

Abstract
The Vascular Endothelial Growth Factor (VEGF) family of secreted proteins and their receptors are major regulators of blood vessel development (hemangiogenesis) and lymphatic vessel development (lymphangiogenesis). VEGF acts through a complex system of receptor tyrosine kinases, which can be membrane-bound or soluble. New data concerning the receptor system are still emerging, thus contributing to the complexity of the system. Very recently a soluble form of VEGFR-2, termed sVEGFR-2, that is a result of alternative splicing has been discovered. It has been shown earlier that a secreted/soluble form of VEGFR-1, termed sVEGFR-1, is produced by alternative splicing and exerts an anti-hemangiogenic effect by binding VEGF-A. The newly discovered spliced variant of sVEGFR-2 binds the lymphangiogenic growth factor VEGF-C and thus inhibits VEGF-C-induced activation of VEGFR-3, consequently inhibiting lymphatic endothelial cell proliferation. Its inactivation in murine embryos permits hyperplasia of dermal lymphatics and invasion of lymphatics into the cornea. Tumor lymphangiogenesis seems to influence the metastatic behavior of malignant cells. A correlation has been found between the downregulation of sVEGFR-2 and the malignant progression of neuroblastoma, which is characterized e.g. by lymphogenic metastases in progressed stages. Data show that lymphangiogenesis is regulated by both activators and inhibitors, and its balance is crucial in health and disease.
Introduction
Angiogenesis and lymphangiogenesis, processes which are associated with tumor growth and metastasis, are regulated by several different growth factors and their associated receptors. The most potent growth factors in these processes are members of the vascular endothelial growth factor (VEGF) family of proteins,1 which consists of VEGF-A, -B, -C, -D, -E and PlGF (Placenta Growth Factor). VEGF-A is a key regulator of angiogenesis and vasculogenesis, VEGF-C and VEGF-D are key regulators of lymphangiogenesis.
The VEGF Family
VEGF-A occurs in several isoforms due to alternative splicing: VEGF-A121, VEGF-A165, VEGF-A189 and VEGF-A206 being the major ones. All except VEGF-A121 have heparin binding domains, which enables anchoring of VEGF-A to heparin sulphate proteoglycans in the extracellular matrix and their presentation to VEGF receptors.2-3 Each VEGF isoform preferentially binds to specific receptor subtypes that activate distinct signaling pathways.4 VEGF-A is functional as a homodimeric glycoprotein and is able to build heterodimers with other members of the VEGF family, such as PlGF and VEGF-B,5-6 thereby not losing its mitogenic ability. In fact, by heterodimerization with VEGF-A some ligands even attain mitogenic abilities. For example, PlGF as a homodimer does not bind VEGFR-2, but as a heterodimer with VEGF-A165 gains the ability to bind both membrane-bound and soluble murine vegfr-2 and induce its activation.7
VEGF-C and VEGF-D are specific ligands of VEGFR-3, which is exclusively expressed on lymphatic endothelial cells. They are both involved in lymphangiogenesis. Proteolytically processed forms of VEGF-C also bind VEGFR-2 thereby stimulating endothelial cell proliferation and migration, indicating that besides its role in lymphangiogenesis, it also plays a role in hemangiogenesis.8 VEGF-D is structurally and functionally related to VEGF-C8 and the unprocessed full-length form preferentially binds VEGFR-3, thus inducing lymphangiogenesis, i.e. cell survival and migration of lymphatic endothelial cells.9-11 The proteolytically-cleaved, mature form of VEGF-D has an equally strong affinity to VEGFR-2 and demonstrates multiple biological responses including eNOS activation, cell survival, migration, tubulogenesis of human umbilical vein endothelial cells (HUVECs), and stimulates angiogenesis in vivo.12
VEGF-E is a VEGF-related protein derived from the Orf-virus genome. It binds exclusively to VEGFR-2 and induces angiogenesis with less pronounced edema and inflammation in mice.13
Although multiple VEGF ligands bind the same receptors, several studies have shown that the activation of the same receptor by different ligands are differentially regulated and exert differential biological effects (differential effects on receptor phosphorylation and signaling, gene expression, proliferation, migration, survival, tubulogenesis and angiogenesis in vivo).12
VEGF Receptors
VEGF activity is mediated through three protein-tyrosine kinase receptors (VEGFR-1, -2 and -3) and two non-protein kinase co-receptors (neuropilin-1 and -2).14 Heparan sulphate proteoglycans, which are situated in the extracellular matrix, are also considered co-receptors of VEGF. All protein-tyrosine kinase receptors have an extracellular domain consisting of seven immunoglobulin-like loops (D1-D7), a single transmembrane domain and an intracellular split catalytic tyrosine kinase domain.15 VEGFR signaling is initiated upon ligand dimer binding to the extracellular region, homo- or heterodimerization of the receptor, phosphorylation of the intracellular domain and cell signaling.16 Ligand binding occurs at the second and third Ig-like domain of VEGF receptors (D2, D3), domains 5-7 (D5-7) are important for stabilizing the ligand-receptor complex, whereas the membrane-proximal domain D7 is crucial for ligand-induced tyrosine phosphorylation and cell signaling. Ligand binding promotes receptor homo- or heterodimerization, phosphorylation of the intracellular domain and signal transduction.17 VEGF receptors require dimerization in order to be activated and one VEGF-A dimer binds two molecules of the extracellular domain of VEGFR-2. The main mechanisms of receptor dimerization, however, remain unclear: the receptors may be present as inactive dimers, thus being activated upon the binding of a ligand, or a ligand may bind receptor monomers on the cell surface, which then dimerize and complete activation.17-18 VEGF-A has been shown to have approximately 100-fold higher affinity towards VEGFR-2 dimers in comparison to receptor monomers, supporting the hypothesis that VEGF-A binds to a pre-dimerized VEGFR-2.19 A recent study using receptor constructs carrying artificial dimerization-promoting transmembrane domains also demonstrates that the latter is most probable. Furthermore, this study shows that although dimerization is necessary for receptor activation, it is not sufficient, due to distinct orientation of receptor monomers, which is required to instignate transmembrane signaling and that activation and signaling of VEGFR is dependent on co-receptors (neuropilins and heparin sulphate proteoglycans). Furthermore, as mentioned above, VEGF-VEGFR interactions differ with each VEGF isoform.
The VEGF ligands A, B and PlGF-1 and -2 bind the VEGFR-1 (Flt-1), which is expressed on vascular endothelial cells, macrophages and on certain types of tumor cells.20 VEGF-A binds this receptor with high affinity, but induces weak tyrosine kinase activity.13 This led to the hypothesis that VEGFR-1 is primarily a “decoy” receptor, which traps VEGF-A and in this manner prevents binding to, and activation of the VEGFR-2, thereby suppressing the pro-angiogenic effect of VEGF-A.21 VEGFR-1 knock-out mice demonstrated a severe disorganization of the vasculature with excessive proliferation of hemangioblasts resulting in intra-uterine death of mice embryos.22 This observation that VEGFR-1 negatively regulates physiological vasculogenesis and angiogenesis in the embryo supports the hypothesis of VEGF-A trapping. That VEGFR-1 is important for VEGF-A trapping (in its extracellular domain) is shown in a further report demonstrating that VEGFR-1 lacking its tyrosine-kinase domain is sufficient for normal angiogenesis in mice.23
In adulthood, VEGFR-1 stimulates inflammation, tumor growth and metastasis in an, at least to a degree, macrophage-dependent manner.13 For VEGF-A-induced chemotaxis of monocytes, the activation of the tyrosine domain of VEGFR-1 is required. VEGFR-1 signaling has been furthermore implied in tumor progression due to releasing of increased amounts of matrix metalloprotease-9 from human vascular smooth muscle cells 24 and has through this mechanism been implied in lung-specific metastasis.25
Due to alternative splicing of VEGFR-1 mRNA,12, 26-27 but also by proteolytic shedding of the extracellular domain,28 this receptor exists as a soluble, truncated form (sVEGFR-1), and is composed of the first six of seven extracellular immunoglobin-like domains. The soluble form retains its affinity for VEGF-A ligands, the neuropilin-1 (NRP-1) co-receptor and extracellular heparin sulphate proteogycans, as well its capacity for homo/heterodimerization with mbVEGFR-1 and VEGFR-2.12, 29-32 By VEGF-A trapping and formation of inactive heterodimers with VEGF receptors, sVEGFR-1 acts predominately negative on VEGF-A action.32 Indeed, sVEGFR-1 has been shown to have potent anti-hemangiogenic features, e.g., it is essential for the avascularity of the cornea during mouse development.33 Furthermore, sVEGFR-1 has been implicated in the pathophysiology of eclampsia.34-35 Soluble VEGFR-1 is secreted by endothelial cells in vitro.36
Through binding and activating the VEGFR-2 (KDR), which is expressed primarily on endothelial cells, VEGF-A acts predominantly and potently pro-angiogenic. Lower expression levels of VEGFR-2 are observed on hematopoietic cells, neuronal cells, osteoblasts, pancreatic duct cells and retinal progenitor cells, all of which the biological significance remains unclear.37 VEGF-C and -D are also ligands of VEGFR-2, through which they may direct their hemangiogenic activity.
Within tumors, tumor cells express high levels of VEGF-A and low levels of VEGFR-2, whereas endothelial cells within the tumors demonstrate a 3.5 fold higher expression of VEGFR-2, compared to normal vasculature.38 This suggests a paracrine loop of VEGF-A and VEGFR-2 between tumor cells and vascular endothelial cells for the stimulation of angiogenesis.1, 39-40 A truncated form of VEGFR-2 (Flk-1) consisting of the complete extracellular domain (seven Ig-like domains) and a small portion of intracellular domain was firstly described by Wen and coworkers in the rat retina. This short form was shown to be functional as demonstrated by the increase of cytoplasmatic calcium, an in vitro test of VEGF-A activity.41
Furthermore, a naturally occurring soluble form of VEGFR-2 (sVEGFR-2) present in mouse and human plasma has been described. This extracellular domain of the VEGFR-2 was detected by immunoprecipitation and ELISA, using anti-VEGFR-2 antibodies raised against the extracellular domain of mouse or human VEGFR-2. Furthermore, sVEGFR-2 was found in conditioned media of mouse and human endothelial cells, suggesting that this soluble receptor form may be secreted by endothelial cells or be proteolytically cleaved.42 The mouse-derived svegfr-2 described by this group had a weight of 160 kDa in its glycosylated form, and approximately 90 kDa when deglycosylated, and was capable of binding VEGF-A-coated plates. The human sVEGFR-2 was not further characterized.
Recombinant murine svegfr-2 and recombinant human sVEGFR-2, obtained by cloning of the complete extracellular domain of the membrane-bound receptor was described by Huang et al. and Roeckl et al. Using far-western blotting and solid-phase assays Roeckl et al. have shown that VEGF-A165 binds to recombinant human sVEGFR-2 only if either the ligand or the receptor is immobilized in the presence of heparin. Unlike sVEGFR-1, sVEGFR-2 did not efficiently antagonize binding of VEGF-A165 to human microvascular endothelial cells (MVE) (which express both mbVEGFR-1 and -2 in culture) and consequently did not inhibit VEGF-A-induced mitogenesis.43-44
Most recently, Albuquerque and coworkers45 described a naturally occurring truncated, soluble form of mouse and human VEGFR-2. They discovered that each consists of six of the seven Ig-like extracellular domains and are the result of alternative splicing. The mouse svegfr-2 contains a 13 amino acid carboxy-terminal sequence and the human sVEGFR-2 contains a unique 16-amino acid carboxy-terminal sequence, both of which are not present in mbvegfr-2/mbVEGFR-2. They are the result of partial intron 13 retention with in-frame termination of 39 nucleotides downstream from the exon 13-intron 13 junction in the mature mRNA. A rabbit polyclonal antibody that binds the C-terminus of svegfr-2/sVEGFR-2 and therefore selectively recognizes the soluble receptor form and does not cross-react with membrane-bound vegfr-2/VEGFR-2 or with svegfr-1/sVEGFR-1, was raised and used to identify the localization of svegfr-2/sVEGFR-2 in tissues. Soluble VEGFR-2 mRNA has been identified in HUVE cells in vitro and found to be present in the human cornea by immunostaining using the aforementioned antibodies.45 We have shown the presence and differential distribution of the sVEGFR-2 protein and mbVEGFR-2 in the human foreskin (Figures 1, 2). Soluble vegfr-2 protein and/or mRNA was found to be present in various tissues of the wild-type mouse, such as the skin, heart, spleen, kidney, ovary and, as previously described, in plasma. Furthermore, immunostaining and western blotting with the aforementioned antibodies revealed the presence of the svegfr-2 protein throughout the cornea (produced in corneal epithelium) of newborn and adult wild-type mice, but the absence of mbvegfr-2. No svegfr-2 was present in the conjunctiva.45
Figure 1.

Immunostaining of membrane-bound VEGFR-2 (mbVEGFR-2) in human foreskin (green) shows its localization on blood vascular and lymphatic endothelial cells (the latter are red by anti-Prox-1 nuclear staining). Dapi staining (blue) was used to demonstrate cell nuclei. A) Bar = 100μm. B) Bar = 50μm.
Figure 2.

Immunostaining of sVEGFR-2 in human foreskin. A) Dapi staining of nuclei. B) Anti-sVEGFR-2. C) Merged picture. D) Negative control. Soluble VEGFR-2 is primarily expressed in the epidermis and in blood vessels in the dermis. Non-specific fluorescence is visible in the hornifying layer of the epidermis. Bar = 200μm.
In order to determine the function of svegfr-2, LeCre;vegfr-2loxP/loxP mice were created, using the Cre-loxP approach.46-47 These mice express Cre recombinase in the cornea under the control of a paired box-6 promoter and specifically lack svegfr-2 in the cornea. Vegfr-2 knock-out mice were not used, as such mice die in utero at an early stage.48 It was found that the LeCre; vegfr-2loxP/loxP mouse corneas were invaded by lymphatic vessels at postnatal day 0, suggesting an anti-lymphangiogenic activity of svegfr-2 (Figure 3). Lymphatics were identified by Lyve-1 staining of CD31 weakly positive vessels, by co-expression of the lymphatic endothelial cell marker Prox-1 and by the presence of typical morphology in transmission electron microscopy. Blood vessels were not present, as confirmed by the lack of MECA-32, a blood vessel-specific marker. All control corneas lacked both blood and lymphatic vessels. These results indicated that the developing mouse cornea is exposed to lymphangiogenic stimuli that are counterbalanced by svegfr-2. A hypothesis evolved that svegfr-2 traps vegf-c and disables its binding and activation of vegfr-3, thus inhibiting lymphangiogenesis. To support this hypothesis, Albuquerque et al. have demonstrated the presence of vegf-c in wild-type newborn mouse cornea. Soluble vegfr-2 contains the vegf-c binding site and interacts with vegf-c. In vitro experiments demonstrated that svegfr-2 inhibits vegf-c-induced vegfr-3 activation and proliferation of lymphatic endothelial cells. Intra-corneal administration of a svegfr-2 plasmid in wild-type mice inhibited vegf-c-induced lymphangiogenesis, demonstrating the anti-lymphangiogenic effect of svegfr-2 in vivo.
Figure 3.
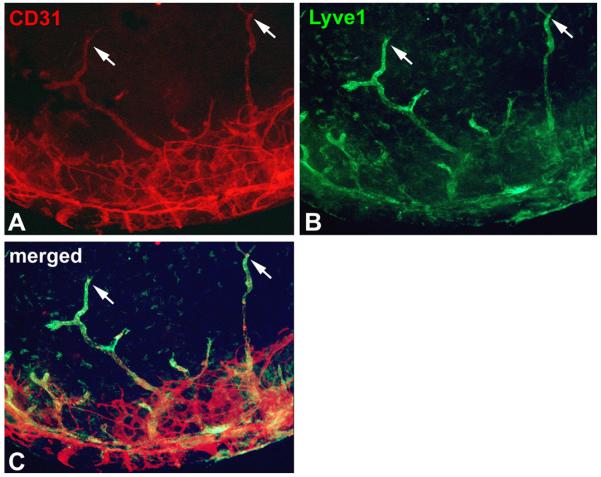
Immunostaining of the cornea of a LeCre;svegfr-2loxP/loxP newborn mouse, which selectively lacks svegfr-2 in the cornea. A) CD31, a pan-endothelial cell marker is stained red corneal vessels. B) A specific marker for lymphatic endothelial cells, Lyve-1 (lymphatic vascular endothelial hyaluronan receptor-1) stains corneal lymphatics in green. C) Merged image of CD31 and Lyve-1 staining demonstrates that all vessels within the cornea are lymphatic vessels, whereas in the limbus both types of vessels are present. The arrows point to typical blind-ended morphology of lymphatic vessels.
Vegf-c (protein and RNA) is not present in adult wild-type mouse corneas. Augmentation of svegfr-2 expression in wild-type mouse corneas using in vivo transfection with psvegfr-2, reduced suture injury-induced lymphangiogenesis and did not influence hemangiogenesis, confirming a selective anti-lymphangiogenic effect of svegfr-2 in vivo. Besides the cornea, svegfr-2/sVEGFR-2 has been shown to be present in the epidermis, which lacks mbvegfr-2/mbVEGFR-2 (Figure 2). In the skin of P0 K14Cre;vegfr2loxP/loxP mice, which specifically lack svegfr-2 in the epidermis and hair follicules, marked enlargement of lymphatic vessels compared to those of controls was found. These findings furthermore support the concept that svegfr-2 is an in vivo antagonist of vegf-c.45 The distribution of sVEGFR-2 in the human skin and cornea is similar to that in mice, suggesting it has a similar role in maintaining an alymphatic state. Since human lymphangioma endothelial cells (LaECs) produce VEGF-C and express VEGFR-3, the potential anti-lymphangiogenic properties of sVEGFR-2 were tested in vitro on these cells. Soluble VEGFR-2 was found to abolish VEGF-C-induced proliferation of LaECs. These results give rise to the possibility of an anti-lymphangiogenic treatment of lymphangiomas.45
To summarize, Albuquerque et al. revealed that the previously described, naturally occurring truncated soluble form of human and murine sVEGFR-242 is the result of alternative splicing and consists of six Ig-like domains with a unique 13 or 16 amino acid carboxy-terminal sequence. Furthermore, the authors show that sVEGFR-2 secreted by human embryonic kidney cells, mouse corneal epithelial cells and Chinese hamster ovary cells are present in monomeric and not in dimeric form. Soluble VEGFR-2 does not abolish VEGF-A mitogenic activity in vitro nor inhibits VEGF-A-mediated hemangiogenesis in vivo after suture injury of the cornea. These data corroborate the previously reported in vitro differential VEGF-A binding avidity between monomeric and dimeric forms of VEGFR-2, indicating that sVEGFR-2 does not play a role in hemangiogenesis.19, 44
Based on the novel finding that sVEGFR-2 is an inhibitor of VEGF-C-induced lymphangiogenesis, which is a crucial process in tumor progression, the role of sVEGFR-2 in progressed stages of neuroblastoma versus regional neuroblastoma was investigated. Neuroblastoma is the most common malignant childhood solid tumor. It arises from the sympathic nervous system, and frequently originates in the adrenal medulla or sympathetic abdominal ganglia, but also in the neck, chest and pelvis. The International Neuroblastoma Staging System (INSS) differentiates 4 stages of neuroblastoma. Stages 1 and 2A are characterized by localized tumors, stage 2B is a localized tumor with ipsilateral lymph node involvement, stage 3 is characterized by tumor progression, with tumor expanding over the midline and invading regional lymph nodes, and stage 4 includes disseminated metastases into distant organs and lymph nodes. The unique neuroblastoma stage 4s is characterized by its occurrence in infants (<1 year of age), its dissemination and capability of spontaneous regression. Five-year survival rates of the progressed, stage 4 neuroblastoma is approximately 30%, compared to 95% of non-disseminated stages 1 and 2. Besides the child’s age and stage of neuroblastoma, several molecular features, such as the amplification of the proto-oncogene MYCN and segmental chromosome alterations such as 1p and 11q deletions, and 1q gain represent risk factors and are associated with poor survival.49 Progressed neuroblastoma is highly vascularized and several studies have demonstrated the significance of angiogenesis and lymphangiogenesis in neuroblastoma progression, correlating high expression levels of VEGF-A, C and D with advanced neuroblastomas and poor outcome.50-53 However, the expression of VEGF-A and C did not correlate to established risk-factors, for e.g. MYCN.
In order to further elucidate the significance of lymphangiogenesis and hemangiogenesis in the progression of neuroblastoma, we have investigated the expression of VEGFs and their receptors, including the naturally occurring splice variant of VEGFR-2 (sVEGFR-2), in primary untreated neuroblastomas.54 There were no significant differences in the expression levels of VEGF-A, VEGF-C and VEGF-D in advanced neuroblastoma (stages 3, 4, 4S) compared to localized neuroblastoma (stages 1 and 2). However, a significant difference between localized and metastatic stages was found for sVEGFR-2. Soluble VEGFR-2 was highly expressed in stages 1 and 2, and barely detectable in stages 3, 4 and 4s (Fig. 4). These data imply that sVEGFR-2 besides being an inhibitor of lymphangiogenesis in normal (mouse) tissues, also seems to play a role in malignant tumor progression in humans. Furthermore, we have investigated how sVEGFR-2 is expressed in stage 4 neuroblastoma with MYCN amplification, a predictor of poor survival. We found that sVEGFR-2 and VEGFR-1 are mostly downregulated in tumors and VEGF-A and VEGF-D mostly upregulated in neuroblastoma tissue. However, this was statistically not significant.54
Figure 4.

Expression of sVEGFR-2 in 49 neuroblastoma (NB) tissue samples, as determined by real-time RT-PCR. The expression levels (bars, and standard error of the mean) are shown separately for each tumor stage. Tumor stages 3, 4 and 4s (progressed NB) demonstrate statistically (p < 0.05) significantly lower levels of sVEGFR-2 compared to localized stages 1 and 2. Primers used for RT-PCR were: sVEGFR-2 fwd: 5′- GCCTTGCTCAAGACAGGAAG -3′; sVEGFR-2 rev: 5′- CAACTGCCTCTGCACAATGA -3′. β-Actin fwd: 5′- GCATCCCCCAAAGTTCACAA -3′; β-Actin rev: 5′- AGGACTGGGCCATTCTCCTT -3′.
In summary, recent data have elucidated the molecular identity and cellular source of sVEGFR-2 and demonstrated its role as an endogenous inhibitor of lymphangiogenesis in the human and mouse cornea. The data also imply a role for sVEGFR-2 in the progression of neuroblastoma, a malignant tumor. Figure 5 schematically presents an overview of VEGFs and VEGF receptors in light of the latest data.
Figure 5.

Schematic presentation of the VEGF ligands and their interaction with membrane-bound and soluble (secreted) VEGF receptors. Notably, inhibitory effects are exerted by sVEGFR-1 (anti-hemangiogenesis) and sVEGFR-2 (anti-lymphangiogenesis). ). Neuropilin-2 (NRP-2) also interacts with VEGFR-3 on lymphatic endothelial cells, which is not shown in this scheme . NRP-1: Neuropilin-1. Modified from: http://www.rosenthallab.com/gallery/images/VEGF_VEGFR.jpeg
References
- 1.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 2.Tischer E, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 3.Houck KA, et al. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- 4.Grunewald FS, et al. Structure-function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim Biophys Acta. 2010;1804:567–580. doi: 10.1016/j.bbapap.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 5.DiSalvo J, et al. Purification and characterization of a naturally occurring vascular endothelial growth factor.placenta growth factor heterodimer. J Biol Chem. 1995;270:7717–7723. doi: 10.1074/jbc.270.13.7717. [DOI] [PubMed] [Google Scholar]
- 6.Olofsson B, et al. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci U S A. 1996;93:2576–2581. doi: 10.1073/pnas.93.6.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao Y, et al. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J Biol Chem. 1996;271:3154–3162. doi: 10.1074/jbc.271.6.3154. [DOI] [PubMed] [Google Scholar]
- 8.Joukov V, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–298. [PMC free article] [PubMed] [Google Scholar]
- 9.Veikkola T, et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001;20:1223–1231. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stacker SA, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–191. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 11.Makinen T, et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001;20:4762–4773. doi: 10.1093/emboj/20.17.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia H, et al. Vascular endothelial growth factor (VEGF)-D and VEGF-A differentially regulate KDR-mediated signaling and biological function in vascular endothelial cells. J Biol Chem. 2004;279:36148–36157. doi: 10.1074/jbc.M401538200. [DOI] [PubMed] [Google Scholar]
- 13.Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–478. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 14.Roskoski R., Jr. VEGF receptor protein-tyrosine kinases: structure and regulation. Biochem Biophys Res Commun. 2008;375:287–291. doi: 10.1016/j.bbrc.2008.07.121. [DOI] [PubMed] [Google Scholar]
- 15.Shibuya M, Ito N, Claesson-Welsh L. Structure and function of vascular endothelial growth factor receptor-1 and -2. Curr Top Microbiol Immunol. 1999;237:59–83. doi: 10.1007/978-3-642-59953-8_4. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, et al. Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc Natl Acad Sci U S A. 2010;107:1906–1911. doi: 10.1073/pnas.0914052107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuttfeld E, Ballmer-Hofer K. Structure and function of VEGF receptors. IUBMB Life. 2009;61:915–922. doi: 10.1002/iub.234. [DOI] [PubMed] [Google Scholar]
- 18.Mac Gabhann F, Popel AS. Dimerization of VEGF receptors and implications for signal transduction: a computational study. Biophys Chem. 2007;128:125–139. doi: 10.1016/j.bpc.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuh G, et al. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem. 1998;273:11197–11204. doi: 10.1074/jbc.273.18.11197. [DOI] [PubMed] [Google Scholar]
- 20.Herold-Mende C, et al. Expression and functional significance of vascular endothelial growth factor receptors in human tumor cells. Lab Invest. 1999;79:1573–1582. [PubMed] [Google Scholar]
- 21.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 22.Fong GH, et al. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development. 1999;126:3015–3025. doi: 10.1242/dev.126.13.3015. [DOI] [PubMed] [Google Scholar]
- 23.Hiratsuka S, et al. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998;95:9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gille H, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 25.Hiratsuka S, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- 26.Olsson AK, et al. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 27.Cai J, et al. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J Biol Chem. 2006;281:3604–3613. doi: 10.1074/jbc.M507401200. [DOI] [PubMed] [Google Scholar]
- 28.Rahimi N, Golde TE, Meyer RD. Identification of ligand-induced proteolytic cleavage and ectodomain shedding of VEGFR-1/FLT1 in leukemic cancer cells. Cancer Res. 2009;69:2607–2614. doi: 10.1158/0008-5472.CAN-08-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barleon B, et al. Mapping of the sites for ligand binding and receptor dimerization at the extracellular domain of the vascular endothelial growth factor receptor FLT-1. J Biol Chem. 1997;272:10382–10388. doi: 10.1074/jbc.272.16.10382. [DOI] [PubMed] [Google Scholar]
- 30.Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem. 2000;275:26690–26695. doi: 10.1074/jbc.M003955200. [DOI] [PubMed] [Google Scholar]
- 31.Cebe-Suarez S, Zehnder-Fjallman A, Ballmer-Hofer K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell Mol Life Sci. 2006;63:601–615. doi: 10.1007/s00018-005-5426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kendall RL, Wang G, Thomas KA. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun. 1996;226:324–328. doi: 10.1006/bbrc.1996.1355. [DOI] [PubMed] [Google Scholar]
- 33.Ambati BK, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koga K, et al. Elevated serum soluble vascular endothelial growth factor receptor 1 (sVEGFR-1) levels in women with preeclampsia. J Clin Endocrinol Metab. 2003;88:2348–2351. doi: 10.1210/jc.2002-021942. [DOI] [PubMed] [Google Scholar]
- 35.Maynard SE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A. 1993;90:10705–10709. doi: 10.1073/pnas.90.22.10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE. 2001;2001:re21. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 38.Plate KH, et al. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer. 1994;59:520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- 39.Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 2002;1:219–227. doi: 10.1016/s1535-6108(02)00051-x. [DOI] [PubMed] [Google Scholar]
- 40.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 41.Wen Y, et al. Two functional forms of vascular endothelial growth factor receptor-2/Flk-1 mRNA are expressed in normal rat retina. J Biol Chem. 1998;273:2090–2097. doi: 10.1074/jbc.273.4.2090. [DOI] [PubMed] [Google Scholar]
- 42.Ebos JM, et al. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Mol Cancer Res. 2004;2:315–326. [PubMed] [Google Scholar]
- 43.Huang X, et al. Expression of soluble VEGF receptor 2 and characterization of its binding by surface plasmon resonance. Biochem Biophys Res Commun. 1998;252:643–648. doi: 10.1006/bbrc.1998.9717. [DOI] [PubMed] [Google Scholar]
- 44.Roeckl W, et al. Differential binding characteristics and cellular inhibition by soluble VEGF receptors 1 and 2. Exp Cell Res. 1998;241:161–170. doi: 10.1006/excr.1998.4039. [DOI] [PubMed] [Google Scholar]
- 45.Albuquerque RJ, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med. 2009;15:1023–1030. doi: 10.1038/nm.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu H, et al. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 47.Ashery-Padan R, et al. Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 2000;14:2701–2711. doi: 10.1101/gad.184000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shalaby F, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 49.Schwab M, et al. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol. 2003;4:472–480. doi: 10.1016/s1470-2045(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 50.Meitar D, et al. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J Clin Oncol. 1996;14:405–414. doi: 10.1200/JCO.1996.14.2.405. [DOI] [PubMed] [Google Scholar]
- 51.Rossler J, et al. Angiogenesis as a target in neuroblastoma. Eur J Cancer. 2008;44:1645–1656. doi: 10.1016/j.ejca.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 52.Eggert A, et al. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6:1900–1908. [PubMed] [Google Scholar]
- 53.Lagodny J, et al. Lymphangiogenesis and its regulation in human neuroblastoma. Biochem Biophys Res Commun. 2007;352:571–577. doi: 10.1016/j.bbrc.2006.11.062. [DOI] [PubMed] [Google Scholar]
- 54.Becker J, et al. Neuroblastoma progression correlates with downregulation of the lymphangiogenesis inhibitor sVEGFR-2. Clin Cancer Res. 2010;16:1431–1441. doi: 10.1158/1078-0432.CCR-09-1936. [DOI] [PMC free article] [PubMed] [Google Scholar]