

Abstract
The sterol regulatory element-binding factor-2 (SREBF2) gene is a bifunctional locus encoding SREBP-2, a well-known transcriptional regulator of genes involved in cholesterol biosynthesis, and microRNA-33a, which has recently been shown to reduce expression of proteins involved in export of cholesterol and β-oxidation of fatty acids, thus adding an unexpected layer of complexity and fine-tuning to regulation of lipid homeostasis.
Introduction
Fatty acids, cholesterol and their lipid derivatives play essential roles in normal cellular function, and serve as structural components, signaling molecules and/or as storage forms of energy (Maxfield and Tabas, 2005). In multicellular organisms, cellular lipid metabolism is regulated to match the needs both of individual cells and of the entire organism. A sedentary lifestyle coupled with overconsumption of calories, as common with a Western-style diet, stresses the regulatory circuits of lipid homeostasis. Although lipids are essential for life, their excessive accumulation and/or abnormal distribution lead to significant health issues such as type 2 diabetes and atherosclerosis, which are responsible for significant morbidity and mortality. As such, understanding the regulation of lipid metabolism may help define novel targets for future therapeutic intervention. Lipid metabolism is regulated at many levels, including transcriptional control of fatty acid synthesis, storage and oxidation by peroxisome proliferator-activated receptors, and cholesterol homeostasis by liver-x-receptors (Pyper et al.; Zhao and Dahlman-Wright, 2010). Several recent publications point to an important role of miRNAs (Gerin et al., 2010b; Horie et al., 2010; Krutzfeldt et al., 2005; Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). Here we review how cholesterol and fatty acid homeostasis is regulated by products of the SREBF bifunctional loci, which encode SREBP transcription factors and the microRNA (miR)-33 family.
SREBPs regulate cellular lipid homeostasis
Sterol regulatory element binding proteins (SREBPs) act as a central hub in the regulation of cellular lipid levels. These basic helix-loop-helix-leucine zipper transcription factors bind to specific cis elements and regulate transcription of target genes, whose products play key roles in lipid metabolism. They are encoded by genes that carry the name sterol regulatory element binding factor (SREBF). While lower organisms possess one SREBF gene, vertebrate genomes contain SREBF1 and SREBF2, which code for SREBP-1 and SREBP-2, respectively. The locus encoding SREBP-1 gives rise to two major mRNA species, SREBP-1a and SREBP-1c, which are transcribed from distinct promoters (Shimomura et al., 1997). SREBP-1a, SREBP-1c and SREBP-2 differ significantly with regard to their transcriptional activity, tissue distribution and mode of regulation (Shimano et al., 1997; Shimomura et al., 1997). Collectively, SREBPs can activate the transcription of virtually all genes involved in cholesterol, fatty acid and phospholipid synthesis (Horton et al., 2002), although a few instances of transcriptional repression by SREBPs have also been described (Zeng et al., 2004). Gene expression studies support the notion that SREBP-1a and 1c preferably activate transcription of genes involved in fatty acid synthesis, whereas SREBP-2 displays some specificity for genes involved in cholesterol biosynthesis (Horton et al., 1998), However, sets of target genes are by no means mutually exclusive, since SREBP-1a also activates genes critical for cholesterol synthesis, and SREBP-2 regulates transcription of genes involved in fatty acid synthesis (Horton et al., 2003). For example, both SREBP-1a and SREBP-2 activate transcription of the genes encoding fatty acid synthase and acetyl-CoA carboxylase 1, which are essential for fatty acid synthesis. Likewise, both factors activate transcription of the gene encoding the rate limiting enzyme of cholesterol synthesis, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase (Horton et al., 2003). It should also be noted that cellular cholesterol and fatty acid levels are not only the result of de novo biosynthesis, but are strongly influenced by uptake and export from cells (Nabel, 2003). To deliver fatty acids and cholesterol to peripheral cells, the liver synthesizes very low density lipoprotein (VLDL) particles. Removal of triacylglycerol and cholesteryl esters eventually leads to the transformation of VLDL into LDL particles, which are cleared from circulation via hepatic LDL receptor (LDLR). LDL cholesterol concentrations are highly correlated with cardiovascular risk, and oxidation and/or impaired clearance of LDL particles increase the risk of foam cell and atheroma formation. In this regard, SREBP-1a and −2 directly activate hepatic transcription of the low density lipoprotein (LDL) receptor, which allows the liver to increase its cellular cholesterol content, and in parallel clears LDL particles from the circulation(Horton et al., 2002; Yokoyama et al., 1993).
The activity of transcription factors is controlled by a myriad of mechanisms, including ligands and post-translational modifications. However, the control of SREBPs is unusual in that they initially reside in an inactive form, inserted into the membrane of the endoplasmic reticulum via two transmembrane domains (Figure 1A). To fulfil their transcriptional function, SREBPs are proteolytically cleaved, freeing an N-terminal fragment that translocates to the nucleus and activates transcription of target genes (Goldstein et al., 2006; Horton et al., 2002). The generation of transcriptionally active SREBP-2 is exquisitely dependent upon content of cholesterol in endoplasmic reticulum (reviewed in (Osborne and Espenshade, 2009)). The activating proteolytic cleavage in the Golgi network only occurs if SREBP-2 is transported from the endoplasmic reticulum (Osborne and Espenshade, 2009). Export to the Golgi apparatus is mediated by a SREBP-binding protein called SREBP cleavage-activating protein (SCAP), which also binds to COPII-coated vesicles, the cellular machinery that exports material from endoplasmic reticulum to the Golgi. However, binding of cholesterol to SCAP leads to conformational changes that masks the binding site for COPII proteins and favors the interaction with INSIG proteins, which reside exclusively within the endoplasmic reticulum. Hence, high cholesterol levels retain SCAP and SREBP in the endoplasmic reticulum and inhibit the production of transcriptionally active SREBP from the Golgi. In turn, the transcriptional targets of SREBP-1 and SREBP-2 act to increase cellular cholesterol levels, which in a negative feedback loop, inhibit proteolytic activation and transcription of SREBPs. Hence, through an elegant mechanism, the activity of transcription factors that regulate transcription of genes involved in cholesterol synthesis is coupled to cellular cholesterol levels (Figure 1A).
Figure 1. miR-33a collaborates with SREBP-2 to maintain cellular lipid homeostasis.
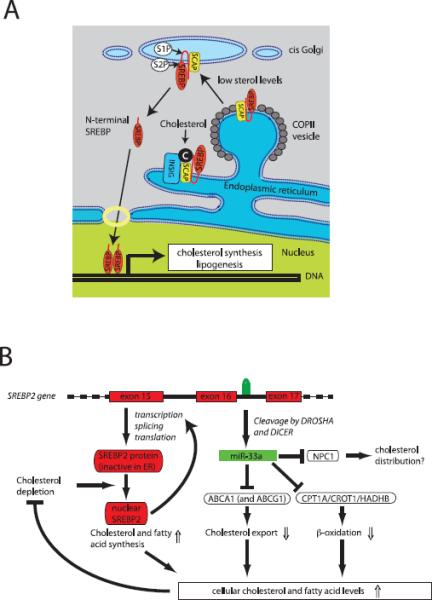
(A) Traditional model of SREBP-2 function. SREBP-2 contains two transmembrane domains and is inserted in the membrane of the endoplasmic reticulum (ER). Under conditions of abundant cholesterol (or phosphatidylethanolamine), SREBP2 is retained in the ER by a complex of SCAP and INSIG proteins. When cholesterol levels fall, the interaction between INSIG and SCAP is disrupted, allowing SCAP to interact with COPII-coated vesicles and target SREBP-2 for export to the Golgi apparatus. Here, in two consecutive cleavages by S1P and S2P, a transcriptionally competent N-terminal fragment is released. After translocation to the nucleus, SREBP-2 induces expression of genes involved in cholesterol and fatty acid synthesis, which serve to normalize cellular lipid levels (modified from (Ikonen, 2008)). (B) Integrated model for how the bifunctional SREBF2 locus maintains lipid homeostasis. After processing from an intron of SREBF2, miR-33a reduces cellular cholesterol export by inhibiting expression of ABCA1 (and in the mouse ABCG1). In addition, miR-33a reduces mitochondrial fatty acid β-oxidation via inhibition of HADHB, CROT and CPT1A to increase intracellular lipid levels. Thus the SREBF2 locus uses two distinct mechanisms to maintain lipid homeostasis: regulated transcriptional activity of SREBP-2 and translational repression by miR-33a.
In addition to regulation by cholesterol, activation of SREBPs by proteolysis is also dependent on hormones and other lipids. For example, in the liver, insulin activates SREBP-1c, even in the presence of high cholesterol levels, by suppressing expression of the endoplasmic reticulum docking protein, INSIG2a (Yabe et al., 2003). In addition, interactions of oxysterols with INSIG proteins stabilize binding to SCAP and thereby prevent SREBP-1 and SREBP-2 movement to the Golgi for proteolysis (Radhakrishnan et al., 2007).
An additional layer of complexity is added by the transcriptional regulation of SREBF genes. SREBP-1c transcription is activated by insulin signaling and the nuclear receptors for oxysterols, liver-x-receptor α and β (Shimomura et al., 1999); (Repa et al., 2000). In turn, SREBPs increase the transcription of SREBF2 in a positive feedback loop (Sato et al., 1996). Post-translational activation of SREBP-2 therefore is paralleled by induction of SREBP-2 mRNA (Figure 1A). Recent studies have shown that the primary transcript of SREBP-2 also encodes miR-33a, a microRNA that regulates cholesterol and fatty acid metabolism, suggesting that transcriptional regulation of the SREBF2 gene modulates the cellular capacity for producing not only an active transcription factor, but also the expression of miR-33a (Gerin et al., 2010b; Horie et al., 2010; Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). Notably, the SREBF1 locus of primates and a limited number of other species (excluding mice) encodes a closely related family member to miR-33a called miR-33b. Experimental data so far is mainly available for miR-33a. We will therefore limit our discussion to the function of miR-33a and will speculate how the lack of miR-33b in mice might affect the lessons we can learn from mouse models.
Role of miR-33a in control of cholesterol metabolism
miRNAs are 21–24 nucleotide, non-coding RNAs, which are important regulators of mRNA stability and translation. By binding to partially complementary sites in the 3' untranslated region (3'UTR) of mRNA transcripts, miRNAs reduce translation of these transcripts and/or lead to their degradation (Bartel, 2009). Recognition of target sequences by miRNA is promiscuous, and in some instances pairing of the six- to eight-nucleotide seed region of the miRNA to target transcripts is sufficient for silencing (Bartel, 2009). Complementarity between miRNAs and target genes, in conjunction with evolutionary conservation of binding sites, is the basis for target site prediction algorithms (Rajewsky, 2006), which support the notion that the majority of protein-coding mRNA transcripts are regulated by miRNAs. In fact, mRNA transcripts are often targeted by several different miRNAs (Bartel, 2009), and proteomic studies demonstrate that miRNAs fine-tune translation from many different target transcripts (Selbach et al., 2008).
The human genome codes for more than 1000 miRNAs. Although produced from distinct genomic loci, some of these miRNAs form families with similar or identical sequences. miRNA families are predicted to bind and repress largely overlapping sets of target transcripts, with functionality influenced by tissue- and stimulus-specific expression of individual members. In general, the potential for functional redundancy and combinatorial complexity in miRNA-mRNA regulatory circuits has hampered the unequivocal assignment of physiological functions to distinct miRNAs.
miRNAs are synthesized by RNA polymerase II as long precursor transcripts containing hairpin loops, and two consecutive cleavages of these hairpins give rise to the mature form of a miRNA (Bartel, 2009). A significant fraction of these precursor transcripts also contain open reading frames. In some instances, proteins encoded by these open reading frames have related functions to the miRNAs. For example, the PPARGC1B locus encodes both PGC-1β, a transcriptional coactivator that regulates cellular energy metabolism, and intronic miR-378/378*, which regulate lipid metabolism in adipocytes and respiration in breast cancer cells (Eichner et al., 2010; Gerin et al.). Recently, five independent groups identified the SREBF2 gene as another example of a bifunctional locus (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010) (Figure 1B). In addition to generation of SREBP-2 protein, the SREBF2 locus contributes to lipid homeostasis by production of miR-33a, which is processed from an intron within the SREBF2 primary transcript (Figure 1B), and which has been highly conserved during evolution from primitive chordates and flies to vertebrates.
Although the recent focus on miR-33a was mainly sparked by its interesting genomic localization (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al., 2010), miR-33a was studied by Rayner and colleagues after observing its elevated expression in an unbiased screen for miRNAs regulated by depletion of cholesterol from human macrophages (Rayner et al., 2010). As confirmed by other groups, they also reported reduced expression of miR-33a when macrophages were loaded with cholesterol using acetylated LDL (Marquart et al.; Rayner et al., 2010). In each case, changes in miR-33a expression were closely paralleled by changes in SREBP-2 mRNA, lending further support for derivation of protein and miRNA from the SREBF2 primary transcript. Whether under certain circumstances miR-33a transcription, maturation and/or stability are regulated independently of SREBP-2 is clearly an interesting question.
Another potential link to cholesterol was established when investigators realized that several prediction algorithms suggested binding sites for miR-33a in the 3'UTRs of genes involved in cholesterol transport, namely ATP binding cassette A1 (ABCA1), ATP binding cassette G1 (ABCG1) and Niemann-Pick C1 (NPC1) (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010). The ABCA1 and ABCG1 cholesterol transporters are membrane proteins that mediate the export of cholesterol from peripheral tissues to HDL particles (Figure 2A) and are critical for reverse cholesterol transport to the liver (Rader et al., 2009; Yvan-Charvet et al., 2007). In the liver and intestine, ABCA1 plays an additional role in de novo HDL synthesis by exporting cholesterol onto apolipoprotein AI (ApoAI)-containing nascent HDL particles, which represents the first step in HDL biogenesis (Brunham et al., 2006) (Figure 2A). Inactivation of both ABCA1 alleles in humans leads to Tangier's disease, which is characterized by absence of HDL cholesterol and accumulation of cholesterol in peripheral tissues (reviewed in (Tang and Oram, 2009)). In addition, variants of the ABCA1 locus are not only associated with alterations in HDL levels, but also cardiovascular risk (Brunham et al., 2008; Brunham et al., 2009; Frikke-Schmidt et al., 2008). Thus, ABCA1 and ABCG1 are key regulators of cholesterol homeostasis.
Figure 2. Influence of miR-33a on HDL cholesterol levels.

miR-33a is generated from an intron of the SREBF2 primary transcript and inhibits translation of ABCA1 and ABCG1 mRNAs, and perhaps NPC1. Putative flux of cholesterol is denoted with red arrows under conditions of A) low sterol concentrations (A) or high sterol concentrations/miR-33a deficiency (B). De novo HDL synthesis requires ABCA1-dependent cholesterol secretion from the liver and intestine into nascent HDL particles. In peripheral organs, HDL-particles accept cholesterol from ABCA1 and ABCG1 transporters. Hepatic uptake of HDL particles via the scavenger receptor-B1 (SR-B1) completes the process of reverse cholesterol transport. To deliver fatty acids and cholesterol to peripheral organs, the liver synthesizes VLDL particles. Delivery of fatty acids and cholesterol eventually leads to the transformation of VLDL into LDL particles, which are cleared from circulation via hepatic LDL receptor (LDLR). Oxidation and/or impaired clearance of LDL increase risk of foam cell formation and atheroma formation. Upon reduction of hepatic cholesterol levels, SREBP-2 increases transcription of genes involved in cholesterol synthesis (e.g. β-HMG CoA reductase), as well as LDLR to increase uptake of LDL. (A) Low sterol concentraions promote the expression of miR-33a, which inhibits the expression of ABCA1 and ABCG1 to suppress cholesterol export from liver, intestine and macrophages and thereby prevents further decreases in intercellular sterol concentrations. miR-33a may also influence intercellular cholesterol distribution by inhibiting NPC1 expression. (B) Suppression of miR-33a expression by either high sterol concentrations or experimental intervention results in increased ABCA1 and ABCG1 expression, which potentially increases not only de novo synthesis of HDL particles but also loading with cholesterol in the periphery. For simplicicty, we did not include a schematic representation of SREBP-2 expression in peripheral tissues, and do not show effects of cholesterol synthesis and cholesterol secretion.
The coordinate regulation of SREBP-2 mRNA and miR-33a expression observed upon cholesterol depletion or loading of macrophages is paralleled by strong inverse regulation of ABCA1 mRNA and protein levels (Rayner et al., 2010). Although transcriptional repression of ABCA1 by SREBP-2 protein may contribute to this process (Zeng et al., 2004), overexpression of miR-33a in a variety of in vitro systems suppresses ABCA1 mRNA and protein levels (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010). In fact, endogenous miR-33a clearly plays a critical regulatory role, because lentiviral expression of competitive inhibitors or transfection with antisense oligonucleotides specific for miR-33a increases ABCA1 protein levels (Figure 1B; (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010). Regulation by miR-33a is not limited to ABCA1 because overexpression of this miRNA also decreases NPC-1, and in mouse cells, ABCG1, albeit to a lesser extent than ABCA1 (Najafi-Shoushtari et al.; Rayner et al., 2010). miRNAs generally regulate cellular processes through relatively small changes to a great number of proteins (Baek et al., 2008; Selbach et al., 2008); thus, it is important to note whether the modest effects of miRs on protein levels are functionally relevant. In this case, ectopic expression of miR-33a in vitro robustly inhibits ApoAI-dependent cholesterol export in a variety of cell culture models, whereas inhibition of endogenous miR-33a increases cholesterol export (Figure 1B; (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010))
Many of these observations have been confirmed and extended using mice. Notably, inhibition of miR-33a by locked-nucleic acid antisense oligonucleotides or by competitive antagonists expressed from lentiviruses led to significant increases in hepatic and macrophage ABCA1 expression, as well as a 30 – 50% increase in serum HDL cholesterol (Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010) (Figure 2B). Support for these findings came from Horie and colleagues, who observed that miR-33a −/− mice have elevated macrophage and hepatic expression of ABCA1, and increased baseline HDL cholesterol levels (Horie et al., 2010). Taken together, these studies provide compelling evidence that miR-33a plays a complementary but independent role to SREBP-2 in the maintenance of cellular cholesterol homeostasis, with a potentially important role in the regulation of HDL cholesterol levels (Figure 1B). It should be noted that although increased HDL cholesterol levels are consistent with increased reverse cholesterol transport (Figure 1B), flux experiments are needed to rigorously prove this point since it is the reverse cholesterol transport and not steady-state HDL levels that are modulating cardiovascular risk (Rader et al., 2009).
Although miR-33a has been studied almost exclusively in hepatocytes and macrophages, its expression in most cell types at readily detectable levels suggests that additional functions remain to be discovered. Despite some disagreement in the published works (Gerin et al.; Horie et al.; Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010), miR-33a and SREBP-2 also appear to be highly expressed in brain, adipose tissue and intestine. Organ-specific differences of target gene regulation and expression may prove to be important. For example, the 3'UTRs for mouse ABCG1 and NPC1 are targets of miR-33a in luciferase reporter assays (Marquart et al.; Najafi-Shoushtari et al.; Rayner et al., 2010), but fairly extensive investigation in mouse and human cell types revealed repression of ABCG1 (and perhaps NPC1) protein by endogenous miR-33a only in mouse macrophages (Rayner et al., 2010). Thus, a more significant regulation of ABCG1 and NPC1 by miR-33a in other cell types or organs should not yet be discounted. In this context, regulation of ABCG1 is expected to cooperate with alterations in ABCA1 to facilitate reverse cholesterol transport (Yvan-Charvet et al., 2007). Although it is clear that NPC1 is involved in recovery of cholesterol from lysosomes, and that its loss leads to cholesterol accumulation in liver, macrophages and brain (Vance, 2010), the contribution of NPC1 to the maintenance of intracellular cholesterol distribution, as well as its role in miR-33a function remains speculative.
Potential roles for miR-33 in fatty acid metabolism
It is curious that most invertebrate animals contain a conserved miR-33, even though many of them are sterol auxotrophs (Karlson, 1970). Furthermore, the lipids that control SREBP processing are different between invertebrates and mammals. For example, in Drosophila melanogaster, SREBP activation is regulated by intermediates of the phospholipid synthesis pathway (i.e. phosphatidylethanolamine rather than cholesterol) (Dobrosotskaya et al., 2002), and the primary transcriptional targets of SREBP are genes involved in fatty acid and phospholipid synthesis. It therefore seems reasonable to hypothesize that the ancestral miR-33 might have regulated other aspects of lipid metabolism, and that miR-33a may have retained some of these functions. In fact, several genes involved in β-oxidation of fatty acids contain predicted binding sites for miR-33a (Gerin et al., 2010b). The mRNA transcripts targeted by miR-33a in reporter assays include enzymes involved in β-oxidation of fatty acids (i.e. the β-subunit of the mitochondrial trifunctional enzyme HADHB) and proteins involved in the import of fatty acids into mitochondria (i.e. carnitine palmitoyl-CoA transferase; CPT1A, and carnitine octanoyl-CoA transferase; CROT) (Figure 1B). Since the rate of β-oxidation is primarily controlled by supply of fatty acid substrates to mitochondria, it is notable that miR-33a targets two transporters responsible for this rate-limiting step. Overexpression of miR-33a in HepG2 cells inhibits expression of CPT1A and HADHB proteins; decreases CROT mRNA; reduces β-oxidation of fatty acids; and increases cellular nonesterified fatty acids and triacylglycerol (Figure 1B) (Gerin et al., 2010b). Notably, a predicted miR-33 binding site is conserved in the transcript of the single CPT1 gene of Drosophila melanogaster, and reporter assays suggest that this site is functional (Gerin et al., 2010b). In mouse models, no significant changes of serum triacylglycerol levels have been observed upon inactivation or inhibition of miR-33 (Horie et al., 2010; Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). However, in these studies cellular triacylglycerol levels in hepatocytes and macrophages were not measured. Hence, further loss-of-function experiments in vitro and in vivo will be needed to evaluate the physiological relevance of these regulatory circuits.
What about miR-33b?
Mammalian genomes contain two SREBF genes encoding SREBP-1a/c and SREBP-2, respectively, and in all of them, SREBF2 contains a miR-33 family member (either miR-33 or miR-33a). In contrast, only a species contain a miR-33 family member (called miR-33b) in the SREBF1 gene. Similar to miR-33b and SREBP-2, levels of miR-33b in human tissues seem to be well correlated with levels of SREBP-1 mRNA levels. Since miR-33a/SREBF2 and miR-33b/SREBF1 show differences in organ distribution and mode of regulation, it is possible that when compared to rodents, the miR-33 family exerts additional functions in the humans. Hence, results obtained in vivo in mice, whose genome does not contain miR-33b, may not be directly transferable to the human system. It is interesting that miR-33 and a single SREBF gene are conserved in some cholesterol auxotroph animals, where the regulation of SREBP protein and its transcriptional targets are centered around phospholipid and fatty acid metabolism. Given the preference of SREBP-1 to activate genes involved in fatty acid metabolism, it is tempting to speculate that the function of ancestral miR-33 may have been more related to miR-33b than miR-33a.
With regard to human disease, it should be noted that patients with metabolic syndrome and peripheral insulin resistance, show increased expression of SREBP-1c in the liver (Kohjima et al., 2008). If miR-33b in these cases is similarly increased, a consecutive down-regulation of ABCA1 could explain a reduced HDL/LDL cholesterol ratio in these patients. In addition, a potential reduction of β-oxidation by miR-33b might contribute to the development of hepatic steatosis. Future studies will explore whether miR-33b plays a role in human disease and whether it represents a target for therapeutic intervention.
Conclusion
Given the integral role of HDL particles to reverse cholesterol transport and the strong association between circulating HDL and cardioprotection, the recent mechanistic insights into miR-33a and cholesterol homeostasis could conceivably open avenues for development of novel therapeutics. However, some caveats to be noted include the fact that only a few miR-33a targets have been identified, and many targets are likely. Thus, pharmacological inhibition of miR-33a to influence metabolic processes may lead to currently unanticipated side effects. Moreover, although increased HDL cholesterol levels are associated with reduced cardiovascular mortality, elevating HDL levels by pharmacological approaches (e.g. by cholesterol ester transfer protein inhibitor) is not easily translated into cardiovascular protection (Tall et al., 2008). Thus, the potential therapeutic utility of altering miR-33a activity or levels awaits further in vivo experimentation in large animal models where rates of reverse cholesterol transport and development of atherosclerosis are included as endpoints.
Acknowledgements
The authors appreciate the comments on this manuscript from Will Cawthorn, Charles Burant and Isabelle Gerin. Work in the laboratory of GTB is supported by the Region Bruxelles Capitale (Brains-Back-to-Brussels), the FNRS, Université Catholique de Louvain and the de Duve Institute. Work in the laboratory of OAM is supported by NIH grants DK51563 and DK62876.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham LR, Kastelein JJ, Hayden MR. ABCA1 gene mutations, HDL cholesterol levels, and risk of ischemic heart disease. JAMA. 2008;300:1997–1998. doi: 10.1001/jama.2008.539. author reply 1998. [DOI] [PubMed] [Google Scholar]
- Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham LR, Singaraja RR, Duong M, Timmins JM, Fievet C, Bissada N, Kang MH, Samra A, Fruchart JC, McManus B, Staels B, Parks JS, Hayden MR. Tissue-specific roles of ABCA1 influence susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:548–554. doi: 10.1161/ATVBAHA.108.182303. [DOI] [PubMed] [Google Scholar]
- Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, Rawson RB. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science. 2002;296:879–883. doi: 10.1126/science.1071124. [DOI] [PubMed] [Google Scholar]
- Eichner LJ, Perry MC, Dufour CR, Bertos N, Park M, St-Pierre J, Giguere V. miR-378(*) mediates metabolic shift in breast cancer cells via the PGC-1beta/ERRgamma transcriptional pathway. Cell Metab. 2010;12:352–361. doi: 10.1016/j.cmet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, Schnohr P, Grande P, Tybjaerg-Hansen A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008;299:2524–2532. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- Gerin I, Bommer GT, McCoin CS, Sousa KM, Krishnan V, MacDougald OA. Roles for miRNA-378/378* in adipocyte gene expression and lipogenesis. Am J Physiol Endocrinol Metab. 2010a;299:E198–206. doi: 10.1152/ajpendo.00179.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, Macdougald OA, Bommer GT. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010b;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci U S A. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–2339. doi: 10.1172/JCI2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Karlson P. Terpenoids in insects. Biochem Soc Symp. 1970;29:145–156. [PubMed] [Google Scholar]
- Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Enjoji M, Takayanagi R, Nakamuta M. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. 2008;21:507–511. [PubMed] [Google Scholar]
- Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Marquart TJ, Allen RM, Ory DS, Baldan A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- Nabel EG. Cardiovascular disease. N Engl J Med. 2003;349:60–72. doi: 10.1056/NEJMra035098. [DOI] [PubMed] [Google Scholar]
- Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev. 2009;23:2578–2591. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. 8:e002. doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50(Suppl):S189–194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato R, Inoue J, Kawabe Y, Kodama T, Takano T, Maeda M. Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J Biol Chem. 1996;271:26461–26464. doi: 10.1074/jbc.271.43.26461. [DOI] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96:13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99:838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Tang C, Oram JF. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta. 2009;1791:563–572. doi: 10.1016/j.bbalip.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Vance JE. Transfer of cholesterol by the NPC team. Cell Metab. 2010;12:105–106. doi: 10.1016/j.cmet.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci U S A. 2003;100:3155–3160. doi: 10.1073/pnas.0130116100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, Brown MS. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187–197. [PubMed] [Google Scholar]
- Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Liao H, Liu Y, Lee TS, Zhu M, Wang X, Stemerman MB, Zhu Y, Shyy JY. Sterol-responsive element-binding protein (SREBP) 2 down-regulates ATP-binding cassette transporter A1 in vascular endothelial cells: a novel role of SREBP in regulating cholesterol metabolism. J Biol Chem. 2004;279:48801–48807. doi: 10.1074/jbc.M407817200. [DOI] [PubMed] [Google Scholar]
- Zhao C, Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010;204:233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]