector (Applied Biosystem). The cycling conditions comprised 2 min at 50°C, 10 min of polymerase activation at 95°C, 40 cycles at 95°C for 20 s, 60°C for 30 s, and 74°C for 30 s. The amplification cycles were followed by a melting curve cycle. Each assay included (in duplicate) a no-template-control. Data of the amplified products was exported as a tab-delimited text file to a Microsoft Excel spread sheet for further analysis. The efficiencies of the reactions were determined by a linear regression method as described54 using the LinReg program. A midpoint threshold was then established as suggested,55 for each of the amplicons arising from a given cDNA. The expression levels (Q) were then determined according to the equation Q=E−Ct, where E is the reaction efficiency and Ct is the cycle threshold. The amplification efficiency of the reference genes was determined using the standard curve method. Results from at least three independent experiments were averaged, and the standard deviations are indicated.
RNA extraction from fractionated nuclear supernatants of HeLa cells
Nuclear supernatants enriched in supraspliceosomes were prepared from HeLa cells (CILBIOTECH) as described.45, 46 Briefly, nuclear supernatant was prepared from clean cell nuclei by microsonication of the nuclei and precipitation of the chromatin in the presence of tRNA. The nuclear supernatant was fractionated on a 10–45% (vol/vol) glycerol gradient. Centrifugations were carried out at 4°C in an SW41 rotor run at 41 krpm for 90 min (or an equivalent w2t). For RNA extraction, fractions of the glycerol gradients (520 μl) were mixed with 150 μl of extraction buffer [50 mM Tris–HCl (pH 7.5), 300 mM NaCl, 1 mM EDTA] and 50 μl of 10% (w/v) SDS, and the RNA was recovered by extraction with phenol and precipitation in ethanol.
Transfections
Transient transfections of human ADAR2 WT cDNA construct, and a construct expressing a cDNA of an inactive mutant ADAR2 E397A,41 (kindly provided by M. O’Connell), were performed with jetPEI (Polyplus-transfections) according to the manufacture’s instructions, transfecting 1 μg of DNA and 2 μl of jetPEI to HeLa cells in 6 cm plates. Twenty-four hours later, the medium was replaced by a fresh one, and RNA and proteins were extracted 48 h after transfection. Plasmids of β-globin, either wt or Ter39 (kindly provided by L. Maquat), were transiently transfected (2 μg per plate) into 6 cm plates of HeLa cells and cotransfected with GFP-EA1 (1 μg per plate) using the calcium phosphate method.56 Twenty four h posttransfection, the medium was replaced and the cells were treated with CHX, (50 μg/ml) for 2 hours, and RNA was then extracted.
Small interfering RNA-mediated down regulation of hUpf1
siRNAtargetedtohUpf157 and siGENOME Non-Targeting siRNA (Dharmacon) were transfected into HeLa cells with DharmaFECT 1 (Dharmacon) according to the manufacturer’s instructions with some modifications. Cells grown in 6-cm plates were transfected with 150 nM siRNA by using 0.28% DharmaFECT 1. After 72 h, total proteins and RNA were extracted.
Western blots
Western blot analyses were performed as previously described,52 using ADAR2 monoclonal antibodies (kindly provided by K. Nishikura), hUpf1 antibodies (kindly provided by J. Lykke-Andersen) and Y12 anti-Sm monoclonal antibodies.
GluR-6 editing levels
GluR-6 editing levels were analyzed by direct sequencing. RT-PCR analysis of the region surrounding the Q/R site of the GluR-6 mRNA was performed (primer sequences were derived from ref. 58). The PCR products were extracted with the HiYield Gel/PCR DNA fragment extraction kit (RBC bioscience). The extracted DNA was sequenced, and the editing level was derived from the ratio between the peak areas of the A and G nucleotides occurring at identical positions in the DNA sequence chromatograms.14
Primers
To amplify both transcripts, including and excluding exon 7a, we used primers from exons 7 (sense) and 8 (antisense).
ADAR2 ex7 s - 5′ GGA ACG TGG TGG GCA TCC AGG G 3′ (sense)
ADAR2 ex8 as - 5′ CCA GCG ACA GTA CAA CGC GTG CT 3′ (antisense)
For quantitative real-time PCR we used the following primers:
For the transcript excluding exon 7a ADAR2 ex7-8 s - 5′ CAA GCC TTT GCT CAG TGG CAT CAG C 3′ (sense) and ADAR2 ex8 as (above). For the transcript including exon 7a:
ADAR2 ex7-7a s - 5′ CAC CCT CAA CAA GCC TTT GCT CAG TGA ATT T 3′ (sense) and
ADAR2 ex7a-8 as - 5′ CGT GCT TCT GCA TTG CTG ATG CCT TAA C 3′ (antisense)
For endogenous ADAR2 (in ADAR2 over expression experiments)
ADAR2 ex-2-1 s - 5′ CGG CTG CGG CTG AGA GTG G 3′ (sense) and
ADAR2 ex-1 as - 5′ TTC GGT GCG AGC GGA CTG AGA G 3′ (antisense)
Other primers used:
For GAPDH
GAPDH s - 5′ TGC ACC ACC AAC TGC TTA GC 3′ (sense) and
GAPDH as - 5′ GGC ATG GAC TGT GGT CAT GAG 3′ (antisense)
For β globin (either WT or Ter38)
globin s - 5′ AGG AGA AGT CTG CCG TTA C 3′ (sense) and
globin as - 5′ CCT GAA GTT CTC AGG ATC C 3′ (antisense)
For GluR-6
GluR6 sense - 5′ ACT TGG AAT AAG TAT TTT GTA CCG C 3′ (sense) and
GluR6 antisense - 5′ CAA ATG CCT CCC ACT ATC CTG 3′ (antisense).58
For the transcript including exon 7a in mouse
rADAR2 ex7-7a s - 5′ AAC AAG CCC CTG CTC AGC GAG TGT 3′ (sense) and
mADAR2 ex7a-8 as - 5′ TGC CTC TGC ATT GCT GAT GCC TTA GC 3′ (antisense)
For the transcript including exon 7a in rat
rADAR2 ex7-7a s - 5′ (above) and
rADAR2 ex7a-8 as - 5′ CGT GCC TCT GCA TTG CTG ATA CCT TAA C 3′ (antisense)
Acknowledgments
We thank Aviva Pecho and Avital Michaeli for excellent technical assistance, and Naama Sebbag-Sznajder for help with the supraspliceosome experiment. We are grateful to Mary O’Connell for the ADAR2 constructs, to Kazuko Nishikura for anti-ADAR2 antibodies, to Jens Lykke-Andersen for anti-hUpf1 antibodies and to Lynne Maquat for the globin constructs. We thank the US NIH, grant GM079549 to R. S. and J.S., and the Helen and Milton Kimmelman Center for Biomolecular Structure and Assembly at the Weizmann Institute of Science (J.S.) for financial support.
Abbreviations
- ADAR
adenosine deaminase that acts on RNA
- NMD
nonsense mediated mRNA decay
- PTC
premature termination codon
- CHX
cycloheximide
References
- 1.Valente L, Nishikura K. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- 2.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–46. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jepson JE, Reenan RA. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta. 2008;1779:459–70. doi: 10.1016/j.bbagrm.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Hanrahan CJ, Palladino MJ, Bonneau LJ, Reenan RA. RNA editing of a Drosophila sodium channel gene. Annals of the New York Academy of Sciences. 1999;868:51–66. doi: 10.1111/j.1749-6632.1999.tb11273.x. [DOI] [PubMed] [Google Scholar]
- 5.Paul MS, Bass BL. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998;17:1120–7. doi: 10.1093/emboj/17.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan BZ, Huang H, Lam R, Soong TW. Dynamic regulation of RNA editing of ion channels and receptors in the mammalian nervous system. Mol Brain. 2009;2:13. doi: 10.1186/1756-6606-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–3. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 8.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–5. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 9.Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–25. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bass BL. How does RNA editing affect dsRNA-mediated gene silencing? Cold Spring Harb Symp Quant Biol. 2006;71:285–92. doi: 10.1101/sqb.2006.71.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishikura K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7:919–31. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–9. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 14.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 15.Seeburg PH, Hartner J. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol. 2003;13:279–83. doi: 10.1016/s0959-4388(03)00062-x. [DOI] [PubMed] [Google Scholar]
- 16.Maas S, Kawahara Y, Tamburro KM, Nishikura K. A-to-I RNA editing and human disease. RNA Biol. 2006;3:1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slavov D, Gardiner K. Phylogenetic comparison of the pre-mRNA adenosine deaminase ADAR2 genes and transcripts: conservation and diversity in editing site sequence and alternative splicing patterns. Gene. 2002;299:83–94. doi: 10.1016/s0378-1119(02)01016-8. [DOI] [PubMed] [Google Scholar]
- 18.Lai F, Chen C-X, Carter KC, Nishikura K. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol Cell Biol. 1997;17:2413–24. doi: 10.1128/mcb.17.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerber A, O’Connell MA, Keller W. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA. 1997;3:453–63. [PMC free article] [PubMed] [Google Scholar]
- 20.Mittaz L, Scott HS, Rossier C, Seeburg PH, Higuchi M, Antonarakis SE. Cloning of a human RNA editing deaminase (ADARB1) of glutamate receptors that maps to chromosome 21q22.3. Genomics. 1997;41:210–7. doi: 10.1006/geno.1997.4655. [DOI] [PubMed] [Google Scholar]
- 21.Villard L, Tassone F, Haymowicz M, Welborn R, Gardiner K. Map location, genomic organization and expression patterns of the human RED1 RNA editase. Somat Cell Mol Genet. 1997;23:135–45. doi: 10.1007/BF02679972. [DOI] [PubMed] [Google Scholar]
- 22.Kawahara Y, Ito K, Ito M, Tsuji S, Kwak S. Novel splice variants of human ADAR2 mRNA: skipping of the exon encoding the dsRNA-binding domains, and multiple C-terminal splice sites. Gene. 2005;363:193–201. doi: 10.1016/j.gene.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 23.Maas S, Gommans WM. Novel exon of mammalian ADAR2 extends open reading frame. PLoS One. 2009;4:e4225. doi: 10.1371/journal.pone.0004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 25.McGlincy NJ, Smith CW. Alternative splicing resulting in nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends Biochem Sci. 2008;33:385–93. doi: 10.1016/j.tibs.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–92. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green RE, Lewis BP, Hillman RT, Blanchette M, Lareau LF, Garnett AT, Rio DC, Brenner SE. Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics. 2003;19 (Suppl 1):i118–21. doi: 10.1093/bioinformatics/btg1015. [DOI] [PubMed] [Google Scholar]
- 28.Pan Q, Saltzman AL, Kim YK, Misquitta C, Shai O, Maquat LE, Frey BJ, Blencowe BJ. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–8. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100. doi: 10.1016/s1097-2765(03)00502-1. [DOI] [PubMed] [Google Scholar]
- 30.Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446:926–9. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 31.Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M., Jr Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21:708–18. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, Blencowe BJ. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol Cell Biol. 2008;28:4320–30. doi: 10.1128/MCB.00361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sperling J, Azubel M, Sperling R. Structure and Function of the Pre-mRNA Splicing Machine. Structure. 2008;16:1605–15. doi: 10.1016/j.str.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–9. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 35.Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem. 1995;270:28995–9003. doi: 10.1074/jbc.270.48.28995. [DOI] [PubMed] [Google Scholar]
- 36.Rajavel KS, Neufeld EF. Nonsense-mediated decay of human HEXA mRNA. Mol Cell Biol. 2001;21:5512–9. doi: 10.1128/MCB.21.16.5512-5519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Sun X, Qian Y, Maquat LE. Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA. 1998;4:801–15. doi: 10.1017/s1355838298971849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 39.Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–56. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 40.Wahlstedt H, Daniel C, Enstero M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;19:978–86. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O’Connell MA. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009;28:3145–56. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markus MA, Heinrich B, Raitskin O, Adams DJ, Mangs H, Goy C, Ladomery M, Sperling R, Stamm S, Morris BJ. WT1 interacts with the splicing protein RBM4 and regulates its ability to modulate alternative splicing in vivo. Exp Cell Res. 2006;312:3379–88. doi: 10.1016/j.yexcr.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Heinrich B, Zhang Z, Raitskin O, Hiller M, Benderska N, Hartmann AM, Bracco L, Elliott D, Ben-Ari S, Soreq H, Sperling J, Sperling R, Stamm S. Heterogeneous Nuclear Ribonucleoprotein G Regulates Splice Site Selection by Binding to CC(A/C)-rich Regions in Pre-mRNA. J Biol Chem. 2009;284:14303–15. doi: 10.1074/jbc.M901026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azubel M, Habib N, Sperling J, Sperling R. Native spliceosomes assemble with pre-mRNA to form supraspliceosomes. J Mol Biol. 2006;356:955–66. doi: 10.1016/j.jmb.2005.11.078. [DOI] [PubMed] [Google Scholar]
- 45.Sperling R, Sperling J. The lnRNP particle - A naturally assembled complex of pre-mRNA and splicing factors. In: Schenkel J, editor. RNP Particles, Splicing and Autoimmune Diseases. Springer; 1998. pp. 29–47. [Google Scholar]
- 46.Spann P, Feinerman M, Sperling J, Sperling R. Isolation and visualization of large compact ribonucleoprotein particles of specific nuclear RNAs. Proc Natl Acad Sci USA. 1989;86:466–70. doi: 10.1073/pnas.86.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baek D, Green P. Sequence conservation, relative isoform frequencies, and nonsense-mediated decay in evolutionarily conserved alternative splicing. Proc Natl Acad Sci USA. 2005;102:12813–8. doi: 10.1073/pnas.0506139102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zetoune AB, Fontaniere S, Magnin D, Anczukow O, Buisson M, Zhang CX, Mazoyer S. Comparison of nonsense-mediated mRNA decay efficiency in various murine tissues. BMC Genet. 2008;9:83. doi: 10.1186/1471-2156-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendell JT, Medghalchi SM, Lake RG, Noensie EN, Dietz HC. Novel Upf2p orthologues suggest a functional link between translation initiation and nonsense surveillance complexes. Mol Cell Biol. 2000;20:8944–57. doi: 10.1128/mcb.20.23.8944-8957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heale BS, Keegan LP, O’Connell MA. ADARs have effects beyond RNA editing. Cell Cycle. 2009;8:4011–2. doi: 10.4161/cc.8.24.10214. [DOI] [PubMed] [Google Scholar]
- 51.Yitzhaki S, Miriami E, Sperling J, Sperling R. Phosphorylated Ser/Arg-rich proteins: Limiting factors in the assembly of 200S large nuclear ribonucleoprotein particles. Proc Natl Acad Sci USA. 1996;93:8830–5. doi: 10.1073/pnas.93.17.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raitskin O, Cho DS, Sperling J, Nishikura K, Sperling R. RNA editing activity is associated with splicing factors in lnRNP particles: The nuclear pre-mRNA processing machinery. Proc Natl Acad Sci USA. 2001;98:6571–6. doi: 10.1073/pnas.111153798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agranat L, Raitskin O, Sperling J, Sperling R. The editing enzyme ADAR1 and the mRNA surveillance protein hUpf1 interact in the cell nucleus. Proc Natl Acad Sci USA. 2008;105:5028–33. doi: 10.1073/pnas.0710576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neuroscience Letters. 2003;339:62–6. doi: 10.1016/s0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 55.Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003:31. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kingston RE. Introduction of DNA into mammalian cells. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Short protocols in molecular biology. NewYork: John Wiley and Sons; 1992. pp. 9.1–9.16. [Google Scholar]
- 57.Mendell JT, ap Rhys CMJ, Dietz HC. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science (New York, NY) 2002;298:419–22. doi: 10.1126/science.1074428. [DOI] [PubMed] [Google Scholar]
- 58.Barbon A, Vallini I, La Via L, Marchina E, Barlati S. Glutamate receptor RNA editing: a molecular analysis of GluR2, GluR5 and GluR6 in human brain tissues and in NT2 cells following in vitro neural differentiation. Brain Res Mol Brain Res. 2003;117:168–78. doi: 10.1016/s0169-328x(03)00317-6. [DOI] [PubMed] [Google Scholar]