

Abstract
The generation of strong, virus-neutralizing antibody responses to the HIV-1 envelope spike (Env) is a major goal in HIV-1 vaccine research. To try to enhance the Env-specific response, we displayed oligomeric gp140 on a virus-like scaffold provided by the lambda phage capsid. To do this, an in vitro complementation system was used to “decorate” phage particles with glycosylated, mammalian cell-derived envelope oligomers. We compared the immune response to lambda phage particles displaying HIV-1 Env to that elicited by soluble oligomeric gp140 in rabbits. Env-binding antibody titers were higher in animals that received oligomeric gp140 as compared to Env decorated phage particles, as were virus neutralizing antibody responses. The Env decorated phage particles were, however, able to efficiently boost a protein-primed humoral response to levels equivalent to those elicited by high-dose adjuvanted Env oligomers. These results show that display of HIV-1 envelope spikes on the bacteriophage lambda capsid does not result in an improved, Env-specific humoral immune response.
Keywords: HIV-1, AIDS, vaccine, envelope spike, virus-like particle, bacteriophage lambda, humoral immunity, antibody
1. Introduction
Structural studies of human and simian immunodeficiency viruses have revealed that the envelope spikes on the virion surface are sparse and irregularly distributed. It has been estimated that normal HIV-1 virus particles may contain as few as 14 ±7 spikes, clustered together [1]. This may contribute to low spike immunogenicity, and further complicate the generation of broadly neutralizing antibodies [2–4], which remains one of the most important goals in HIV-1 vaccine design and development. The display of proteins in an ordered, repetitive array can result in greatly increased immune responses, compared to immunization with soluble protein antigens. This is exemplified by the success of virus-like particles (VLPs) as recombinant vaccine platforms, both for hepatitis B virus [5] and more recently for human papillomavirus (HPV) [6–8]. Phage vectors are also being explored as VLP-like scaffolds for vaccine applications, especially in situations in which antibody responses are desirable [9–11]. A phage-based vaccine for smokers, which contains the hapten nicotine coupled to a virus-like particle derived from the coat protein of bacteriophage Qβ, is already in clinical trial [11]. Bacteriophage have been experimentally administered to animals and safely used in humans for several decades, both for the treatment of bacterial infections [12, 13] and also for the assessment of humoral immune responses in immunocompromised subjects [14, 15]. More recently, the development of phage display technology has made it possible to display short, exogenous peptides at high copy number and surface density on the capsid of filamentous bacteriophage [16, 17], leading to the evaluation of phage display vectors as potential vaccine delivery platforms [13, 18–20]. In general, phage vectors have considerable advantages over other vaccine platforms, such as mammalian virus vectors, due to genetic tractability, inexpensive production, and suitability for scale-up [21], as well as their physical stability. Importantly, vaccination with filamentous phage particles that display antigen on their surface results in strong humoral immune responses in experimental animals [22–24].
Phage capsids are used to display not only short peptides but also intact proteins. Display systems developed for lambda phage permit display of foreign proteins by fusing them to the gpD major coat protein [25–31]. gpD is a trimeric, 109 amino acid protein that is required for the packaging of full-length genomes [30]. It is unusual among phage display scaffolds in being highly tolerant of large peptides or protein fusions, which can therefore be displayed at high copy number and surface density on the phage capsid [18, 29, 30]. This differentiates lambda from filamentous phage vectors, in which only short peptides can generally be displayed at high copy number.
A second key difference between filamentous and lambda phage vectors is the fact that gpD-deficient λ phage capsids can be “decorated” in vitro with exogenously supplied gpD [18, 29, 30]. This permits considerable flexibility with respect to the surface display of complex antigens such as the HIV-1 envelope spike. In the present work, we used a simple in vitro complementation system to decorate lambda phage capsids with glycosylated, mammalian cell-derived HIV-1 envelope trimers. We hypothesized that the immunogenicity of HIV-1 envelope spikes is limited, in part, as a result of their sparse and irregular distribution on the virion surface. Therefore, the high-density, repeating array of the HIV-1 envelope antigen on the surface of the phage capsid would result in enhanced humoral immune responses. The Env-binding antibody titers, as well as the neutralizing antibody responses, were not higher in those groups that received Env decorated phage particles as compared to soluble oligomeric gp140. The Env decorated phage, however, were able to efficiently boost a protein-primed humoral response that was comparable to that elicited by high-dose adjuvanted Env oligomers. Overall, these results suggest that Env decorated phage particles alone do not significantly improve the humoral immune response as compared to soluble oligomeric protein.
2. Materials and Methods
2.1 Envelope glycoprotein and gpD expression plasmids
The strategy employed by Wyatt and Stamatatos [32–36] was used to generate mammalian expression constructs that encode a cleavage deficient, trimeric HIV-Envgp140. To do this, a human codon-optimized derivative of the R5 HIV-1 isolate YU2 gene was generated synthetically (GeneArt, Regensburg, Germany). The Env construct encodes the complete gp120 and gp41 ectodomain with alterations in the gp120/gp41 cleavage site (arginines at amino acid positions 508 and 511 changed to serine) [37] fused in frame to the human tissue plasminogen activator (TPA) leader sequence. The trimeric motif derived from T4 bacteriophage fibritin (FT) was positioned after lysine 683 followed by a His6 tag and stop codon. To produce gp140:gpD fusion protein, a short flexible linker peptide [Gly4Ser]2 was added following the FT domain and gpD was fused to the C-terminus along with the His6 tag. Both gp140 and gp140:gpD fusion constructs were subsequently cloned into pcDNA3 vector (Invitrogen) for expression in 293 Freestyle cells. Amino acid residue numbers correspond to those of the prototypic HXBc2 HIV envelope glycoprotein.
In order to derive a construct that expressed gpD alone, a human codon-optimized derivative of the wild type λ gpD gene was generated synthetically (GeneArt, Regensburg, Germany) with a short flexible linker peptide [Gly4Ser]1 followed by a His6 tag and stop codon. The gene was cloned into pcDNA3 vector (Invitrogen) for expression in 293 FreeStyle cells.
2.2 Expression and purification of Envgp140:gpD, Envgp140 and gpD proteins
All proteins were expressed in serum-free medium by transient transfection of suspension-adapted FreeStyle HEK 293-F cells (Invitrogen). Briefly, 293-F cells cultured in FreeStyle 293 Expression medium (Invitrogen) were seeded at a density of 7.5 × 105 cells per ml the day before transfection. After overnight incubation, just prior to transfection, culture cell density was adjusted to 1.0 × 106 cells/ml by addition of fresh medium. FreeStyle MAX reagent (Invitrogen) was used to transfect pcDNA3 constructs per manufacturer’s instructions. Five to 6 days post-transfection the cell culture supernatants were collected and centrifuged at 3,500 × g to remove cell debris. All proteins were purified by metal affinity chromatography using Ni-NTA resin (Qiagen). Columns were washed with increasing concentrations of imidazole (20 mM and 40 mM) followed by elution in the presence of 250 mM imidazole. Fractions containing purified protein were pooled, dialyzed against phage buffer (50 mM Tris-HCl, pH 8.0, 10 mM NaCl, 10 mM MgCl2) for YU2gp140:gpD fusion and gpD protein or against PBS (pH 7.4) for YU2gp140 protein and concentrated with Amicon Ultra centrifugal filter devices (Millipore). Elution fractions were analyzed for yield and purity by performing SDS-PAGE with Coomassie blue staining (described below) and Bradford assay. Protein aliquots were stored at −80°C until further use. Protein yields were between 1–2 mg of purified protein per liter of culture.
2.3 SDS-PAGE, Size exclusion chromatography and Blue Native PAGE analyses
Affinity purified YU2gp140 and YU2gp140:gpD fusion proteins were subjected to SDS-PAGE analysis. Reduced and nonreduced samples were prepared by boiling for 2 min in sample buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 0.1% bromophenol blue) in the presence or absence of 100 mM dithiothreitol (DTT). Purified proteins were further analyzed by size exclusion chromatography on a Superdex 200 column (Amersham Pharmacia) using phosphate buffered saline (PBS) as the running buffer. The flow rate was set to 1 ml/min, which allowed separation of the oligomeric states and protein retention was determined by monitoring the UV absorption at a wavelength of 280 nm. The column was calibrated using thyroglobulin, ferritin, and YAD as molecular protein markers that exist in oligomeric states of 669, 440, and 150 kDa, respectively.
Blue Native (BN)-PAGE was carried out using Invitrogen NativePAGE Novex Bis-Tris Gel system according to the manufacturer’s instructions. Briefly, purified protein samples were diluted to a final 1X concentration using 4X Native PAGE sample buffer containing BisTris buffer, pH 7.2, NaCl, glycerol, and Ponceau S just prior to loading onto a 3 to 12% Bis-Tris NuPAGE gel (Invitrogen). Typically, gel electrophoresis was performed for 2 h at 150V using NativePAGE anode buffer and NativePAGE dark blue cathode buffer (0.02% G-250) in the upper buffer chamber. The gel was destained using 40% methanol, 10% acetic acid. Typically, 5 μg of purified protein was loaded per lane. HMW Native Mark (GE Healthcare) was used as a protein standard for BN PAGE. Recombinant HIV-1 IIIB Glycoprotein gp120 (ImmunoDiagnostics Inc) was run on SDS-PAGE and BN-PAGE as a monomeric control Env glycoprotein.
2.4 Generation of mosaic Env decorated phage particles
Lysogens of TOP10 cells (Invitrogen) containing λ D1180 [25], a generous gift from Dr. Mahito Nakanishi, which is deficient in gpD, were grown overnight at 32°C and the resulting culture was then used to inoculate 4 × 1L of fresh NZCYM medium the next day (at a dilution of 1:100). Cultures were grown at 32°C with vigorous shaking (300 rpm) until an OD600 of between 0.5–0.6 was reached. Lysogen was induced by transferring the bacteria to a water bath set between 51 and 53°C, followed by incubation with gentle shaking for 15 min. After thermal induction, the cultures were vigorously shaken for an additional 3 hr at 38°C. Bacteria were then pelleted and resuspended in phage suspension media (SM: 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10 mM MgSO4, 0.01% gelatin) and lysed with the addition of 12% chloroform. After chloroform treatment, bacterial DNA was digested with DNase I at a final concentration of 10 μg/ml. The lysate was then cleared of cellular debris by low-speed centrifugation. SM was added to the phage pellet along with recombinant proteins (WT gpD, gp140:gpD) and decoration was allowed to occur overnight at 4°C with gentle shaking. For high density-display phage (HI Env Phage), gpD and Env:gpD fusion proteins were mixed at a 1:1 molar ratio. For low density-display phage (LO Env Phage), gpD and Env:gpD fusion proteins were mixed at a 20:1 molar ratio. Decorated phage were subsequently purified by cesium chloride equilibrium density gradient centrifugation and phage was dialyzed against dialysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM NaCl, 10 mM MgCl2) prior to storage at 4°C. Serial dilutions were then titered on LE392 E.coli bacteria. Wildtype phage preparations were generated similarly except that lysogen λ D1180 transformed with a gpD expression plasmid was used [38] and only SM buffer was added to pelleted phage after low-speed centrifugation. Typical titers for WT phage were 1×1012 PFU/ml, while Env decorated phage titers were 5×1011 PFU/ml. Endotoxin was removed from phage preparations using EndoTrap Red endotoxin removal system (Profos AG) prior to use in in vivo studies. The final endotoxin (ET) content was measured using the limulus amebocyte lectin (LAL) assays performed by Associates of Cape Cod.
2.5 Phage stability and immunoblot analysis of purified phage preparations
1×109 PFU (plaque-forming units) of gpD-deficient bacteriophage lambda was decorated with varying molar ratios of WT gpD protein and YU2gp140:gpD fusion protein. The decorations were performed by incubating the desired molar ratios of proteins with the gpD-deficient phage at 30°C for 20 minutes. The stability of the decorated phage samples was then tested using EDTA. Decorated phage samples were diluted into either 10 mM Tris, pH 7.5/10 mM MgCl2 (TM) or 10 mM Tris, pH 7.5/100mM EDTA (TE) and incubated for 30 min at 37°C. Serial dilutions were then titered on LE392 E.coli bacteria and phage titers reported as PFU per ml. Decorated phage preparations (1:1, 20:1, and gpD only) were tested for stability in serum by incubating 1×1010 PFU phage particles in phage suspension media (SM) or normal rabbit serum for 25 min at 37°C. Serial dilutions were then titered as above.
To determine whether gpD or gp140:gpD fusion proteins were present on the phage capsid, 1×109 PFU of phage decorated with 1:1 or 20:1 ratio of WT gpD to gp140:gpD protein, or WT gpD protein alone, was denatured and structural proteins separated by 12.5% SDS-PAGE. Phage proteins were then detected by western blotting with a rabbit antiserum for gpD as described [39].
2.6 ELISA analysis of soluble gp140:gpD and phage displaying gp140:gpD fusion protein
Gp140 and gp140:gpD fusion proteins produced in 293F cells were tested by ELISA for binding reactivity to a panel of Env-specific antibodies. Microtiter plates were coated overnight at 4°C with 100 ng of each respective protein diluted in 1X phosphate-buffered saline (PBS). After blocking with 3% BSA in 1X PBS, antibodies were added to wells in serial three fold dilutions (0.0015–3.3 μg/ml) in high salt (HS) PBS containing a final concentration of 300 mM sodium chloride and 1% BSA. After four washes with PBS/0.025% Tween-20, a secondary anti-Human-IgG-HRP antibody (Sigma) was added in washing buffer at a 1:5000 dilution for 1 hr at RT. Following four washes, the ELISAs were developed with 100 μl TMB substrate. The reaction was stopped by adding 100 μl 1N sulfuric acid to each well. The optical density at 450 nm was read on a microplate reader (Beckman Coulter, DTX800).
For ELISA using phage displaying gp140:gpD protein, plates were coated overnight at 4°C with various Env-specific antibodies at a concentration of 0.05 μg/ml diluted in 1X PBS. After blocking with 3% BSA in 1X PBS, WT or HI Env phage were added to wells in serial three fold dilutions (1.5 × 105 – 1.1 × 108 PFU) in 1X PBS/1% BSA. After four washes with PBS/0.025% Tween-20, anti-gpV antiserum was added at a 1:1000 dilution in PBS/3% BSA to detect phage particles [38]. Following four washes, a secondary anti-rabbit-IgG-HRP (GE Healthcare) was added in washing buffer at a 1:3000 dilution for 1 hr at RT. Following four washes, the ELISAs were developed with 100 μl TMB substrate. The reaction was stopped by adding 100 μl 1N sulfuric acid to each well. The optical density at 450 nm was read on a microplate reader. The following HIV-1 gp120 and gp41 monoclonal antibodies (mAbs) were obtained through the NIH AIDS Research and Reference Reagents Program, Division of AIDS, NIAID, NIH: b12, 2G12, 4E10, 126-7, 17b, Z13, 447-52D, and 257-D. Polyclonal anti-gpV recognizes the major tail protein of bacteriophage lambda and has been previously described [38].
2.7 Rabbit Immunization protocol
Groups of four New Zealand White rabbits (female, ~12 weeks of age) were inoculated intradermally with each respective antigen. A total of 0.5 ml volume was delivered as follows: (1) 5 × 1010 PFU high density Env phage (HI Env), (2) 5 × 1010 PFU low density Env phage (LO Env), (3) 2 × 1011 PFU wild-type phage, (4) 20 μg (high dose) trimeric YU2gp140 protein, (5) 20 μg (high dose) trimeric YU2gp140:gpD fusion protein, (6) 1.5 μg (low dose) trimeric YU2gp140 protein, (7) 1.5 μg (low dose) trimeric YU2gp140:gpD fusion protein, (8) prime with 5 × 1010 PFU high density Env phage, boost with 20 μg trimeric YU2gp140 protein, and (9) prime with 20 μg trimeric YU2gp140 protein and boost with 5 × 1010 PFU high density Env phage. For animals that received 5 × 1010 PFU, this represents approximately 1.0 μg of Env for the HI Env phage, which displays 30 copies of Env trimer per capsid, and approximately 100 ng of Env for the LO Env phage. We chose to use two doses for our soluble Env and Env:gpD: (1) a high dose of 20 μg that is a standard dose expected to elicit a strong humoral response [40] and (2) a low dose of 1.5 μg that is comparable to the amount of Env displayed on HI Env phage. All protein antigens were emulsified in Titer Max Gold adjuvant (Sigma); phage preparations were not adjuvanted. Pre-bleeds were collected prior to immunization to be used as controls. Boosting inoculations occurred 5, 10, and 15 weeks after the initial inoculation. Ear bleeding was performed 4 weeks after the first and 7 days after the second, third, and fourth inoculations. Animal housing and immunization procedures were performed by ProSci Inc. and adhered to IACUC (Institutional Animal Care and Use Committee). To isolate serum, blood was incubated at 37°C for 2 h and then overnight at 4°C to allow clot formation before centrifuging to separate the liquid phase from the clotted components. The serum was collected and incubated at 55°C for 1 h to heat-inactivate complement and stored at −20°C until subjected to analysis.
2.8 Determination of IgG antibody binding titers by ELISA for anti-gp140 and anti-gpD reactivity in rabbit sera
To determine the anti-gp140 reactivity in sera from immunized animals, 100 ng of purified mammalian-expressed YU2 gp140 in PBS was absorbed into each well of a high protein-binding microwell plate (Dynex Technologies, Chantilly, VA) overnight at 4°C. After blocking the plates with 100 μl blocking buffer (PBS with 2% dry milk and 5% HIFBS), serial serum dilutions in ELISA blocking buffer were incubated in each well for 1 hr at RT. After five washes with PBS/0.2% Tween-20, a secondary anti-Rabbit-IgG-HRP antibody (Sigma) was added in washing buffer at a 1:5000 dilution for 1 hr at RT. Following five washes, the ELISAs were developed with 100 μl TMB substrate. The reaction was stopped by adding 100 μl 1N sulfuric acid to each well. The optical density at 450 nm was read on a microplate reader (Beckman Coulter, DTX800). Endpoint titers were defined as the last reciprocal serum dilution at which the absorption at 450 nm was greater than two-fold over the signal detected with pre-immune serum. ELISA analysis for phage capsid binding titers was performed as described above except 100 ng of purified mammalian-expressed gpD in PBS was absorbed into each well of a microwell plate overnight at 4°C. In addition, washes were performed with PBS/0.5% Tween-20.
2.9 Env-pseudotyped virus neutralization and luciferase reporter cell assay
Virus-neutralizing activity was measured in TZM-bl cells against HIV-1 MN.3, SF162 and YU2 strains of Env-pseudotyped viruses in a validated luciferase reporter gene assay as previously described [41] by David Montefiori at Duke University. In the assays, the level of HIV-1 infection was quantified by measuring the relative light units (RLU) of luminescence, which are directly proportional to the amount of virus input. Neutralizing antibody titers are expressed as the serum dilution required to reduce luminescence by 50%.
3. Results
3.1. Production of recombinant gp140 and gpD proteins for phage decoration
In order to display HIV-1 Env on lambda phage capsid, we constructed mammalian expression vectors encoding translational fusions, in which the major coat protein, gpD, was fused to the C-terminus of HIV-1 Env glycoprotein. GpD is known to accept large protein inserts on both its N- and C- termini, and the presence of these exogenous sequences does not interfere with the ability of gpD to bind the phage capsid [29]. It has also been shown that one can fuse HIV-1 Env to heterologous proteins without compromising its functional/structural integrity [42–44]. We generated Env:gpD fusion proteins in which the Env component was analogous to Env oligomer constructs described by Wyatt and Stamatatos [32–36, 45]. YU2 Envgp140 with a human tissue plasminogen activator (tPA) leader sequence and a synthetic trimerization domain derived from the T4 bacteriophage FT was translationally fused to a short flexible linker peptide ([Gly4Ser]2) at its C-terminus, and then to a human codon-optimized version of gpD followed by a terminal His6 tag for purification. Additional constructs without the gpD domain were made, in order to produce wild type YU2gp140 trimer that was used as a positive control immunogen.
The gp140, gp140:gpD, and gpD proteins were purified from 293F cell supernatants to >90% homogeneity as determined by SDS-PAGE (Fig. 1A). Under nonreducing conditions, both gp140 and gp140:gpD migrated above the 250 kDa molecular weight marker, indicative of oligomers, with no low molecular weight monomers observed. The retention of complete oligomeric structure under nonreducing conditions in a SDS-PAGE gel is not unexpected, due to the T4 fibritin trimerization domain fused to the gp140 proteins. Upon reduction with DTT, gp140 migrated as a predominant 140-kDa band slightly above gp120. Similarly, gp140:gpD migrated as a predominant 150kDa band slightly above gp140 due to the additional gpD moiety. Recombinant HIV-1 IIIB gp120 protein (ImmunoDiagnostics) was run in parallel as a monomeric control Env glycoprotein. Purified gpD protein was also analyzed by SDS-PAGE and migrated as a predominant 12 kDa band (data not shown) as expected.
Figure 1. Biophysical analysis of purified YU2gp140 and gp14:gpD fusion proteins.

(A) SDS-PAGE analysis of 293F cell expressed proteins. 1 μg of gp120 (ImmunoDiagnostics), gp140, or gp140:gpD fusion proteins in sample buffer with (reduced) or without (non-reduced) 100 mM DTT were resolved on a 7.5% polyacrylamide gel. (B) Analytical size exclusion chromatography. Purified gp140 and gp140:gpD proteins were resolved on a Superdex 200 column in phosphate-buffered saline, and their retention times were compared with those of known molecular mass standard proteins of 150, 440, and 669 kDa (arrows). The main peak retention time of gp140 (18.05 min) and gp140:gpD (18.042 min) is consistent with both proteins migrating as a trimer (ferritin marker protein (440 kDa), 17.95min). (C) Blue native (BN)–PAGE analysis of gp120, and 293F cell-derived, purified gp140 and gp140:gpD proteins (5 μg).
Purified gp140 and gp140:gpD were also examined by size exclusion chromatography. Purified gp140 eluted as a major peak with a retention time of 18.05 min and an apparent molecular mass of ~440 kDa (Fig. 1B). The retention time (18.042 min) and apparent molecular mass (~440 kDa) of gp140:gpD protein paralleled that of gp140 protein, consistent with it being a trimer that is slightly larger than gp140 due to the gpD fusion. A small peak at 669 kDa was detected in both gp140 and gp140:gpD preparations; this most likely represents aggregates or a dimer of trimers, as previously been reported [46]. There was also a minor peak detected at 150 kDa in gp140:gpD protein, presumably representing monomer. Gel elution profiles containing both protein and molecular weight markers can be found in Supplemental Fig. 1. Overall, 90–99% of gp140 and gp140:gpD proteins, respectively, were found to be trimeric by size exclusion chromatography analysis.
We further analyzed both gp140 and gp140:gpD proteins in parallel with gp120 monomer and known molecular weight calibration markers (Amersham) on Blue Native (BN) PAGE (Fig. 1C). Based on the apparent molecular weight of the monomer and the gel mobility positions relative to the molecular weight markers, both gp140 and gp140:gpD proteins migrate at about 669 kDa, with gp140:gpD migrating slightly more slowly - as expected due to the gpD fusion. There can be difficulties estimating the molecular mass of large glycoproteins by electrophoresis and this may explain the difference in molecular size estimates determined using the gel filtration and BN- PAGE analyses [47]. Taken as a whole, the SDS-PAGE, gel filtration chromatography, and BN-PAGE analyses suggest that our purified gp140 and gp140:gpD proteins are indeed trimeric in nature and the addition of gpD to the C-terminus of Envgp140 does not effect the oligomeric conformation of the protein.
Finally, in order to further investigate the conformational integrity of our gp140 and gp140:gpD proteins, we tested their reactivity with a panel of anti-HIV-1 Env antibodies, including conformationally sensitive antibodies (b12, 2G12, 4E10, 126-7, 17b, Z13, and 447-52D). As shown in Fig. 2, the gp140 and gp140:gpD reacted very similarly with all of the antibodies tested.
Figure 2. Reactivity of gp140 and gp140:gpD proteins to a panel of Env-specific antibodies.

Antigens were coated directly onto ELISA wells at 100 ng per well and serial dilutions of the antibodies were tested at concentrations of 0.0015–3.3 μg/ml (antibodies tested were: b12, 126-7, Z13, 447-52D and 257-D). After washing, bound antibodies were detected by colorimetric ELISA. Results are expressed as optical density (OD, at 450 nm). Top panel: Control gp140 protein coated onto ELISA plate wells; bottom panel: gp140:gpD protein.
3.2. Generation of lambda phage particles that display HIV-1 envelope spikes
GpD is required for the generation of stable lambda phage particles containing wild type genomes. However, it is possible to package genomes of sub-genomic length in the absence of gpD, provided that (i) the genome is between approximately 78% and 82% of wild-type length, and (ii) exogenous Mg2+ ions are provided to negate the charge interactions that otherwise lead to head instability [48]. To produce phage particles that display HIV-1 envelope spikes, purified gpD-deficient phage particles were mixed with purified His6-tagged gpD and gp140:gpD fusion proteins at various molar ratios (Fig. 3). Under these conditions, the exogenous gpD is rapidly incorporated into the phage head, resulting in the generation of EDTA-resistant phage particles that can be purified by CsCl density gradient centrifugation [30].
Figure 3. Schematic diagram of in vitro decoration of gpD deficient lambda phage particles with recombinant WT gpD and gp140:gpD fusion proteins.

gpD-deficient bacteriophage lambda procapsids were decorated with recombinant mammalian derived WT gpD protein and gp140:gpD fusion protein. The stability of the decorated phage samples was then tested by treatment with EDTA. Exposing gpD-deficient phage to EDTA disrupts the phage capsid, resulting in loss of infectivity (EDTA sensitive). In contrast, a successfully fully decorated phage particle will remain infectious in the presence of EDTA (EDTA resistant).
Using this in vitro decoration approach, we first tested the ability of recombinant mammalian derived gp140:gpD fusion protein to successfully decorate gpD-deficient phage capsids, either when added alone or in the form of a mixture with wild-type gpD protein. The effectiveness of the decoration reaction was assessed by measuring the titers of the resulting phage preparations before and after exposure to a high concentration (100 mM) of EDTA, which inactivates gpD-deficient capsids but has minimal effect on gpD-bearing particles [29, 30]. Phage titers remained stable in the presence of EDTA if particles were decorated with wild type gpD alone or with various molar ratios of wild type gpD to gp140:gpD (from 20:1 to 1:1) (Fig. 4A). Phage were not stable if they were decorated with 20 fold molar excess gp140:gpD fusion protein (1:20) or with gp140:gpD fusion protein alone, as exemplified by the 2 log drop in phage titer in Fig. 3A. We hypothesize that gp140:gpD fusion protein fails to efficiently decorate the gpD-deficient phage capsids when it is the only available form of gpD, possibly due to steric hindrance, since the envelope protein is considerably larger than wild-type gpD.
Figure 4. Stability of Env decorated bacteriophage particles.

(A) 1×109 PFU (plaque-forming units) of gpD-deficient bacteriophage lambda procapsid were decorated with varying ratios of WT gpD protein and gp140:gpD fusion protein. The decorations were performed by incubating the desired molar ratios of proteins with the gpD-deficient phage (1× 109 PFU) at 30°C for 20 minutes. The stability of the decorated phage samples was then tested by the addition of 100mM EDTA (see Fig. 2) and titering on LE392 bacteria. Undecorated phage was analyzed as a control (EDTA sensitive). The decorations were performed in triplicate within the same assay. (B) 1L preparations of phage decorated with 1:1 or 20:1 WT gpD to gp140:gpD, or WT gpD only were prepared by CsCl-banding and dialysis, titered on LE392 E.coli host cells and then loaded on a 12.5% SDS-PAGE gel, (109 PFU/lane). Phage protein content was examined by immunoblot analysis, using antiserum directed against gpD. Incorporation of both wild-type gpD and gp140:gpD fusion protein in the phage preparations decorated with 1:1 or 20:1 WT gpD to gp140:gpD is indicated by the arrows. (C) 1:1, 20:1 and wild-type (WT) phage (1 × 1010 PFU/ml) were incubated for 30 min at room temperature in SM buffer or normal rabbit serum and subsequently titered on LE392 bacteria to determine stability. The data are represented as means ± SEM.
We next proceeded to large scale preparations of the various decorated phage to verify the integrity of the gp140:gpD fusion proteins displayed on the phage surface. In order to confirm that the gp140:gpD fusion proteins were incorporated onto the phage capsid, equal amounts (PFU) of CsCl-gradient purified decorated phage particles were subjected to SDS-PAGE and immunoblot analysis using gpD-specific antiserum. Western blot analysis showed successful incorporation of both gpD and gp140:gpD fusion proteins when added at molar ratios of 20:1 or 1:1 (gpD to gp140:gpD fusion) to gpD-deficient phage (Fig. 4B). These two molar ratios were used to produce phage particles with both low and high density Env display, referred to hereafter as “HI Env” and “LO Env” phage particles. As shown in Fig. 4B, the relative ratios of wild type gpD to gp140:gpD fusion protein that were detected in the purified high density and low density phage display preparations were approximately 1:1 and 20:1, as expected. The 1:1 decorated phage appear to contain slightly more gp140:gpD fusion protein than wildtype gpD protein. To eliminate the possibility that the gp140:gpD fusion protein simply co-purifies with phage during processing, we analyzed cesium chloride density gradients loaded with gp140:gpD soluble protein alone (no phage particles) versus gradients loaded with phage particles that were decorated with gp140:gpD and WT gpD soluble proteins (1:1). Fractions were pulled from the same area in the CsCl gradients and analyzed by western blot analysis with anti-gpD antiserum. As shown in Fig. S2B, gp140:gpD protein was only detected in the gradient fraction that was loaded with decorated Env phage particles suggesting that soluble gp140:gpD protein does not sediment in the same area as purified phage particles. Thus, it seems unlikely that non-phage-bound soluble protein could have co-purified with phage due to co-sedimentation. A more plausible explanation for the greater than 1:1 ratio of gp140:gpD to gpD protein in our HI Env decorated phage particles may relate to the fact that the soluble gp140:gpD protein is trimeric due to the FT domain, while soluble gpD is monomeric. As a result, gp140:gpD protein interacts with the phage capsids in an oligovalent fashion, and may bind more efficiently that the monomeric soluble gpD protein.
In addition to assessing phage stability in EDTA, we also evaluated the stability of our decorated phage in serum, since we wished to know if they would be stable under conditions likely to be encountered in vivo. As shown in Fig. 4C, both the LO Env and HI Env decorated phage were stable in serum, as were WT phage.
We performed an experiment to measure the amount of Env present on HI Env phage particles. To do this, HI Env phage and various amounts of gp140:gpD fusion protein (50, 100, 150, and 200 ng) were separated by SDS/PAGE, and then subjected to immunoblot analysis with a gpD-specific antibody to detect the gp140:gpD fusion protein (Fig. S2A). This revealed that 3 × 109 HI Env phage particles contained approximately 50 ng of soluble gp140:gpD protein - suggesting that 1 × 1010 phage particles contain approximately 0.2 μg of soluble protein. Since our immunization experiments used 5 × 1010 PFU of phage per dose, we therefore estimate that animals immunized with HI Env phage particles received approximately 1.0 μg of Env protein; this is closely comparable to the amount of Env administered to animals that received the “low dose” soluble Env protein (1.5 μg).
The amount of trimeric gp140:gpD per HI Env phage particle determined from Fig. S2A is equivalent to about 30 copies of gp140:gpD trimers per phage capsid. Since these particles contain more gp140:gpD fusion protein than WT gpD protein (Fig. 4B), we conclude that majority of the 405 gpD binding sites on the phage capsid must be unoccupied. This is unexpected, in light of the physical stability of the HI Env phage particles in EDTA (Fig. 4A) – and presumably reflects the fact that gp140:gpD can stabilize phage particles even when it occupies only a fraction of the available gpD binding sites on the capsid.
In order to investigate the conformational integrity of our displayed gp140:gpD protein, we tested the reactivity of our HI Env phage with a panel of anti-HIV-1 Env antibodies including conformationally sensitive antibodies (b12, 126-7, Z13, 257-D and 447-52D). As shown in Fig. 5, HI Env phage reacted strongly with all of the antibodies tested (Fig. 5, top panel) while WT Phage did not (bottom panel). These data show that the presentation of antibody epitopes within gp140 is unaffected by display on the phage particles.
Figure 5. Reactivity of Env-displaying phage particles with a panel of Env-specific antibodies.

b12, 126-7, Z13, 447-52D and 257-D antibodies were coated directly onto ELISA wells at 0.05 μg/ml and serial dilutions of the phage particles in plaque forming units (PFU), were then added. After washing, bound phage were detected by colorimetric ELISA. Results are expressed as optical density (OD, at 450 nm). Top panel: Env+ HI phage particles; bottom panel: WT phage particles (no Env).
3.3 Immunization of rabbits with Env phage particles
The immunogenicity of both low density and high density-display Env phage particles was tested in rabbits. The immunization protocol consisted of a priming inoculation followed by subsequent boosting inoculations at 5, 10, and 15 weeks after priming. Sera were collected 7 days post-injection except for the first bleed, which was 4 weeks post the priming inoculation. Control groups were immunized with: (a) wild-type phage that displayed only gpD on its surface, (b) trimeric YU2gp140 or trimeric YU2gp140:gpD fusion proteins at doses of either 20 μg, a standard dose expected to elicit a strong humoral response [40], or 1.5 μg, a low dose that is a comparable to the amount of Env displayed on our HI Env phage. Protein immunogens were emulsified in Titer Max (Sigma) adjuvant prior to injection. We also included two prime/boost groups: (8) prime with high density Env phage followed by a boost with trimeric YU2gp140 or (9) prime with trimeric YU2gp140 and boost with high density Env phage (Table 1).
Table 1.
Rabbit Immunization Groups
| Group | Animal | Immunogen | Group | Animal | Immunogen |
|---|---|---|---|---|---|
| 1 | 27 | HI Env phage (1.0μg/dose) | 6 | 55 | YU2gp140 (1.5μg/dose) |
| 28 | 56 | ||||
| 29 | 57 | ||||
| 30 | 58 | ||||
| 2 | 35 | LO Env Phage (est.100ng/dose) | 7 | 59 | YU2gp140:gpD (1.5μg/dose) |
| 36 | 60 | ||||
| 37 | 61 | ||||
| 38 | 62 | ||||
| 3 | 43 | WT Phage | 8 | 63 | Prime HI Env phage/boost YU2gp140a |
| 44 | 64 | ||||
| 45 | 65 | ||||
| 46 | 66 | ||||
| 4 | 47 | YU2gp140 (20μg/dose) | 9 | 67 | Prime YU2gp140/boost HI Env phagea |
| 48 | 68 | ||||
| 49 | 69 | ||||
| 50 | 70 | ||||
| 5 | 51 | YU2gp140:gpD (20μg/dose) | |||
| 52 | |||||
| 53 | |||||
| 54 |
For prime/boost groups, immunizations were at 0 weeks and boosting immunizations were at 5,10, 15 weekds′
3.4 ELISA analysis of gp140 antibody response in sera of immunized rabbits
After each bleed, sera were collected and tested for binding activity to YU2gp140 protein by ELISA. All of the animals that received both low and high dose adjuvanted Env protein oligomers achieved peak titers of anti-gp140 IgG after three to four inoculations, with endpoint titers ranging from 12,500 to 62,500 (Fig. 6A–C). This is consistent with previously published results in rabbits using analogous trimeric YU2gp140 protein preparations [35, 40]. Both the low and high density-display Env phage groups, however, generated lower titers of anti-gp140 IgG as compared with the Env oligomer control groups. The high and low density-display phage groups had endpoint titers ranging from 100 to 12500 after the fourth inoculation (Fig. 6C). The Wilcoxon rank sum test was used to compare final Env-binding antibody titers between the HI Env phage group and a composite group comprising all animals that received immunogens containing a high dose of gp140 protein (i.e. the Phage/Protein, Protein/Phage and the HI gp140 groups). This analysis revealed a strong statistical difference between these two groups (p-value=0.0005), indicating that Env-specific Ab titers were higher in animals that received soluble gp140 protein as compared to animals that received only Env-displaying phage particles (HI Env phage).
Figure 6. Envelope glycoprotein reactivity immunized rabbit sera.
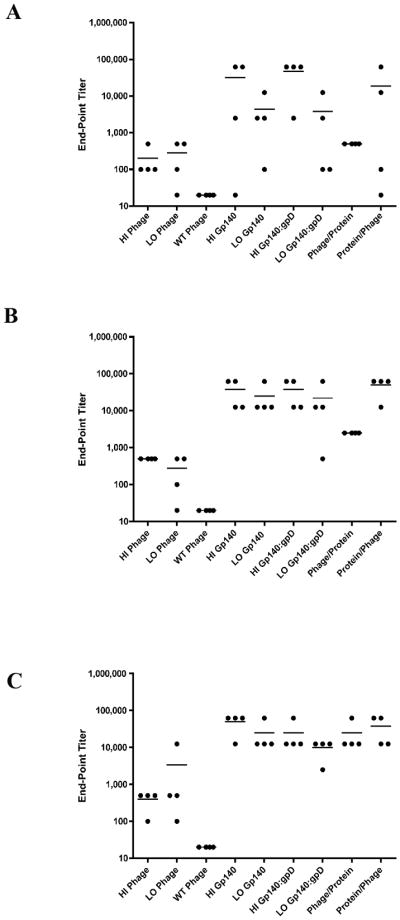
(A–C) Endpoint titers for Env-binding IgG reactivity from bleeds 2–4 (panel A = bleed 2, panel B = bleed 3, panel C = bleed 4). Sera were collected 7 days after the 2nd, 3rd and 4th immunizations, serially diluted 5-fold, and analyzed by IgG ELISA against immobilized gp140 oligomers. End-point ELISA titers were defined as the last reciprocal serum dilution at which the optical density signal was greater than two-fold over the signal detected with the preimmune serum. Dots denote end-point titers for individual animals, while horizontal lines denote mean end-point titers for all 4 animals in each group.
We did not observe a progressively increasing humoral immune response to the displayed Env antigen after the second, third and fourth inoculations with HI Env phage (data not shown). However, the Env decorated phage particles were able to efficiently boost a protein-primed humoral response to levels equivalent to those elicited by high-dose, adjuvanted, soluble Env oligomers (Fig. 6, Protein/Phage group). Thus, there was no statistically significant difference in final Env-specific Ab titers between the HI gp140 protein group and a composite group comprising all animals receiving immunization regimens that combined soluble gp140 protein and Env-decorated phage (i.e. the Phage/Protein and Protein/Phage groups) (p-value=0.54).
3.5 ELISA analysis of phage immune response
The humoral immune response to the lambda phage itself was also evaluated using ELISA assays with immobilized recombinant gpD as the target antigen. ELISA results revealed a low level of anti-lambda phage antibodies after the secondary inoculation that increased gradually following each immunization for those groups that received phage (Fig. 7). As shown in Fig. 5, the phage-specific humoral immune response was greatest in animals that received the HI dose of gp140:gpD fusion protein or wild-type phage particles. A much weaker phage-specific humoral response was detected in animals that received Env-decorated phage particles. We attribute this to the fact that animals immunized with the WT phage received 2 × 1011 PFU of phage per inoculation, whereas animals immunized with Env-decorated phages particles all received only 5 × 1010 PFU of phage per inoculation. As expected, only minimal phage-specific humoral immune reactivity was detected in animals that received unmodified gp140 protein (HI and LO gp140 protein groups). Note that one animal in each of these groups exhibited an unexplained phage-specific response that was not present in preimmune sera.
Figure 7. ELISA analysis of phage specific immune response.

Equal amounts of purified gpD protein (lambda phage capsid protein) were coated onto ELISA plates and reacted with fivefold serial dilution of sera collected after the 2nd, 3rd and 4th immunizations. Data represent mean values for OD450 reading for 1:100 dilution sera for all four animals in each group; error bars denote the standard error of the mean.
3.6 Assessment of virus neutralizing activity
To assess the quality of the Env-specific antibody response, we analyzed virus neutralizing activity in serum specimens obtained after both the third and fourth inoculations. In collaboration with Dr. David Montefiori, neutralization assays were performed in TZM-bl cells against the homologous YU2 virus and two other heterologous Tier 1A viruses (MN.3 and SF162). The data show that the quality and magnitude of the virus-neutralizing antibody response in animals that were immunized by bacteriophage particles displaying HIV-1 Env (HI phage) was not greater than that elicited in animals immunized with soluble recombinant Env oligomers (HI gp140) (Table 2). IC50 neutralization values were <20 for all animals immunized with Env-decorated bacteriophage particles. In contrast, virus neutralizing antibody titers were detected in 3 out of 4 animals that received soluble recombinant Env oligomers. Three animals in this group had significant neutralizing activity against heterologous MN.3 and SF162 viruses after the third and fourth inoculations, and two had detectable neutralizing responses to homologous Tier 2 YU2 virus after the fourth inoculation. The quality and magnitude of the virus-neutralizing antibody response in animals that received a priming inoculation with soluble recombinant Env oligomers followed by boost inoculation with Env-displaying phage particles was similar to that elicited by soluble recombinant protein oligomers alone. In three of the four animals, virus-neutralizing responses were detected against two heterologous Tier 1A viruses (including a very strong response versus MN.3 in one animal). In addition, one animal mounted a robust neutralizing response to the homologous Tier 2 virus, YU2. IC50 neutralization values of 1667 and 1524 were detected after the third and fourth inoculations against heterologous MN.3 virus and one animal had a neutralizing titer of 216 against YU2 virus after the fourth inoculation. When the Env binding endpoint titers elicited in those animals that had neutralization titers >20 (Neut +) and those that were <20 (Neut −) were statistically compared by a nonparametric Mann Whitney test, it was apparent that animals with positive neutralization titers also had higher Env binding endpoint titers (p-value = 0.0001) (Fig. S3). This suggests that there is a correlation between high Env binding titers and the presence of neutralizing antibodies.
Table 2.
Neutralizing activity of sera from rabbits after fourth inoculation
| Group | # Animals Positive | Individual Animals | MN.3 (Tier1) | SF162.LS (Tier 1A) | YU2 (Tier 2) |
|---|---|---|---|---|---|
| HI Phage | 0/4 | 27–30 | neg | neg | neg |
| LO Phage | 0/4 | 35–38 | neg | neg | neg |
| WT Phage | 0/4 | 43–46 | neg | neg | neg |
| HI Gp140 | 3/4 | 47 48 49 50 |
neg 181 27 neg |
neg 94 59 96 |
neg 32 neg 424 |
| HI Gp140:gpD | 3/4 | 51 52 53 54 |
neg 96 neg neg |
314 37 80 neg |
neg neg neg neg |
| LO Gp140 | 2/4 | 55,56 57 58 |
neg neg 81 |
neg 37 286 |
neg neg neg |
| LO Gp140:gpD | 1/4 | 59–61 62 |
neg 35 |
neg neg |
neg neg |
| Phage/Protein | 2/4 | 63,65 64 66 |
neg 21 neg |
neg neg 212 |
neg neg neg |
| Protein/Phage | 3/4 | 67 68 69 70 |
1524 22 neg 29 |
41 192 neg neg |
neg neg neg 216 |
HIV-1 single round in vitro neutralization in TZM-bl cells using rabbit serum collected alter four inoculations. Numbers under viral isolates indicate the sample dilution at which relative luminescence units (RLUs) were reduced 50% compared to virus control wells. All pre bleed samples were negative. Neg denotes a value <20.
These findings show that Env-displaying phage particles can efficiently boost the humoral response primed by soluble Env oligomers. The quality and magnitude of the virus-neutralizing antibody response in animals that received a priming inoculation with soluble recombinant Env oligomers followed by boost inoculations with Env-displaying phage particles was similar to that elicited by soluble Env oligomers alone.
4. Discussion
The generation of broadly neutralizing antibodies against HIV-1 envelope glycoprotein is one of the key goals in HIV-1 vaccine development. HIV-1 Env presents a recalcitrant target in part due to the extensive glycosylation that hides antibody epitopes, and also because conserved domains which can serve as potential targets for such antibodies are physically sequestered [49–52]. An additional factor that may contribute to low Env spike immunogenicity is the scarcity of envelope spikes on the virion surface, and their irregular, clustered distribution [1].
Here, we tested the ability of lambda phage particles to act as a structural scaffold to display HIV-1 envelope spikes in a highly immunogenic context. To produce phage particles that displayed HIV-1 Env, we used a simple in vitro complementation system to decorate gpD-deficient phage particles with glycosylated envelope trimers that were translationally fused to gpD. This was achieved by producing recombinant Envgp140:gpD fusion protein (as well as gpD alone) in mammalian cells, and then comparing the biochemical properties of the resulting fusion protein to wild-type (WT) Envgp140 oligomers. Both native gel electrophoresis and gel filtration analysis demonstrated the purity and oligomeric integrity of our Envgp140:gpD fusion protein, and showed that it was essentially indistinguishable from WT gp140 protein.
With this material in hand, we then generated Env decorated phage particles by mixing gpD-deficient phage particles with purified gpD and Envgp140:gpD fusion proteins at various molar ratios, followed by CsCl-gradient purification. The effectiveness of the decoration reaction was assessed by measuring the titers of the resulting phage preparations before and after exposure to high concentrations of EDTA, which inactivates gpD-deficient capsids but has minimal effect on gpD-bearing particles [29, 30]. We were able to generate stable mosaic Env decorated phage particles using various molar ratios of soluble wild type gpD and gp140:gpD fusion protein (20:1, 5:1, and 1:1). Phage that were decorated with Env protein alone were not stable in the presence of EDTA. In contrast, phage decorated with a 1:1 ratio of gp140:gpD to wild type gpD, were stable in the presence of EDTA. Unexpectedly, however, when we measured the amount of gp140:gpD actually incorporated onto these mosaic phage capsids, we determined that only a fraction of the available binding sites for gpD on the phage capsid were in fact occupied.
The high density Env decorated phage capsids incorporated about 30 copies of Env trimers per particle, which occupied 90 (30 trimers × 3 molecules of gpD per trimer) of the 405 available gpD binding sites on the phage surface – or roughly 25% of the total. Since gp140:gpD was more abundant than wild type gpD on these mosaic particles (Fig. 4B), we conclude that the Env-displaying phage capsids were EDTA stable even though most of the available binding sites for gpD were unoccupied.
Native HIV-1 virions display only 14 ± 7 spikes per virion (138) and have a diameter of approximately 145 nm (15). In contrast, the lambda phage head has a diameter of 50 nm and displays ~30 spikes per capsid in the case of phage particles prepared using a 1:1 ratio of wild-type gpD to gp140:gpD fusion protein. Thus, the expected density of Env spikes on the much smaller surface of the phage head will be roughly 18 to 20-fold greater than that on native HIV-1 particles.
We anticipated that the more dense, array of displayed envelope trimers on the surface of the phage capsid would result in enhanced humoral immune responses against the Env antigen. We therefore assessed the immunogenicity of our lambda phage particles displaying HIV-1 Env, using immunization experiments in rabbits that employed a very similar design and dose regimen to that described by Wyatt and colleagues in their studies of recombinant, oligomeric HIV-1 gp140 [35]. In this experiment, the Env decorated phage particles elicited Env-binding antibody titers and virus neutralizing responses that were no higher than those in those animals that received adjuvanted, conventional oligomeric gp140 protein.
This unexpected result may be related to the fact that sequential immunizations with lambda phage particles displaying an exogenous antigen have been shown to result in progressively diminishing humoral immune response to the displayed antigen – with no immunologic boosting of the response [53]. This has been attributed to a strong and immunodominant response to the phage-capsid, which is efficiently boosted upon sequential immunization, at the expense of the response to the exogenous antigen [53]. Our experiments showed a similar boosting of the humoral response to the phage capsid (gpD coat protein) in those animals that received Env-displaying phage particles, suggesting that this may have contributed to the poor Env-specific response that was elicited by the Env-displaying particles. These findings are consistent with the phenomenon of carrier induced epitopic suppression (CIES) [54]. For uncertain reasons, this phenomenon of CIES was less pronounced when Env-displaying phage particles were used to boost a heterologous gp140 protein prime. Neutralization titers elicited by the protein prime/phage boost group were comparable to those elicited by homologous prime-boost immunization with a high dose of adjuvanted, soluble gp140 - supporting the idea of phage being a poor prime but possibly effective boost.
Collectively, these results show that display of HIV-1 envelope spikes on the VLP-like scaffold provided by phage lambda capsids does not result in an improved humoral immune response as compared to adjuvanted, soluble oligomeric protein.
Supplementary Material
Acknowledgments
This work was supported by the following grants from the National Institutes of Health (NIH): R21AI074351 (SD), T32DE007202 (JM). We gratefully acknowledge Dr. David Montefiori (Duke University) for performing the neutralizing antibody assays, through the Comprehensive Antibody Vaccine Immune Monitoring Consortium. We also thank Dr. Ron Hoess for very helpful suggestions and advice, as well as phage reagents, Dr. Andreas Plückthun and Dr. Patrick Forrer (University of Zurich) for providing gpD-encoding plasmids, and Dr. Mahito Nakanishi and DNAVEC Corp. for providing us with phage vectors [D1180]. Finally, we thank Drs. Rick Wyatt and Jon T. Warren of NIH for their advice, assistance and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006 Jun 15;441(7095):847–52. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 2.Burton DR. Structural biology: images from the surface of HIV. Nature. 2006 Jun 15;441(7095):817–8. doi: 10.1038/441817a. [DOI] [PubMed] [Google Scholar]
- 3.Pantophlet R, Burton DR. GP120: target for neutralizing HIV-1 antibodies. Annu Rev Immunol. 2006;24:739–69. doi: 10.1146/annurev.immunol.24.021605.090557. [DOI] [PubMed] [Google Scholar]
- 4.Zolla-Pazner S. Identifying epitopes of HIV-1 that induce protective antibodies. Nat Rev Immunol. 2004 Mar;4(3):199–210. doi: 10.1038/nri1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984 Jan 12–18;307(5947):178–80. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 6.Evans TG, Bonnez W, Rose RC, Koenig S, Demeter L, Suzich JA, et al. A Phase 1 study of a recombinant viruslike particle vaccine against human papillomavirus type 11 in healthy adult volunteers. J Infect Dis. 2001 May 15;183(10):1485–93. doi: 10.1086/320190. [DOI] [PubMed] [Google Scholar]
- 7.Harper DM, Franco EL, Wheeler C, Ferris DG, Jenkins D, Schuind A, et al. Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomised controlled trial. Lancet. 2004 Nov 13–19;364(9447):1757–65. doi: 10.1016/S0140-6736(04)17398-4. [DOI] [PubMed] [Google Scholar]
- 8.Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, et al. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol. 2005 May;6(5):271–8. doi: 10.1016/S1470-2045(05)70101-7. [DOI] [PubMed] [Google Scholar]
- 9.Sathaliyawala T, Rao M, Maclean DM, Birx DL, Alving CR, Rao VB. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: a novel in vitro approach to construct multicomponent HIV vaccines. J Virol. 2006 Aug;80(15):7688–98. doi: 10.1128/JVI.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spohn G, Guler R, Johansen P, Keller I, Jacobs M, Beck M, et al. A virus-like particle-based vaccine selectively targeting soluble TNF-alpha protects from arthritis without inducing reactivation of latent tuberculosis. J Immunol. 2007 Jun 1;178(11):7450–7. doi: 10.4049/jimmunol.178.11.7450. [DOI] [PubMed] [Google Scholar]
- 11.Maurer P, Jennings GT, Willers J, Rohner F, Lindman Y, Roubicek K, et al. A therapeutic vaccine for nicotine dependence: preclinical efficacy, and Phase I safety and immunogenicity. Eur J Immunol. 2005 Jul;35(7):2031–40. doi: 10.1002/eji.200526285. [DOI] [PubMed] [Google Scholar]
- 12.Merril CR, Scholl D, Adhya SL. The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov. 2003 Jun;2(6):489–97. doi: 10.1038/nrd1111. [DOI] [PubMed] [Google Scholar]
- 13.Barrow PA, Soothill JS. Bacteriophage therapy and prophylaxis: rediscovery and renewed assessment of potential. Trends Microbiol. 1997 Jul;5(7):268–71. doi: 10.1016/S0966-842X(97)01054-8. [DOI] [PubMed] [Google Scholar]
- 14.Fogelman I, Davey V, Ochs HD, Elashoff M, Feinberg MB, Mican J, et al. Evaluation of CD4+ T cell function In vivo in HIV-infected patients as measured by bacteriophage phiX174 immunization. J Infect Dis. 2000 Aug;182(2):435–41. doi: 10.1086/315739. [DOI] [PubMed] [Google Scholar]
- 15.Bearden CM, Agarwal A, Book BK, Vieira CA, Sidner RA, Ochs HD, et al. Rituximab inhibits the in vivo primary and secondary antibody response to a neoantigen, bacteriophage phiX174. Am J Transplant. 2005 Jan;5(1):50–7. doi: 10.1111/j.1600-6143.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- 16.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990 Jul 27;249(4967):386–90. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 17.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985 Jun 14;228(4705):1315–7. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 18.Hoess RH. Bacteriophage lambda as a vehicle for peptide and protein display. Curr Pharm Biotechnol. 2002 Mar;3(1):23–8. doi: 10.2174/1389201023378481. [DOI] [PubMed] [Google Scholar]
- 19.Monaci P, Urbanelli L, Fontana L. Phage as gene delivery vectors. Curr Opin Mol Ther. 2001 Apr;3(2):159–69. [PubMed] [Google Scholar]
- 20.Sulakvelidze A, Alavidze Z, Morris JG., Jr Bacteriophage therapy. Antimicrob Agents Chemother. 2001 Mar;45(3):649–59. doi: 10.1128/AAC.45.3.649-659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen BY, Lim HC. Bioreactor studies on temperature induction of the Q- mutant of bacteriophage lambda in Escherichia coli. J Biotechnol. 1996 Oct 18;51(1):1–20. doi: 10.1016/0168-1656(96)01571-4. [DOI] [PubMed] [Google Scholar]
- 22.Greenwood J, Willis AE, Perham RN. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J Mol Biol. 1991 Aug 20;220(4):821–7. doi: 10.1016/0022-2836(91)90354-9. [DOI] [PubMed] [Google Scholar]
- 23.de la Cruz VF, Lal AA, McCutchan TF. Immunogenicity and epitope mapping of foreign sequences via genetically engineered filamentous phage. J Biol Chem. 1988 Mar 25;263(9):4318–22. [PubMed] [Google Scholar]
- 24.Willis AE, Perham RN, Wraith D. Immunological properties of foreign peptides in multiple display on a filamentous bacteriophage. Gene. 1993 Jun 15;128(1):79–83. doi: 10.1016/0378-1119(93)90156-w. [DOI] [PubMed] [Google Scholar]
- 25.Eguchi A, Akuta T, Okuyama H, Senda T, Yokoi H, Inokuchi H, et al. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J Biol Chem. 2001 Jul 13;276(28):26204–10. doi: 10.1074/jbc.M010625200. [DOI] [PubMed] [Google Scholar]
- 26.Gupta A, Onda M, Pastan I, Adhya S, Chaudhary VK. High-density functional display of proteins on bacteriophage lambda. J Mol Biol. 2003 Nov 21;334(2):241–54. doi: 10.1016/j.jmb.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 27.Maruyama IN, Maruyama HI, Brenner S. Lambda foo: a lambda phage vector for the expression of foreign proteins. Proc Natl Acad Sci U S A. 1994 Aug 16;91(17):8273–7. doi: 10.1073/pnas.91.17.8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piersanti S, Cherubini G, Martina Y, Salone B, Avitabile D, Grosso F, et al. Mammalian cell transduction and internalization properties of lambda phages displaying the full-length adenoviral penton base or its central domain. J Mol Med. 2004 Jul;82(7):467–76. doi: 10.1007/s00109-004-0543-2. [DOI] [PubMed] [Google Scholar]
- 29.Yang F, Forrer P, Dauter Z, Conway JF, Cheng N, Cerritelli ME, et al. Novel fold and capsid-binding properties of the lambda-phage display platform protein gpD. Nat Struct Biol. 2000 Mar;7(3):230–7. doi: 10.1038/73347. [DOI] [PubMed] [Google Scholar]
- 30.Sternberg N, Hoess RH. Display of peptides and proteins on the surface of bacteriophage lambda. Proc Natl Acad Sci U S A. 1995 Feb 28;92(5):1609–13. doi: 10.1073/pnas.92.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santi E, Capone S, Mennuni C, Lahm A, Tramontano A, Luzzago A, et al. Bacteriophage lambda display of complex cDNA libraries: a new approach to functional genomics. J Mol Biol. 2000 Feb 18;296(2):497–508. doi: 10.1006/jmbi.1999.3471. [DOI] [PubMed] [Google Scholar]
- 32.Yang X, Farzan M, Wyatt R, Sodroski J. Characterization of stable, soluble trimers containing complete ectodomains of human immunodeficiency virus type 1 envelope glycoproteins. J Virol. 2000 Jun;74(12):5716–25. doi: 10.1128/jvi.74.12.5716-5725.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Florin L, Farzan M, Kolchinsky P, Kwong PD, Sodroski J, et al. Modifications that stabilize human immunodeficiency virus envelope glycoprotein trimers in solution. J Virol. 2000 May;74(10):4746–54. doi: 10.1128/jvi.74.10.4746-4754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Lee J, Mahony EM, Kwong PD, Wyatt R, Sodroski J. Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J Virol. 2002 May;76(9):4634–42. doi: 10.1128/JVI.76.9.4634-4642.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grundner C, Li Y, Louder M, Mascola J, Yang X, Sodroski J, et al. Analysis of the neutralizing antibody response elicited in rabbits by repeated inoculation with trimeric HIV-1 envelope glycoproteins. Virology. 2005 Jan 5;331(1):33–46. doi: 10.1016/j.virol.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 36.Grundner C, Pancera M, Kang JM, Koch M, Sodroski J, Wyatt R. Factors limiting the immunogenicity of HIV-1 gp120 envelope glycoproteins. Virology. 2004 Dec 5;330(1):233–48. doi: 10.1016/j.virol.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 37.Yuan W, Craig S, Yang X, Sodroski J. Inter-subunit disulfide bonds in soluble HIV-1 envelope glycoprotein trimers. Virology. 2005 Feb 5;332(1):369–83. doi: 10.1016/j.virol.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Zanghi CN, Sapinoro R, Bradel-Tretheway B, Dewhurst S. A tractable method for simultaneous modifications to the head and tail of bacteriophage lambda and its application to enhancing phage-mediated gene delivery. Nucleic Acids Res. 2007;35(8):e59. doi: 10.1093/nar/gkm146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zanghi CN, Lankes HA, Bradel-Tretheway B, Wegman J, Dewhurst S. A simple method for displaying recalcitrant proteins on the surface of bacteriophage lambda. Nucleic Acids Res. 2005;33(18):e160. doi: 10.1093/nar/gni158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Svehla K, Mathy NL, Voss G, Mascola JR, Wyatt R. Characterization of antibody responses elicited by human immunodeficiency virus type 1 primary isolate trimeric and monomeric envelope glycoproteins in selected adjuvants. J Virol. 2006 Feb;80(3):1414–26. doi: 10.1128/JVI.80.3.1414-1426.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mascola JR, D’Souza P, Gilbert P, Hahn BH, Haigwood NL, Morris L, et al. Recommendations for the design and use of standard virus panels to assess neutralizing antibody responses elicited by candidate human immunodeficiency virus type 1 vaccines. J Virol. 2005 Aug;79(16):10103–7. doi: 10.1128/JVI.79.16.10103-10107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bower JF, Yang X, Sodroski J, Ross TM. Elicitation of neutralizing antibodies with DNA vaccines expressing soluble stabilized human immunodeficiency virus type 1 envelope glycoprotein trimers conjugated to C3d. J Virol. 2004 May;78(9):4710–9. doi: 10.1128/JVI.78.9.4710-4719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green TD, Montefiori DC, Ross TM. Enhancement of antibodies to the human immunodeficiency virus type 1 envelope by using the molecular adjuvant C3d. J Virol. 2003 Feb;77(3):2046–55. doi: 10.1128/JVI.77.3.2046-2055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross TM, Xu Y, Green TD, Montefiori DC, Robinson HL. Enhanced avidity maturation of antibody to human immunodeficiency virus envelope: DNA vaccination with gp120-C3d fusion proteins. AIDS Res Hum Retroviruses. 2001 Jun 10;17(9):829–35. doi: 10.1089/088922201750252025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Wyatt R, Sodroski J. Improved elicitation of neutralizing antibodies against primary human immunodeficiency viruses by soluble stabilized envelope glycoprotein trimers. J Virol. 2001 Feb;75(3):1165–71. doi: 10.1128/JVI.75.3.1165-1171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pancera M, Lebowitz J, Schon A, Zhu P, Freire E, Kwong PD, et al. Soluble mimetics of human immunodeficiency virus type 1 viral spikes produced by replacement of the native trimerization domain with a heterologous trimerization motif: characterization and ligand binding analysis. J Virol. 2005 Aug;79(15):9954–69. doi: 10.1128/JVI.79.15.9954-9969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Center RJ, Leapman RD, Lebowitz J, Arthur LO, Earl PL, Moss B. Oligomeric structure of the human immunodeficiency virus type 1 envelope protein on the virion surface. J Virol. 2002 Aug;76(15):7863–7. doi: 10.1128/JVI.76.15.7863-7867.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sternberg N, Weisberg R. Packaging of coliphage lambda DNA II. The role of the gene D protein. J Mol Biol. 1977 Dec 15;117(3):733–59. doi: 10.1016/0022-2836(77)90067-5. [DOI] [PubMed] [Google Scholar]
- 49.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998 Jun 18;393(6686):648–59. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, et al. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998 Jun 19;280(5371):1949–53. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 51.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature. 2003 Mar 20;422(6929):307–12. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 52.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, et al. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998 Jun 18;393(6686):705–11. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 53.Gamage LN, Ellis J, Hayes S. Immunogenicity of bacteriophage lambda particles displaying porcine Circovirus 2 (PCV2) capsid protein epitopes. Vaccine. 2009 Nov 5;27(47):6595–604. doi: 10.1016/j.vaccine.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 54.Jegerlehner A, Wiesel M, Dietmeier K, Zabel F, Gatto D, Saudan P, et al. Carrier induced epitopic suppression of antibody responses induced by virus-like particles is a dynamic phenomenon caused by carrier-specific antibodies. Vaccine. 2010 Jul 26;28(33):5503–12. doi: 10.1016/j.vaccine.2010.02.103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.