

Abstract
The proliferative compartment of stratified squamous epithelia consists of stem and transient amplifying (TA) keratinocytes. Some polypeptides are more abundant in putative epidermal stem cells than in TA cells, but no polypeptide confined to the stem cells has yet been identified. Here we show that the p63 transcription factor, a p53 homologue essential for regenerative proliferation in epithelial development, distinguishes human keratinocyte stem cells from their TA progeny. Within the cornea, nuclear p63 is expressed by the basal cells of the limbal epithelium, but not by TA cells covering the corneal surface. Human keratinocyte stem and TA cells when isolated in culture give rise to holoclones and paraclones, respectively. We show by clonal analysis that p63 is abundantly expressed by epidermal and limbal holoclones, but is undetectable in paraclones. TA keratinocytes, immediately after their withdrawal from the stem cell compartment (meroclones), have greatly reduced p63, even though they possess very appreciable proliferative capacity. Clonal evolution (i.e., generation of TA cells from precursor stem cells) is promoted by the sigma isoform of the 14-3-3 family of proteins. Keratinocytes whose 14-3-3σ has been down-regulated remain in the stem cell compartment and maintain p63 during serial cultivation. The identification of p63 as a keratinocyte stem cell marker will be of practical importance for the clinical application of epithelial cultures in cell therapy as well as for studies on epithelial tumorigenesis.
A population of keratinocyte stem cells in defined locations governs the renewal of mammalian stratified epithelia (1–3). These stem cells generate transient amplifying (TA) cells that terminally differentiate after a discrete number of cell divisions (4). In vivo, keratinocyte stem cells are usually slow-cycling and retain labeled DNA precursors, whereas TA cells divide rapidly and dilute their label quickly (4–8).
Human keratinocyte stem and TA cells when isolated in culture give rise to holoclones and paraclones, respectively (9–11). The great proliferative potential of holoclones (9–12), the capacity of a single holoclone to generate a mature epithelium in vivo (13) and to differentiate into distinct cellular lineages (11), and the permanent epithelial regeneration achieved in burn victims by means of grafts of autologous cultured keratinocytes (14–16), provide compelling evidence that keratinocyte “stem-ness” can be preserved in culture. The proliferative compartment of squamous epithelia also contains a third type of cell, the meroclone (9), which is considered a “young” TA cell endowed with a greater proliferative capacity than the paraclone (11).
Some polypeptides are more abundant in putative epidermal stem cells than in TA cells (17, 18), but no polypeptide confined exclusively to the stem cells has yet been identified.
The p63 transcription factor belongs to a family that includes two structurally related proteins, p53 and p73 (19). Whereas p53 plays a well-established role in tumor suppression, p63 and p73 play unique roles in morphogenesis (20–23). In particular, p63−/− mice have major defects in their limb and craniofacial development, as well as a striking absence of stratified epithelia (20, 21). This phenotype could be explained by either inability of the p63−/− ectoderm to develop into epithelial lineages (20), or by lack of stem cell character necessary to sustain epithelial morphogenesis and renewal (21).
Here we investigate the expression of p63 in epithelial stem and TA cells and show that p63 is a specific marker of human corneal and epidermal stem cells.
Materials and Methods
Cell Culture.
3T3-J2 cells (a gift from Howard Green, Harvard Medical School, Boston) were cultivated as described (16). Donors provided informed consent for biopsy. Permission was obtained for specimens taken from organ donors. Epidermal keratinocytes were obtained from skin biopsies of healthy donors and cultivated on a feeder-layer of lethally irradiated 3T3-J2 cells as described (16). Limbal/corneal keratinocytes were obtained from ocular biopsies taken from organ donors and cultivated as described (11). For serial propagation, cells were passaged at the stage of subconfluence, until they reached senescence (11).
Colony Forming Efficiency (CFE) and Number of Cell Generations.
Cells (100–1000) from each biopsy and from each cell passage were plated onto 3T3-feeder layers and cultured as above. Colonies were fixed 9–12 days later, stained with Rhodamine B, and scored under a dissecting microscope. CFE values are expressed as the ratio of the number colonies to the number of inoculated cells. The number of aborted colonies was calculated as described (9, 11).
The number of cell generations was calculated by using the following formula: x = 3.322 log N/No, where N is the total number of cells obtained at each passage and No is the number of clonogenic cells. Clonogenic cells were calculated from CFE data, which were determined separately in parallel dishes at the time of cell passage.
Clonal Analysis.
Clonal analysis was performed from subconfluent primary cultures as described (9, 11, 16). Briefly, single cells were inoculated onto multiwell plates containing a feeder layer of 3T3 cells. After 7 days of cultivation, clones were identified under an inverted microscope and photographed. Each clone was transferred to three dishes. One dish (1/4 of the clone) was fixed 9–12 days later and stained with Rhodamine B for the classification of clonal type (9, 11). The clonal type was determined by the percentage of terminal colonies (scored as in ref. 9) formed by the progeny of the founding cell. When 0–5% of colonies were terminal the clone was scored as holoclone. When more than 95% of the colonies were terminal the clone was classified as paraclone. When more than 5%, but less than 95% of the colonies were terminal, the clone was classified as meroclone (9, 11). The second dish was used for serial cultivation and evaluation of the number of cell generations. Cells plated in the third dish were cultivated for 4–5 days, then used to prepare cell extracts destined to Western analysis.
Immunohistochemistry and Western Analysis.
Ocular biopsies were obtained from organ donors. Sheets of cultured epithelium were detached from the vessels with Dispase II (24). Specimens were fixed in paraformaldehyde (4% in PBS) 30 min at room temperature and embedded in paraffin. For immunohistochemistry (performed as described in ref. 11), sections were stained with a p63-specific mAb (21, 25), a Keratin-3-specific mAb (AE5, a gift from Tung-Tien Sun, New York University Medical Center, New York) (26), and a proliferating cell nuclear antigen (PCNA)-specific mAb (27) (Santa Cruz Biotechnology).
For immunoblots, mass or clonal cultures were extracted on ice with RIPA buffer (0.15 mM NaCl/0.05 mM Tris⋅HCl, pH 7.5/1% Triton X-100/1% sodium deoxycholate/0.1% SDS) (12). Equal amounts of samples were electrophoresed on 7.5%–12,5% SDS-polyacrylamide gels and transferred to poly(vinylidene difluoride) (PVDF) filters (Immobilon-P, Millipore). Immunoreactions were carried out as described (12), using polyclonal antibodies to 14-3-3ζ and to pan-14-3-3 (Pan) (a gift from Alastair Aitken, University of Edinburgh, Edinburgh), and mAb to p63 (21, 25) and to PCNA (27). Immobilon-bound antibodies were detected by chemiluminescence with ECL (Amersham Pharmacia).
Results
Expression of p63 by Human Limbal/Corneal Stem and TA Cells.
Identification of epithelial stem cells may rely on label-retaining experiments in vivo (4–8) or on clonal analysis of multiplying keratinocytes in vitro (9–13). In corneal epithelium, the two criteria identify the same cells. Slow-cycling corneal cells and human corneal holoclones are confined to the basal layer of the limbus, the transitional zone between the corneal epithelium and the conjunctiva (4, 7, 11). The corneal epithelium is formed exclusively by rapidly dividing TA cells that migrate millimeters away from their precursor limbal stem cells (4, 26).
Fig. 1A shows immunohistochemical analysis of sections of the human ocular surface spanning the peripheral cornea (P) and the limbus (L). Nuclear p63 was abundantly expressed only in the basal layer of limbal epithelium (L, arrows). Within the limbus, patches of cells expressing very low levels of p63 (L, asterisks) were interspersed with the more numerous p63+ cells. The TA cells covering the corneal surface (P) do not express detectable levels of p63 (P). Very low levels of the protein were observed in occasional basal cells of the peripheral cornea, adjacent to the limbal epithelium (P, arrowheads).
Figure 1.

Expression of p63 by human corneal stem and TA cells. (A) Corneal biopsies spanning from the peripheral cornea (P) to the limbus (L) were stained with p63-specific mAb. Eight different inclusions from four different biopsies were analyzed. The transition from limbus to peripheral cornea was identified by the beginning of the Bowman's membrane (double arrowhead, B) and by immunostaining with Keratin-3-specific mAb (not shown). Note that the limbal basal layer (arrows) expresses p63, and that patches of p63− cells (asterisks) stagger p63+ cells. (B) Clonal analysis of subconfluent primary limbal cultures obtained from two different donors (LC3 and LC5). Cell extracts were prepared from cultures generated by holoclones (H), paraclones (P), and meroclones (M). Equal amounts of protein were fractionated on 7.5% SDS-polyacrylamide gels, transferred to PVDF filters, and immunostained with p63-specific mAb (p63) and with antibodies to pan-14-3-3 (Pan). Identical results were obtained with two other limbal strains (LC1 and LC25). (C) Densitometric analysis of the blots shown in B.
To investigate the expression of p63 in isolated stem and TA cells, single keratinocytes were isolated from subconfluent primary limbal cultures. After 7 days of cultivation each clone was transferred to three dishes. One dish was used for the classification of the clonal type (9, 11). The second dishes was used for evaluation of the number of cell generations. Cells plated in the third dish were cultivated for 4–5 days, then used to prepare cell extracts destined to Western analysis.
We analyzed 118 limbal clones. The majority of clones (84.9%) were classified as TA cells, whereas stem cells (holoclones) represented 15.1% of total clones. Limbal holoclones were serially propagated and each holoclone produced approximately 100 generations before senescence, accounting for the entire proliferative capacity of the parental mass culture. In contrast, paraclones underwent only 15–20 cell doublings before mitotic arrest.
Western analysis showed that cultures generated by holoclones possessed high levels of p63 (Fig. 1B, H). In sharp contrast, p63 was undetectable in cultures generated by paraclones (Fig. 1B, P). Although meroclones possess a significant proliferative capacity (an average of approximately 40 cell doublings; ref. 11), p63 was barely detectable in meroclones (Fig. 1B, M). Cultures generated by all clonal types synthesized almost identical amounts of a control protein (pan-14-3-3) (Fig. 1B), suggesting that differences of p63 content between different clonal types were not due to general alterations of protein synthesis.
The relative content of p63 in clones was quantified by densitometric analysis of the blots and is shown in Fig. 1C. Limbal holoclones contained 8–10-fold and ≥200-fold more p63 than meroclones and paraclones, respectively.
Expression of p63 by Stem and TA Cells of Epidermal Cultures.
In the epidermis, stem cells and TA cells are not segregated as they are in the cornea, but are interspersed in the epidermal basal layer (1, 17). Cultured primary keratinocytes generate cohesive sheets of stratified epithelium (24). As in natural epidermis (25), p63 was confined to the basal layer of these sheets (Fig. 2A). Cells with high content of nuclear p63 were located at intervals (Fig. 2A, arrowheads), sometimes in clusters (Fig. 2A, arrows) separated by stretches of p63− cells. At higher magnification, it was quite common to observe cells with abundant p63 (Fig. 2A Inset, arrow) flanked by cells with little or no p63 (Fig. 2A Inset, arrowheads and asterisks). Thus, the expression pattern of p63 is reminiscent of the proposed columnar organization of the epidermis, where each column is thought to be generated by a single stem cell (28).
Figure 2.

Expression of p63 by human epidermal stem and TA cells. (A) Cultured epidermal sheets prepared from primary keratinocytes were stained with p63-specific mAb. p63 was confined to the basal cell layer. Note that p63+ cells were located at intervals as single cells (arrowheads) or as patches (arrows), and were separated by stretches of p63− cells. At higher magnification (Inset), it is possible to observe cells expressing high levels of p63 (arrow) flanked with cells expressing lower levels of the protein (arrowheads). The latter cells are flanked by p63− cells (asterisks). (B) Clonal analysis of subconfluent primary epidermal cultures obtained from two different donors (K71 and K100). Cell extracts were prepared from cultures generated by holoclones (H), meroclones (M), and paraclones (P). Equal amounts of protein were fractionated on 7.5% SDS-polyacrylamide gels, transferred to PVDF filters and immunostained with p63-specific mAb (p63) and with polyclonal antibodies to 14-3-3ζ (ζ). p63 was high in holoclones, nearly absent in meroclones, and undetectable in paraclones; 14-3-3ζ was similar in all.
We analyzed 120 clones obtained from subconfluent primary epidermal cultures (9–11). The vast majority (93.3%) of clones were classified as meroclones and paraclones (TA cells), whereas holoclones (stem cells) represented 6.6% of total clones. Western analysis showed that cultures generated by holoclones expressed high levels of p63 (Fig. 2B, H lanes). In sharp contrast, p63 was undetectable in cultures generated by paraclones (Fig. 2B, P lanes) and barely detectable in cultures generated by meroclones (Fig. 2B, M lanes). Like limbal holoclones, epidermal holoclones possess 8–10-fold and ≥200-fold more p63 than meroclones and paraclones, respectively.
p63 Expression and the Potential for Cell Proliferation.
In human epidermis, hair follicles, and stratified epidermal cultures, p63 is expressed in the nuclei of cells that are either proliferating or possess the ability to multiply (25). PCNA is a DNA polymerase δ-associated protein that is synthesized in G1 and S phases of the cell cycle and is therefore a specific marker of proliferating cells (27). Staining for p63 and PCNA performed on parallel limbal sections revealed that cells forming the basal layer of human limbus express both p63 (Fig. 3 A–C) and PCNA (D–F). Although most cells expressing PCNA also express p63 (see black asterisks in A, B, D, and E), it was possible to observe cells expressing PCNA but not p63 (see red asterisks in B and E). These latter cells often flanked p63+ cells (B and E). Most frequently, cells expressing high levels of p63 (A–C, arrows) did not possess PCNA (D–F, arrows). These observations suggest that p63 is expressed by keratinocytes that possess the ability to proliferate and not simply by keratinocytes that are duplicating their DNA, as already shown in human epidermis (25).
Figure 3.

Expression of p63 and PCNA by keratinocyte stem and TA cells. (A–F) Parallel sections of limbal biopsies were stained with p63-specific mAb (A–C) and PCNA-specific mAb (D–F). Arrows indicate basal cells that expressed p63, but not PCNA. Black asterisks indicate cells that expressed both p63 and PCNA. B and E note a p63+/PCNA+ cell (black asterisks) flanked by cells expressing PCNA, but not p63 (red asterisks). (G and H) Clonal analysis of subconfluent primary corneal (LC25) and epidermal (K71) cultures. Cell extracts were prepared from cultures generated by holoclones (H), meroclones (M), and paraclones (P). Equal amounts of protein were fractionated on 7.5% (G) or on 12.5% (H) SDS-polyacrylamide gels, transferred to PVDF filters and immunostained with p63-specific mAb (p63) and with PCNA-specific mAb (PCNA).
We then sought to investigate the expression of p63 and PCNA in cultured corneal (Fig. 3G) and epidermal (Fig. 3H) stem (H) and TA (M and P) cells. Western analysis showed that cultures generated by holoclones (G and H, H lanes) possessed high levels of p63 and PCNA. In sharp contrast, cultures generated by paraclones (G and H, P lanes) expressed PCNA, but not p63. Corneal meroclones (G, M lanes) possessed high levels of PCNA, but very low levels of p63.
These data confirm that p63 protein is principally restricted to keratinocyte stem cells. The observation that p63+ cells do not necessarily express PCNA in vivo (Fig. 3 A–F and ref. 25), but that all cultured p63+ clones possess PCNA (Fig. 3 G and H), is consistent with the notion that epithelial stem cells are slow-cycling in vivo, but actively proliferating in culture.
Expression of p63 in Cultured Keratinocytes Maintained in the Stem Cell Compartment.
Clonal evolution (i.e., generation of TA cells from a parental stem cell) is a continuous unidirectional process occurring during aging, wound healing, and serial cultivation (1, 4, 9, 11, 29, 30). Replicative senescence occurs when all stem cells have completed their clonal evolution and have generated terminal TA cells (9, 11–13). We have shown that the sigma isoform of the 14-3-3 family of proteins (31, 32) promotes this clonal evolution (12). When expression of 14-3-3σ was stably down-regulated in primary human epidermal keratinocytes by transduction with defective retrovirus carrying a full-length human 14-3-3σ cDNA in an antisense orientation (12), clonal evolution was prevented and the keratinocytes were maintained in the stem cell compartment and immortalized (these cell lines and the procedures to generate them are fully described in ref. 12). We therefore examined expression of p63 in antisense-σ-transduced keratinocytes.
Clonal analysis performed on control keratinocytes (Fig. 4A, yellow columns) showed that 95% of clonogenic cells could be identified as either meroclones (57.5%) or paraclones (37.5%), whereas stem cells represented 5% of total clonogenic cells. In contrast, epidermal cultures generated from antisense-σ-transduced cells were formed mainly by stem cell holoclones, which accounted for about 50% of total clonogenic cells (Fig. 4A, blue columns). As expected, antisense-σ-transduced keratinocytes maintained p63 expression during serial cultivation and after bypass of replicative senescence. The frequency of cells strongly staining for p63 in the transduced keratinocytes shown in Fig. 4B was compared with that of untransduced cells, such as those shown in Fig. 2A. The frequencies were 71.2% and 10.7%, respectively.
Figure 4.
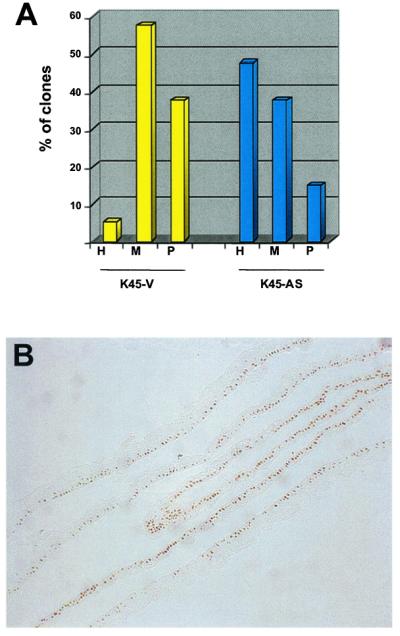
Expression of p63 in keratinocytes immortalized by transduction with antisense RNA for 14-3-3σ. The expression of 14-3-3σ was stably down-regulated in primary keratinocytes by transduction with defective retrovirus carrying a full-length human 14-3-3σ cDNA in antisense orientation (12). These cells maintained telomerase activity and became immortal (12). (A) Clonal analysis was performed on keratinocytes transduced with antisense σ DNA (K45-AS) or with an empty vector (K45-V). Note that the percentage of stem cells in the control K45-V cultures was 5% (H, yellow column), but rose to approximately 50% in K45-AS keratinocytes (H, blue column). (B) Cultured epidermal sheets prepared from antisense-σ-transduced keratinocytes were stained with p63-specific mAb. Note that most of basal cells express high levels of p63.
Discussion
To our knowledge, p63 is the first gene product definitely distinguishing stem cells from their TA progeny in stratified squamous epithelia. Identification of p63 as a stem cell marker is consistent with the phenotype of p63−/− mice (20, 21). It was known that p63−/− mice lack stratified epithelia and contain clusters of terminally differentiated keratinocytes on the exposed dermis (21), and that p63 is expressed in the nuclei of keratinocytes with proliferative potential (25). The finding that p63 is specifically expressed by stem cells of human epidermis and limbal epithelium, and not by TA cells (this paper), strongly suggests that the phenotype of p63−/− mice should be ascribed to a failure to maintain stem cells (21) rather than to inability of the p63−/− ectoderm to form epithelial lineages during development (20).
Newborns start their life with a high content of keratinocyte stem cells, but the number declines during aging (1, 9). Furthermore, division of stem cells gives rise to hierarchy of TA cells with progressively less proliferative potential (4, 8, 9, 11). This has been clearly shown in corneal epithelium where there is a well-established centripetal migration of limbus-derived TA cells, as they progressively decrease their proliferative capacity (4). We show here that p63 is not expressed by the basal TA cells of corneal epithelium. We also show that TA keratinocytes, immediately after their withdrawal from the stem cell compartment (meroclones), already have greatly reduced p63, even though they possess very appreciable proliferative capacity (9, 11, 17). Furthermore, cultures generated by TA cells express a cell-proliferation-associated nuclear antigen (PCNA), but not p63. Taken together, these observations show that possession of p63 is not simply a property of multiplying cells (ref. 25 and this paper), but a property of stem cells as they are identified by clonal analysis.
Human cornea and epidermis display a slightly different pattern of p63 localization from that seen in other mucosal epithelia. For instance, p63 is expressed by all basal and most suprabasal cells of cervical squamous mucosa (33). More experiments are needed to clarify whether differences in p63 expression reflect different functions of the protein in maintaining the proliferative potential of other stratified epithelia. Finally, in the human hair follicle, p63 is expressed by keratinocytes forming the outer root sheath as well as by bulb keratinocytes surrounding the surface of the follicular papilla (25). This finding suggests that keratinocytes endowed with stem cell properties are present not only in specific regions of the outer epithelial sheath (10, 34), but also in the hair bulb, as previously postulated (10, 35).
Our findings have major implications for the study of keratinocytes in two different fields. First, cultured keratinocytes generate sheets of stratified epithelia suitable for transplantation in patients with life-threatening or disabling epithelial defects (14–16). Permanent regeneration of the epithelium obviously requires engraftment of stem cells. This engraftment of stem cells will also be essential for successful gene therapy. Second, stem cells are thought to be involved in the formation of malignant tumors (36, 37). In this respect, it is known that most cells of poorly differentiated squamous cell carcinomas possess p63 (25). Thus, the definition of this long-sought keratinocyte stem cell marker will be of crucial importance for proper clinical application of epithelial cultures in cell therapy as well as for studies on epithelial tumorigenesis.
Acknowledgments
This work was supported by Telethon-Italy (Grants A.106 and B-53) and by Ministero della Sanità, Italy.
Abbreviations
- TA
transient amplifying
- PCNA
proliferating cell nuclear antigen
- PVDF
poly(vinylidene difluoride)
References
- 1.Barrandon Y. Dev Biol. 1993;4:209–215. [Google Scholar]
- 2.Fuchs E, Segre J A. Cell. 2000;100:143–155. doi: 10.1016/s0092-8674(00)81691-8. [DOI] [PubMed] [Google Scholar]
- 3.Watt F M, Hogan B L M. Science. 2000;287:1427–1430. doi: 10.1126/science.287.5457.1427. [DOI] [PubMed] [Google Scholar]
- 4.Lehrer M S, Sun T-T, Lavker R M. J Cell Sci. 1998;111:2867–2875. doi: 10.1242/jcs.111.19.2867. [DOI] [PubMed] [Google Scholar]
- 5.Morris R J, Potten C S. Cell Prolif. 1994;27:279–289. doi: 10.1111/j.1365-2184.1994.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 6.Morris R J, Potten C S. J Invest Dermatol. 1999;112:470–475. doi: 10.1046/j.1523-1747.1999.00537.x. [DOI] [PubMed] [Google Scholar]
- 7.Cotsarelis G, Cheng S-Z, Dong G, Sun T-T, Lavker R M. Cell. 1989;57:201–209. doi: 10.1016/0092-8674(89)90958-6. [DOI] [PubMed] [Google Scholar]
- 8.Taylor G, Lehrer M S, Jensen P J, Sun T-T, Lavker R M. Cell. 2000;102:451–461. doi: 10.1016/s0092-8674(00)00050-7. [DOI] [PubMed] [Google Scholar]
- 9.Barrandon Y, Green H. Proc Natl Acad Sci USA. 1987;84:2302–2306. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rochat A, Kobayashi K, Barrandon Y. Cell. 1994;76:1063–1073. doi: 10.1016/0092-8674(94)90383-2. [DOI] [PubMed] [Google Scholar]
- 11.Pellegrini G, Golisano O, Paterna P, Lambiase A, Bonini S, Rama P, De Luca M. J Cell Biol. 1999;145:769–782. doi: 10.1083/jcb.145.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dellambra E, Golisano O, Bondanza S, Siviero E, Lacal P, Molinari M, D'Atri S, De Luca M. J Cell Biol. 2000;149:1117–1129. doi: 10.1083/jcb.149.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathor M B, Ferrari G, Dellambra E, Cilli M, Mavilio F, Cancedda R, De Luca M. Proc Natl Acad Sci USA. 1996;93:10371–10376. doi: 10.1073/pnas.93.19.10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallico G G, O'Connor N E, Compton C C, Kehinde O, Green H. N Engl J Med. 1984;311:448–451. doi: 10.1056/NEJM198408163110706. [DOI] [PubMed] [Google Scholar]
- 15.Pellegrini G, Traverso C E, Franzi A T, Zingirian M, Cancedda R, De Luca M. Lancet. 1997;349:990–993. doi: 10.1016/S0140-6736(96)11188-0. [DOI] [PubMed] [Google Scholar]
- 16.Pellegrini G, Ranno R, Stracuzzi G, Bondanza S, Guerra L, Zambruno G, Micali G, De Luca M. Transplantation. 1999;68:868–879. doi: 10.1097/00007890-199909270-00021. [DOI] [PubMed] [Google Scholar]
- 17.Jones P H, Harper S, Watt F M. Cell. 1995;80:83–93. doi: 10.1016/0092-8674(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 18.Li A, Simmons P J, Kaur P. Proc Natl Acad Sci USA. 1998;95:3902–3907. doi: 10.1073/pnas.95.7.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lohrum M A E, Vousden K H. Trends Cell Biol. 2000;10:197–202. doi: 10.1016/s0962-8924(00)01736-0. [DOI] [PubMed] [Google Scholar]
- 20.Mills A A, Zheng B, Wang X-J, Vogel H, Roop D R, Bradley A. Nature (London) 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 21.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson R T, Tabin C, Sharpe A, Caput D, Crum C, et al. Nature (London) 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 22.Celli J, Duijf P, Hamel B C J, Bamshad M, Kramer B, Smits A P T, Newbury-Ecob R, Hennekam R C M, Van Buggenhout G, van Haeringen A, et al. Cell. 1999;99:143–153. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 23.Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, et al. Nature (London) 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 24.Green H, Kehinde O, Thomas J. Proc Natl Acad Sci USA. 1979;76:5665–5668. doi: 10.1073/pnas.76.11.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parsa R, Yang A, McKeon F, Green H. J Invest Dermatol. 1999;113:1099–1105. doi: 10.1046/j.1523-1747.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 26.Schermer A, Galvin S, Sun T-T. J Cell Biol. 1986;103:49–62. doi: 10.1083/jcb.103.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bravo R, Frank R, Blundell P A, MacDonald-Bravo H. Nature (London) 1987;326:515–517. doi: 10.1038/326515a0. [DOI] [PubMed] [Google Scholar]
- 28.Potten C S. Int Rev Cytol. 1981;69:271–318. doi: 10.1016/s0074-7696(08)62326-8. [DOI] [PubMed] [Google Scholar]
- 29.Watt F M. Philos Trans R Soc London B. 1998;353:831–837. doi: 10.1098/rstb.1998.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campisi J. J Invest Dermatol. 1998;3:1–5. [PubMed] [Google Scholar]
- 31.Aitken A. Trends Cell Biol. 1996;6:341–347. doi: 10.1016/0962-8924(96)10029-5. [DOI] [PubMed] [Google Scholar]
- 32.Hermeking H, Lengauer C, Polyak K, He T-C, Zhang L, Thiagalingam S, Kinzler K W, Vogelstein B. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 33.Quade, B. J., Yang, A., Wang, Y., Sun, D., Park, J.-J., Sheets, E. E., Cviko, A., Federschneider, J., Peters, R., McKeon, F., et al. (2000) Gynecol. Oncol., in press. [DOI] [PubMed]
- 34.Cotsarelis G, Sun T-T, Lavker R M. Cell. 1990;61:1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 35.Reynolds A J, Lawrence C M, Jahoda C A B. J Invest Dermatol. 1993;101:634–638. doi: 10.1111/1523-1747.ep12366095. [DOI] [PubMed] [Google Scholar]
- 36.Miller S J, Wei Z G, Wilson C, Dzubow L, Sun T-T, Lavker R M. J Invest Dermatol. 1993;101:591–594. doi: 10.1111/1523-1747.ep12366045. [DOI] [PubMed] [Google Scholar]
- 37.Morris R J, Coulter K, Tryson K, Steinberg S R. Cancer Res. 1997;57:3436–3443. [PubMed] [Google Scholar]