

Abstract
Myoblast fusion is an intricate process that is initiated by cell recognition and adhesion, and culminates in cell membrane breakdown and formation of multinucleate syncytia. In the Drosophila embryo, this process occurs asymmetrically between founder cells that pattern the musculature and fusion-competent myoblasts (FCMs) that account for the bulk of the myoblasts. The present studies clarify and amplify current models of myoblast fusion in several important ways. We demonstrate that the non-conventional guanine nucleotide exchange factor (GEF) Mbc plays a fundamental role in the FCMs, where it functions to activate Rac1, but is not required in the founder cells for fusion. Mbc, active Rac1 and F-actin foci are highly enriched in the FCMs, where they localize to the Sns:Kirre junction. Furthermore, Mbc is crucial for the integrity of the F-actin foci and the FCM cytoskeleton, presumably via its activation of Rac1 in these cells. Finally, the local asymmetric distribution of these proteins at adhesion sites is reminiscent of invasive podosomes and, consistent with this model, they are enriched at sites of membrane deformation, where the FCM protrudes into the founder cell/myotube. These data are consistent with models promoting actin polymerization as the driving force for myoblast fusion.
Keywords: Drosophila, Mbc, Rac1, F-actin foci, Asymmetry, Myoblast fusion, Fusion-competent myoblasts, Fusion
INTRODUCTION
Myoblast fusion in the Drosophila embryo requires founder cells and fusion competent myoblasts (FCMs). Founder cells dictate the size, shape, location and pattern of innervation of each muscle fiber through expression of one or more muscle identity genes. The founder cell also seeds the fusion process through interaction with FCMs. The FCMs, by contrast, take on the identity of the founder cell with which they fuse and express the appropriate muscle identity genes (Abmayr et al., 2003; Abmayr et al., 2005; Baylies et al., 1998). The initial fusion event is an asymmetric process between the founder cell and a single FCM. Recognition between these cell types is controlled by members of the immunoglobulin superfamily (IgSF), including Kin-of-IrreC (Kirre; Dumbfounded), Roughest (Rst; IrreC), Sticks-and-stones (Sns) and Hibris (Hbs) (Artero et al., 2001; Bour et al., 2000; Dworak et al., 2001; Ruiz-Gomez et al., 2000; Strunkelnberg et al., 2001). Kirre or Roughest must be present on the surface of the founder cell, while the FCM must express Sns or Hbs (Bour et al., 2000; Shelton et al., 2009). Subsequent rounds of fusion then occur between a syncytial Kirre- or Rst-expressing muscle precursor and additional Sns-expressing FCMs until the proper muscle size is achieved. These later fusion events are also asymmetric, with neither the developing syncitia nor the mononucleate FCMs fusing symmetrically with like cell subtypes. The interactions and relative affinities between these transmembrane proteins at the cell surface help to ensure that fusion remains asymmetric (Galletta et al., 2004). However, the cytoplasmic components of the fusion machinery downstream of these cell surface receptors include proteins that are exclusive to each cell type, as well as proteins that are present in both cell types (Haralalka and Abmayr, 2010; Onel and Renkawitz-Pohl, 2009; Rochlin et al., 2010; Zhang and Chen, 2008).
Recent studies have reported the presence of F-actin foci at points of cell contact (Gildor et al., 2009; Kim et al., 2007; Richardson et al., 2007) that disappear just prior to fusion (Richardson et al., 2007). These dynamic foci are dependent on genes associated with actin polymerization, including the HEM/SCAR, Vrp/WASp and the Arp2/3 complex (Berger et al., 2008; Gildor et al., 2009; Richardson et al., 2007). In addition, Sns and Kirre become organized into a ring-like structure at the sites of FCM-founder cell/syncitia contact that surround an F-actin core, termed the FuRMAS (fusion-restricted myogenic-adhesive structure) (Kesper et al., 2007).
Among those proteins commonly thought to function in both cell types in Drosophila myoblast fusion are the Rho-family monomeric GTPase Rac1 and Myoblast City (Mbc) (Hakeda-Suzuki et al., 2002; Luo et al., 1994; Rushton et al., 1995). Mbc comprises one subunit of a highly conserved bipartite guanine nucleotide exchange factor (GEF), and is the Drosophila ortholog of mammalian Dock180 and C. elegans CED-5 (Cote and Vuori, 2007; Meller et al., 2005; Rushton et al., 1995). The other component of this complex is Elmo in vertebrates and Drosophila, and CED-12 in C. elegans (Cote and Vuori, 2007; Geisbrecht et al., 2008). The Dock180/Elmo complex facilitates exchange of GDP for GTP on Rac1 (Lu and Ravichandran, 2006), and both the Elmo and the putative Rac1-binding domains of Mbc are essential (Balagopalan et al., 2006). The mammalian complex is recruited to receptors at the membrane through interaction with small SH2-SH3 adaptor proteins such as Crk (Albert et al., 2000), where it activates Rac1. Rac1 regulates cell polarity, phagocytosis, migration, vesicle trafficking and axon guidance via regulation of actin dynamics (Bosco et al., 2009; Heasman and Ridley, 2008). Although Drosophila Crk is essential for the action of Mbc/Elmo in the adult thorax (Ishimaru et al., 2004), Mbc in the musculature acts independently of Crk binding (Balagopalan et al., 2006). It remains to be shown whether localization of Mbc can, like mammalian Dock180, also occur through mechanisms that include phosphatidylinositol (3,5)P2 (PIP2) and phosphatidylinositol (3,4,5)P3 (PIP3) binding sites (Cote et al., 2005). Together, these data support a pathway in which the bipartite GEF complex is targeted to sites at the membrane where it activates Rac1. Rac1, in turn, stimulates downstream targets that include actin nucleation promoting factors (NPFs) (Derivery and Gautreau, 2010; Heasman and Ridley, 2008; Lebensohn and Kirschner, 2009).
The present studies clarify and amplify current models for myoblast fusion in several important ways. First, Mbc is explicitly required in the FCMs and is not needed in the founder cells. Thus, the essential function of Rols/Ants cannot be to direct recruitment of Mbc to Kirre in the founder cells (Chen and Olson, 2001). Second, the primary role of Mbc in the musculature is to activate the small GTPase Rac1 and, more importantly, both Mbc and active Rac1 are concentrated in FCMs at their site of contact with founder cells. Consistent with the localization of Rac1, F-actin is highly enriched, if not exclusive to, the FCMs, and FCM-associated structures project into the founder cell and myotube prior to fusion. Last, Mbc plays a crucial role in the formation and organization of F-actin foci and in cytoskeletal structure in the FCMs, presumably via its activation of Rac1 in these cells.
MATERIALS AND METHODS
Fly stocks and genetics
Fly stocks included UAS-lacZ, UAS-RacWT and UAS-RacV12 (Bloomington Stock Center); rP298-lacZ (Nose et al., 1998); UAS-Mbc[HA] and UAS-MbcNPxxP, Δ1807 (Balagopalan et al., 2006); UAS-Kirre (Ruiz-Gomez et al., 2000); UAS-Rols (Menon and Chia, 2001); UAS-Actin-mCherry (Fricke et al., 2009); sxl-GFP, sns-Gal4 (Kocherlakota et al., 2008); rP298-Gal4 (Menon and Chia, 2001); mbcD11.2 (Erickson et al., 1997); the kirre, rst deficiency Df(1)w67k30 (Ruiz-Gomez et al., 2000); rols1728-20L (Menon and Chia, 2001); spg242 (Postner et al., 1992); and rac1J11, rac2Δ (Hakeda-Suzuki et al., 2002). The sns-mCherryNLS reporter was generated by replacing the lacZ region of the sns-lacZ construct (Kocherlakota et al., 2008) with the RFP variant, mCherry (Clontech, Mountain View, CA), and addition of the NLS sequence from the Drosophila transformer gene (Barolo et al., 2000). For live imaging, we derived new transgene insertions by mobilizing UAS-Gap-GFP and twi-Gal4 (Bloomington Stock Center) and selecting reinsertion lines on the basis of increased fluorescent signal.
Germ line clone analysis was carried out as described previously (Balagopalan et al., 2006).
Preparation of primary cells
Drosophila primary myoblast cultures were prepared as described previously (Bai et al., 2009; Bernstein et al., 1978) with the following modifications. Embryos were sterilized for 10 minutes at 25°C in 50% bleach, washed and disrupted at 0.01-0.02 g embryos/ml. After washing, the cells were cultured without a pre-plating step in complete Shields and Sang M3 media (10% FBS, 1% pen strep, 10 mU/ml bovine insulin) on acid-washed coverslips (1 M HCl, 30 minutes, room temperature) at a density of 2.5×105 cells/cm2. Cells were incubated at 25°C for the indicated period of time, fixed in 3.7% formaldehyde for 20 minutes and immunostained as indicated. For mbc mutant myoblasts, embryos at 6-9 hours AEL of the genotype mbcD11.2/TM3, twi-Gal4, UAS-GFP were collected using a CoPas Plus embryo sorter (UnionBiometrica, Holliston, MA). Sorted mbcD11.2 mutant embryos were hand checked for GFP-positive embryos and primary myoblasts prepared as above. Where indicated below, they were co-cultured for 24 hours. FCMs were identified by the presence of mCherry from the sns-mCherryNLS reporter and by their rounded morphology. Founder cells/myotubes were identified by expression of rP298-lacZ and/or by their characteristic elongated morphology by differential interference contrast (DIC) and/or by the number of nuclei, revealed by DAPI, as indicated.
Immunohistochemistry and immunofluorescence
Embryos were collected and processed as described (Erickson et al., 1997). Fixed primary myoblast cultures were permeabilized in PBS, 0.1% Triton X-100 for 5 minutes at 25°C, and blocked in PBS containing 0.05% Tween-20, 5% BSA for 1 hour at 25°C. Primary antibodies, affinity purified where noted (AP), included anti-Sns (AP, 1:1000) (Bour et al., 2000), anti-Kirre (AP, 1:1000) (Galletta et al., 2004), anti-Mbc (AP, 1:1000; embryos, 1:200) (Erickson et al., 1997), anti-Alk (Stute et al., 2004) anti-β-gal (1:200, Abcam), anti-GFP (1:400, Rockland Immunochemicals), anti-active Rac1-GTP (1:200, NewEast Biotech), anti-RFP, for visualization of mCherry (1:200, Clontech), anti-myosin heavy chain (MHC, 1:1000, D. Kiehart), anti-Even-skipped (Eve, 1:1000) (Frasch et al., 1987), anti-Discs Large (Dlg, 1:10, Developmental Studies Hybridoma Bank, clone 4F3), anti-Slouch (Beckett and Baylies, 2007) and anti-Kruppel (Kr, 1:300) (Kosman et al., 1998). Colorimetric detection used biotinylated anti-rat or anti-rabbit secondary antibodies (1:200, Vector Laboratories) and the Vectastain ABC elite kit (Vector Laboratories). Fluorescence detection used Alexa fluor-conjugated secondary antibodies (1:200, Invitrogen). F-actin was visualized in hand-devitellinized embryos incubated with Alexa fluor-conjugated phalloidin (embryos, 1:20, primary myoblasts, 1:50; Invitrogen).
Quantitation of fusion
Nuclei of late stage 15 embryos were visualized using anti-Eve, anti-Slouch or anti-Kr antisera. Eve/Kr-positive (DA1), Kr-positive (DO1, LT2 and LT4), Slouch-positive (VT1 and LO1) and Slouch/Kr-positive (VA2) nuclei from abdominal hemisegments 2-7 were counted manually from z-series images.
Confocal microscopy
A Zeiss LSM-510 Meta microscope was used for collection of confocal z-series images of embryos and primary myoblasts with a Plan-Apochromat 63×/1.40 Oil DIC objective at 2× (Fig. 5A,B) or 5× zoom (Fig. 5C-E), with 0.3 μm steps for actin foci in embryos, a Plan-Apochromat 40×/1.3 Oil DIC with 0.5-0.8 μm steps for other data in embryos, and a Plan-Apochromat 100×/1.46 Oil DIC M27 objective at 3× zoom with 0.2 μm steps for primary myoblasts. Image field resolution was 512×512 pixels and pinhole aperture was 1.0 airy units for all images. Images were processed post collection with ImageJ and Imaris (Bitplane).
Fig. 5.

F-actin foci are present in fusion-competent myoblasts at points of contact with founder cells/myotubes in wild-type embryos, and disrupted in embryos lacking mbc. Phalloidin labels F-actin (green) and Dlg marks the membrane (red). (A-E) Single serial section views of (A,C) wild-type or (B,D,E) mbcD11.2 embryos. Confocal images were collected at a 5× magnification (C-E) or 2× magnification (A,B). Individual fluorescence channels are shown in black and white panels. In all examples, fusion-competent myoblasts (FCMs) were identified by expression of a sns-mCherryNLS transgene (shown only in E). (A,B,E) Many FCMs contact a single myotube/founder cell. (C,D) A single FCM contacts a myotube/founder cell. (A′,B′) Images from 3D reconstructions of z-series data. (E) A ‘rosette’ structure formed in an mbcD11.2 embryo by multiple FCMs contacting a founder cell. Note the apparently ‘larger’ but more diffuse actin focus in this example. Asterisks mark the myotube/founder cell in C-E. See Movies 5, 6 in the supplementary material. Scale bars: 5 μm.
RESULTS
An asymmetric, cell type-specific requirement for Mbc in fusing myoblasts
To determine the cell-type requirement for Mbc in vivo, we targeted expression of UAS-Mbc to founder cells or FCMs of mbcD11.2 mutant embryos using rP298-Gal4 or sns-Gal4, respectively. Surprisingly, a wild-type pattern of myofibers developed when Mbc was expressed exclusively in the FCMs (Fig. 1A,B). By contrast, myoblast fusion remained compromised when Mbc was expressed in the founder cells of these mutants (Fig. 1C). The ability of rP298-Gal4 to direct transgene expression was confirmed by restoration of the wild-type muscle pattern with UAS-Rols in rols/ants mutant embryos (Fig. 1D). These data are consistent with the proposed requirement for rols/ants in founder cells (Chen and Olson, 2001; Menon and Chia, 2001; Rau et al., 2001). More importantly, rP298-Gal4-directed expression of Mbc is readily detectable in mbcD11.2 mutants and is actually higher than that of endogenous Mbc in wild-type embryos (see Fig. S1 in the supplementary material). Finally, we confirmed that sns-Gal4 is specific to the FCMs as reported (Kocherlakota et al., 2008), and is not expressed promiscuously in founder cells, as expression of UAS-Kirre rescued almost no fusion in embryos lacking kirre and rst (Fig. 1E).
Fig. 1.

Mbc is required in the fusion-competent myoblasts for fusion with founder cells. Stage 16 embryos immunostained with anti-MHC antibody. Dorsolateral, lateral and ventrolateral views are provided, with anterior towards the left and dorsal towards the top. (A) Unfused myoblasts are apparent in mbcD11.2 embryos. (B) Fusion is restored to a wild-type pattern upon expression of Mbc in fusion-competent myoblasts (FCMs). (C) Myoblast fusion remains severely impaired when Mbc is expressed in founder cells. (D) Rols in founder cells restores fusion in rols1728-20L. (E) FCM-directed Kirre does not rescue fusion in kirre, rst mutants. (F) FCM-directed Mbc lacking the Crk-binding sites restores fusion in an mbc mutant embryo. (G) FCM-directed Mbc rescues fusion in mbcmat/zyg mutants. (H) FCM-directed RacV12 rescues fusion in mbc mutants. (I) Limited fusion in mbc mutants upon rP298-Gal4 directed RacV12. (J) FCM-directed Rac1 does not rescue fusion in mbc mutants. Scale bar: 50 μm.
We next examined whether myotube formation was rescued by FCM-targeted expression of Mbc lacking the ability to bind Crk. As anticipated from our previous analysis (Balagopalan et al., 2006), a wild-type pattern of muscles was observed (Fig. 1F), indicating that a Crk/Mbc interaction is not essential in the embryonic musculature. Because limited mbc mRNA is provided maternally (Erickson et al., 1997), we also confirmed that FCM-specific Mbc rescued muscle formation in embryos lacking mbc both maternally and zygotically (Fig. 1G). Finally, we examined the ability of activated Rac1 to rescue the mbc mutant phenotype when expressed in FCMs. These studies revealed that constitutively active Rac1 can rescue myogenesis in mbc mutants to a near wild-type pattern (Fig. 1H). By comparison, activated Rac1 was unable to rescue myoblast fusion in mbcD11.2 embryos when directed to the founder cells (Fig. 1I). This severe lack of fusion is not a consequence of excess active Rac1, as described previously (Geisbrecht et al., 2008; Luo et al., 1994), because rP298-Gal4 targeted expression in wild-type embryos causes only very mild defects (data not shown). As expected, its wild-type counterpart fails to rescue fusion as Mbc is not present for its activation (Fig. 1J). Thus, Mbc plays a fundamental role in the FCMs but is dispensable in the founder cells for efficient fusion. Moreover, its primary role is to direct activation of Rac1 in the FCMs in a manner that does not require Crk binding.
Limited Mbc-independent fusion of founder cells
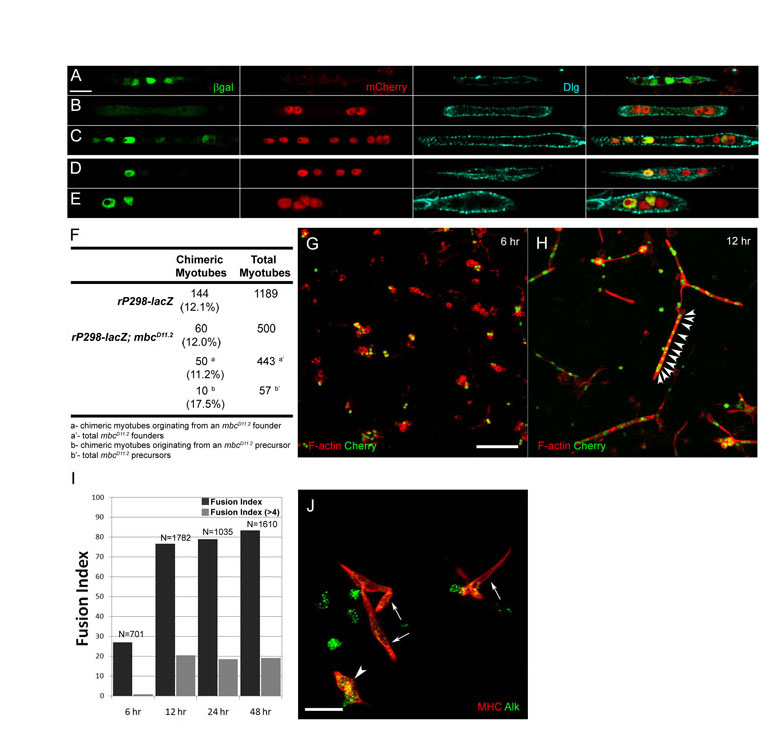
Previous studies describing the mbc mutant phenotype have concluded that myoblast fusion does not occur in its absence (Erickson et al., 1997; Rushton et al., 1995). Using the sensitive nuclear reporters rP298-lacZ and sns-mCherryNLS, we find that limited fusion does occur in these mutants, as binucleate muscle precursors that express both reporters are present (Fig. 2A,B). To determine whether Mbc function is provided maternally, we examined fusion in germline clone embryos (Fig. 2C). The overall level of fusion in the complete absence of mbc was roughly comparable with that in zygotic loss-of-function mutants. Additionally, the level of fusion did not decrease upon removal of sponge, a paralog of mbc (Geisbrecht et al., 2008; Postner et al., 1992) (Fig. 2F; Table 1). Quantitation of fusion in specific muscles using the identity markers Kr and Eve revealed that 72-95% of these founder cells do not fuse in the absence of mbc, whereas 19-28% of the dorsal Kr- and Eve-expressing founder cells undergo a single fusion event (Fig. 2D,E; Table 1). Of note, this amount of fusion is slightly higher than that reported elsewhere (Beckett and Baylies, 2007) but, as mbcD11.2 is a protein null allele, we cannot account for the difference.
Fig. 2.

Limited precursor formation occurs in embryos lacking mbc. (A-C‴) Stage 16 embryos immunostained with anti-βgal (green), anti-MHC (cyan) and anti-RFP (red). (A-B‴) Binucleate precursors expressing rP298-lacZ and sns-mCherryNLS indicate fusion in mbc mutants. (C-C‴) Precursors in mbcmat/zyg embryos. (D-I) Stage 15 embryos immunostained for Eve, Kr and Slouch reveal limited precursor formation in mbc mutants. (D,H) mbcD11.2. (E) mbcmat/zyg. (F) mbcD11.2, spg242. (G,I) rP298-Gal4-driven Mbc in mbc mutants. In D-G, arrows indicate DA1 and arrowheads indicate DO1. In H,I, arrows indicate LO1, open arrowheads indicate LT2, closed arrowheads indicate LT4, dotted circles indicate VT1 and asterisks indicate VA2. (J,K) Leaky expression of rP298-Gal4 in FCMs. Arrows indicate mCherry and βgal FCMs. (L,M) The dorsolateral (L) and ventrolateral (M) founder cells. PC, pericardial cells. Scale bars: 20 μm in A-A‴,C-C‴,D-I; 5 μm in B-B‴,J,K.
Table 1.
Quantitation of precursor formation in embryos

Despite its inability to rescue significant myoblast fusion, we wished to determine whether rP298-Gal4-directed Mbc rescued precursor formation. Fusion was quantitated in founder cells expressing Kr, Eve and Slouch (Fig. 2D,G-I,L,M; Table 1), revealing an approximately twofold increase in precursor formation. These data, at first glance, seem to suggest that founder cell expression of Mbc directs some precursor formation. Upon closer examination, however, we noted limited co-expression of rP298-Gal4 driven UAS-lacZ and sns-mCherry in FCMs in mbcD11.2 embryos (Fig. 2J,K). Whereas most rP298-Gal4-driven UAS-lacZ-expressing founder cells adopted a characteristic elongated morphology, the βgal/mCherry double-positive mononucleate cells exhibited the rounded morphology of sns-mCherry-expressing FCMs. We therefore suggest that rP298-Gal4 drives expression of Mbc in these FCMs, accounting for their fusion with founder cells. We note that similar results have recently been reported (Sens et al., 2010). Most founder cells remain mononucleate and do not undergo a single fusion event with an FCM under these conditions. Thus, expression of Mbc in all founder cells as well as a small number of FCMs is still insufficient for over half of the founder cells to undergo any fusion. By contrast, myoblast fusion approximates that in wild-type embryos upon expression of Mbc in FCMs.
FCM-directed Rac1 rescues muscle formation in rac1, rac2 mutant embryos
We next chose to examine whether expression of Rac1 in the FCMs was sufficient to rescue fusion in rac1, rac2 mutant embryos. Note that, in contrast to the extremely limited fusion in embryos lacking mbc, more fusion occurs in rac1, rac2 mutant embryos due to the perdurance of maternally provided gene product (Fig. 3A) (Hakeda-Suzuki et al., 2002). As for mbc mutant embryos, this fusion was quantified in the dorsal and ventrolateral muscles (Fig. 3D,E; Table 1). Wild-type Rac1 under UAS control was then targeted to founder cells or FCMs of rac1J11, rac2Δ embryos. Clearly, sns-Gal4-directed expression of wild-type Rac1 rescued the myoblast fusion defects of these mutant embryos to a near normal pattern (Fig. 3A,B). By comparison, rP298-Gal4-specific expression rescued fusion much less efficiently (Fig. 3C). These results may imply that fusion occurs if Rac1 is present in either fusing partner but occurs more efficiently when present in both, as in cultured mammalian myoblasts (Vasyutina et al., 2009). Alternatively, Rac1 may play a more important role in the FCMs. Unfortunately, we cannot resolve these possibilities as some fusion has occurred in rac1J11, rac2Δ mutants and we are unable to eliminate FCM contributed protein from the resulting myotube. Nevertheless, if Rac1 is essential in the founder cell for its fusion with FCMs, our data establish that it must be activated by a GEF other than Mbc/Elmo.
Fig. 3.

Rescue of rac1, rac2 mutant embryos by fusion-competent myoblast-specific expression of Rac1. (A-C) Stage 16 embryos immunostained for myosin heavy chain (MHC). All panels are dorsolateral, lateral or ventrolateral views, with anterior towards the left and dorsal towards the top. (A) rac1J11rac2Δ exhibit defects in myoblast fusion. (B) Fusion in rac1J11rac2Δ with fusion-competent myoblast (FCM)-directed Rac. (C) Limited fusion in rac1J11rac2Δ with rP298-Gal4-directed Rac. Scale bar: 50 μm. (D,E) Late stage 15 rac1J11rac2Δ embryos immunostained for Eve (green) and Kr (red) (D) or Slouch (E) to facilitate quantitation of fusion. Arrows and arrowheads in D indicate DO1 and DA1, respectively. Arrows and asterisk in E indicate LO1 and VT1, respectively. Scale bar: 20 μm.
Asymmetric localization of Mbc, active Rac1 and actin foci in primary FCMs
The data described above suggest an asymmetric requirement for Mbc, and Mbc-activated Rac1, in the fusion of FCMs with founder cells and myotubes. We therefore wished to examine these two cell types for the presence of asymmetrically localized proteins. These studies were carried out in isolated primary myoblasts for clear visualization of cell-cell contacts. Cells were isolated as described previously (Bai et al., 2008; Bai et al., 2009; Bernstein et al., 1978) and behave similarly in our hands (see Fig. S2 in the supplementary material). For example, the mean number of nuclei per myotube was 3.5 and 20% of the myotubes contained five or more nuclei at 12 hours. We also confirmed the asymmetric requirement for Mbc in these cells (see Fig. S2 in the supplementary material). In summary, ~12% of wild-type founder cells fused with wild-type FCMs, and 12% of mbcD11.2 founder cells fused with wild-type FCMs.
These primary myoblasts were then labeled for Sns and Kirre to identify points of cell-cell contact and co-stained for Mbc or F-actin to determine their distribution within the cell. The FCM of each cell pair was identified by its rounded morphology visible by DIC optics, and by post-fixation fluorescence of mCherry from sns-mCherryNLS. Founder cells and myotubes were identified by their elongated morphology and the number of nuclei in myotubes.
These analyses revealed strong enrichment of Mbc in the FCM at the Sns:Kirre adhesive junction, but not in the associated founder cell or myotube (Fig. 4A,B; see Movie 1 in the supplementary material). The multinucleate myotubes do contain anti-Mbc reactive material, but it was not enriched at points of contact with FCMs. We then visualized Sns, Kirre and activated Rac1 in these cells. Activated Rac1 is quite noticeably enriched in the FCMs at contacts with either founder cells (Fig. 4C) or myotubes (Fig. 4D; see Movie 2 in the supplementary material). This localization is similar to that of Mbc, consistent with the earlier finding that sns-Gal4 expression of activated Rac1 rescues the mbcD11.2 phenotype. We next examined the distribution of F-actin, as Dock180 and Rac1 have been implicated in actin polymerization (Bosco et al., 2009; Derivery and Gautreau, 2010; Heasman and Ridley, 2008), and because F-actin foci are associated with myoblast fusion (Gildor et al., 2009; Kim et al., 2007; Richardson et al., 2007). Cortical F-actin is uniformly distributed along the surface of the myotube and FCMs. More importantly, a large focus of polymerized F-actin is clearly evident and enriched in the rounded FCM at the Sns-Kirre point of adhesion (Fig. 4E,F; see Movie 3 in the supplementary material).
Fig. 4.

Localization of Mbc, active Rac1 and F-actin in fusion-competent myoblasts at points of contact with founder cells and myotubes in primary myoblasts. (A-G′) Primary myoblasts 14 hours after plating were immunostained for Kirre (red), Sns (blue) and either Mbc (A-B′), active Rac1 (C-D′) or F-actin (E-G′), shown in green. (A-F′) Single confocal sections of wild type; strong enrichment of all three proteins (Mbc, active Rac1, F-actin) in the fusion-competent myoblasts (FCMs). (G) DIC and projection images of four serial confocal sections from mbcD11.2 cells; absence of compact foci. A-G are lower magnification views that include DAPI nuclear staining (light blue). Boxed areas indicate the region shown at higher magnification in A′-G′. (A,C,E,G) Representative FCMs in contact with mononucleate founder cells. (B,D,F) FCMs in contact with syncitia of four to six nuclei. The FCM of each cell pair was identified by its rounded morphology visible by DIC optics and by post-fixation fluorescence of the native protein from an sns-mCherryNLS transgene (not shown). Founder cells/myotubes were identified by their elongated morphology (A,C,E) or the presence of multiple nuclei by DAPI staining (B,D,F). Asterisks indicate myotube nuclei and arrows indicate the FCM. Broken lines in single channel views are provided as a reference for cell boundaries. (A″-G″) Images from 3D reconstructions of z-series data. See Movies 1-4 in the supplementary material. Scale bars: 10 μm in A-G; 2 μm in A′-G′.
Lastly, we examined the distribution of F-actin in myoblasts isolated from mbcD11.2 embryos. Most apparent in the FCMs was the absence of a concentrated F-actin focus. Rather, F-actin is diffuse and foci are fragmented at sites of cell contact (Fig. 4G; see Movie 4 in the supplementary material). From these data, we conclude that F-actin, Mbc and activated Rac1 are asymmetrically localized in the FCM at points of Sns:Kirre-mediated contact with founder cells and myotubes, and that Rac1 activation via Mbc plays a crucial role in formation of the F-actin focus.
Mbc is essential for the integrity of the F-actin foci in FCMs in intact embryos
We next sought to confirm that the FCM-enriched actin foci were present in the intact embryo. Numerous studies have described these foci (Gildor et al., 2009; Kim et al., 2007; Richardson et al., 2007) but are either lacking a membrane marker or obscure cell topology by analyzing confocal projections greater than a cell diameter. Using the membrane marker Dlg and phalloidin staining, F-actin foci were apparent in the FCMs at points of contact with developing myofibers in wild-type embryos (Fig. 5A,C) and visualized in 3D (see Movie 5 in the supplementary material). We then examined the requirement for Mbc in formation of these foci. Early mutant embryos were examined to visualize a single FCM contacting a founder cell before large clusters of unfused cells had formed. Whereas the F-actin foci of wild-type embryos are intense, fairly compact and prominent at cell contacts, the foci of mbcD11.2 FCMs are less dense and somewhat dispersed (Fig. 5B,D; see Movie 6 in the supplementary material). These data suggest that Mbc and Rac1 play a role in organizing the F-actin foci. As this finding contrasts with published studies reporting abnormally large foci in the absence of mbc (Richardson et al., 2007), we examined clusters of unfused cells in later stage embryos. We ensured that data was not collected under saturating conditions for F-actin. In combination with the Dlg membrane marker, this analysis revealed that the large actin foci seen in mbcD11.2 embryos represented a convergence of FCMs on one founder cell (Fig. 5E), with actin foci that were also more dispersed. Thus, as observed in primary myoblasts, Mbc plays a crucial role in organizing the F-actin foci within the FCM.
To address whether the asymmetric distribution of Mbc, active Rac1 and F-actin foci is a transient step in the fusion process, we employed timelapse confocal microscopy to characterize actin dynamics at points of cell contact in live stage 13 embryos. Actin-mCherry and Gap-GFP were expressed pan-mesodermally under control of a twi-Gal4 driver. As shown in primary myoblasts and fixed embryos, actin accumulates in the FCM at the point of contact with the myotube (Fig. 6A-D; see Movie 7 in the supplementary material) just prior to membrane breakdown and fusion (Fig. 6E-G). Relative pixel intensities were plotted across the actin enrichment, including the region of contact between the FCM and myotube, to define the location of this actin focus more precisely (Fig. 6A′-G′). The peak mCherry signal is on the FCM side of the GFP (Fig. 6A′-D′), indicating the actin enrichment is primarily, if not entirely, within the FCM. At the time of fusion (Fig. 6E′), the actin enrichment dissipates (Fig. 6F′-G′). We then used the same basic approach to examine actin organization in mbcD11.2 embryos. Whereas some accumulation of actin was present at the site of cell-cell contact, this enrichment was lower in intensity and more diffuse than that in wild-type FCMs (Fig. 6H-N; see Movie 8 in the supplementary material). This slight enrichment does not increase over time and large actin foci were not observed. These results support the conclusion that F-actin foci are located primarily, if not wholly, within the FCM, and that Mbc is required for their proper assembly.
Fig. 6.

Timelapse imaging reveals actin foci in fusion-competent myoblasts prior to fusion, and their disorganization in embryos lacking mbc. Stage 13 embryos expressed Gap-GFP and Actin-mCherry under twi-Gal4 control. (A-G) Wild type; single optical sections from Movie 7 in the supplementary material. Arrowheads indicate the site of contact between the FCM and myotube, asterisks mark neighboring cells for reference. Broken lines indicate syncytia boundary. (A′-G′) Relative intensities of red (Actin) and green (membrane Gap) for the regions in insets. A dense actin focus in the FCM at its point of myotube contact disappears upon cell fusion and membrane breakdown. Images are at 64-second intervals, with a gap between F and G. Scale bar: 5 μm. (H-N) mbcD11.2; single optical sections. Broken lines mark the founder cell. Actin is weakly polarized (asterisks) in the mutant FCM at the point of contact with the founder cell. Fluorescence signals (shown as black and white panels) were collected separately. Images are at 7-minute intervals, with a gap between M and N. Scale bars: 5 μm. See Movies 7, 8 in the supplementary material.
Mbc is essential for the integrity of the actin cytoskeletal network in FCMs
We returned to primary myoblasts to compare F-actin in wild-type and mbc mutant cells. As described earlier (Fig. 4), actin foci were visible in wild-type FCMs but not in FCMs lacking Mbc (Fig. 7A,B). Additionally, actin fibers were visible in both wild-type myotubes and elongated mbc-mutant founder cells. As the cytoskeleton was visible under saturating conditions, we also examined its integrity in wild-type FCMs and mutant FCMs (Fig. 7; see Fig. S3 and Movies 9, 10 in the supplementary material). Of note, the cytoskeleton of FCMs lacking mbc had a distinctly different organization of F-actin from that in wild-type FCMs. In fact, many mutant cells exhibit a complete collapse of the network that was clearly visible in wild-type FCMs. Thus, Mbc is essential for the integrity of F-actin in the FCMs: in foci at points of cell contact and in the overall cytoskeletal network. As an apparent consequence of these perturbations, the defective FCMs are unable to fuse.
Fig. 7.

The actin cytoskeletal network is also disorganized in FCMs in the absence of mbc in cultured myoblasts. Primary myoblasts 14 hours after plating, labeled with phalloidin to visualize F-actin (green) and Dlg (red) to mark the membrane. Single confocal sections are shown. (A) Wild type; the dense actin focus is apparent in the FCM at the site of myotube contact. (B) mbcD11.2; both the F-actin focus and actin network are disrupted. Signal saturation revealed the actin cytoskeleton. (A′,B′) F-actin was visualized in a 3D reconstruction with iso-surfacing. Scale bar: 5 μm. See Movies 9, 10 in the supplementary material.
Sns, Mbc, active Rac1 and F-actin foci are associated with FCM-driven deformation of the founder cell/myotube membrane prior to fusion
We have demonstrated that Mbc plays a fundamental role in the FCMs in directing their fusion with founder cells, and that Mbc pathway components are distributed asymmetrically at sites in the FCM where it contacts either a founder cell or a myotube. In addition to these features, however, visualization in primary myoblasts revealed distinct cell morphologies prior to fusion (Fig. 8). Fig. 8A is representative of the smallest contact site, in which Sns is present at the tip of an elongated process from the FCM and colocalizes with Kirre where this process contacts the myoblast with no obvious concentration of F-actin at the contact site. In particular, we also observe a more transient stage in which the F-actin focus actually protrudes from the FCM into the myotube and is associated with deformation of the myotube membrane (Fig. 8B,C; see Movie 11 in the supplementary material). This FCM-based protrusion is also readily visible in cells labeled for active Rac1 (Fig. 8D,E; see Movie 12 in the supplementary material). We conclude from these data that the FCM plays an active, dynamic and apparently dominant role in driving deformation of the membrane myotube as a prelude to myoblast fusion.
Fig. 8.

FCM-associated protrusions and deformation of the myotube membrane at cell contact sites in primary cell cultures. (A-E) Primary myoblasts 14 hours after plating were immunostained for Sns (blue), Kirre (red) and F-actin (A-C, green) or active-Rac (D,E, green). DAPI marks the nuclei. All panels are projections of two serial z-sections of an FCM contacting a myotube (six nuclei; A) or protruding into a founder cell/precursor (B-E). (A′-E′) High magnification views of the myoblasts shown in A-E. Arrows in A′-E′ indicate the FCM, which was identified as in Fig. 4. Single channel images are shown as black and white. Dotted lines are provided as a reference for the cell boundaries. Scale bar: 5 μm. (A″-E″) Images from 3D reconstructions of z-series data. See Movies 11 and 12 in the supplementary material.
DISCUSSION
Recent reviews of myoblast fusion have included a common model in which Mbc and Rac1 function downstream of Kirre in the founder cells to direct actin polymerization (Haralalka and Abmayr, 2010; Onel and Renkawitz-Pohl, 2009; Rochlin et al., 2010), and is based on studies showing that Mbc interacts with the Kirre-associated Rols/Ants protein (Chen and Olson, 2001). However, as founder cell-specific expression of Mbc is inadequate to rescue its loss of function fusion phenotype [(Balagopalan et al., 2006) and herein], we reasoned that it must be required in the FCMs. Recent reviews have also suggested that F-actin foci are present symmetrically at points of contact between founder cells and FCMs (Haralalka and Abmayr, 2010; Onel and Renkawitz-Pohl, 2009; Rochlin et al., 2010). This symmetric F-actin-associated adhesive structure has been termed the FuRMAS (fusion-restricted myogenic-adhesive structure), and appears to be a ring of Sns and Kirre surrounding a central core of F-actin (Kesper et al., 2007). Our results amplify these models in several important areas, as depicted in the model in Fig. 9. First, our data reveal that Mbc is explicitly required in the FCMs and is not needed in the founder cells for fusion to occur. Thus, the essential function of Rols/Ants in fusion cannot be to direct recruitment of Mbc to Kirre in the founder cells. Second, high-resolution imaging has revealed that Mbc, active Rac1 and F-actin are concentrated in FCMs near Sns at the point of contact with founder cells, and are therefore localized asymmetrically in the fusion partners. Moreover, FCM-associated structures project into the founder cell and myotube at the Sns:Kirre adhesion site prior to fusion. Consistent with these observations, other approaches have recently reported the enrichment of actin foci in FCMs and the presence of invadopodia visible in EM that extend from the FCM into the founder cell/myotube (Sens et al., 2010). Finally, we also find that Mbc is important for the integrity of the F-actin focus and for the overall integrity of the actin cytoskeleton in FCMs.
Fig. 9.

A revised model for Mbc and F-actin foci in Drosophila myoblast fusion. The FCM protrudes into the founder cell. Sns and Kirre/Duf are present at the adhesion site of the FCM and founder cell, respectively. Mbc, Rac-GTP and F-actin foci are highly enriched in the FCM. Scar is proposed to function downstream of Rac in the FCM. Scar is also present in the founder cell.
Mbc is both necessary and sufficient in the FCMs for fusion with founder cells
Although very limited fusion occurs in embryos lacking Mbc, more than 80% of the founder cells do not undergo even a single fusion event. By contrast, FCM-directed expression of Mbc rescues an almost wild-type pattern of muscle fibers. Thus, Mbc expression in the FCMs is both necessary and sufficient for their fusion with founder cells. We also observe robust fusion when activated Rac1 is expressed in the FCMs of mbcD11.2 embryos, indicating that the primary role of Mbc is to activate Rac1. Our data do not support a mechanism in which Rols/Ants functions in the founder cells to recruit Mbc to the cytodomain of Kirre (Chen and Olson, 2001), a mechanism that is also inconsistent with the ability of Kirre to direct precursor formation in the absence of its cytodomain (Bulchand et al., 2010). Rather, these data support a mechanism in which Rols/Ants stabilizes Kirre (Menon et al., 2005), thereby ensuring that Kirre continues to be present on the myotube surface for fusion with FCMs. Unfortunately, it not possible to determine in the embryo whether Mbc or activated Rac1 is sufficient in the FCMs for later Mbc-dependent fusion between syncitia and FCMs, as the contents of the FCMs become incorporated into the syncitia following fusion. In primary cultures, however, wild-type founder cells as well as founder cells and binucleate precursors lacking Mbc all fuse with wild-type FCMs and at similar rates. Thus, the asymmetric distribution of Mbc in the FCMs is also sufficient for later fusion events, at least in cultured myoblasts.
We are unable to address whether Rac1 and Rac2 are specifically required in the FCMs as the founder cells of rac1J11, rac2Δ embryos are already syncytial owing to the perdurance of maternally provided gene product. Although localization of active Rac1 to points of cell contact in the FCMs may be an indication that, like Mbc, Rac1 is essential only in the FCMs, we note that Rac1 is required in both fusion partners in vertebrates (Vasyutina et al., 2009). In either case, we can conclude that any requirement for Rac1 in the founder cells must involve a GEF other than Mbc/Elmo.
Mbc dependent F-actin foci are present at sites of FCM-driven deformation of the founder cell/myotube membrane
Our data localize Mbc, active Rac1 and F-actin to FCMs at the Sns:Kirre adhesion site with either founder cells or developing myotubes. This asymmetric distribution is independent of whether the FCMs are contacting founder cells or myotubes and, in combination with features of fusion in primary myoblasts, suggests that the first fusion event does not differ from subsequent events with respect to these proteins. These data support a model in which both early and later stages of fusion are highly asymmetric, and that this asymmetry extends beyond recognition and adhesion at the cell surface to cytoplasmic events associated with polymerization of actin. Mbc, which is present but not localized in founder cells/myotubes, may serve a different purpose in these cells such as activating Rac1-dependent myotube guidance or attachment. In addition, though we have not observed large F-actin foci on the founder cell/myotube side of the adhesion site, we do observe a strong layer of cortical actin along the surface of the founder/developing myotube, as previously observed by immunoEM (Kim et al., 2007).
The local accumulation of F-actin in FCMs is reminiscent of dynamic actin foci found at sites of fusion and associated with the WAVE/SCAR, Vrp/WASp and the Arp2/3 complex (Gildor et al., 2009; Richardson et al., 2007). F-actin is also present in the core of the muscle-specific FuRMAS (Kesper et al., 2007). Interestingly, similarities have been noted between the FuRMAS and the immunological synapse (IS), podosome and invadopodia (Onel and Renkawitz-Pohl, 2009). Our data provides important new information in support of this analogy, as invasive podosomes, the IS and invadopodia are actually all associated with asymmetic F-actin (Caldieri et al., 2009a; Caldieri et al., 2009b; Gawden-Bone et al., 2010; Linder, 2007; Linder and Aepfelbacher, 2003; Vignjevic and Montagnac, 2008; Wernimont et al., 2008). As noted earlier, strong support for this analogy has recently been demonstrated by the presence of invadopodia-like invasive structures at the level of EM, that extend multiple finger-like projections into the founder cell/myotube (Sens et al., 2010). Interestingly, the IS, invadopodia and muscle-specific FuRMAS have common F-actin regulators that include WASp, HEM/Kette, SCAR/WAVE and Rho-family GTPases (Caldieri et al., 2009a; Grakoui et al., 1999; Linder and Aepfelbacher, 2003; Sens et al., 2010). Thus, the FCM appears to provide the primary F-actin associated force for myoblast fusion.
Mbc affects the F-actin foci and cytoskeletal network
Previous studies have reported that F-actin foci in mbc mutants are enlarged and increased in number (Richardson et al., 2007). However, careful 3D reconstruction of F-actin in embryos and in primary cells suggests that the tight actin foci found in wild-type FCMs are less organized and more dispersed in mbc mutants. Our analysis also revealed that the cytoskeletal network at the periphery of the FCM has collapsed in the absence of mbc. Thus, it appears that Mbc positively regulates organization of the actin cytoskeleton and actin polymerization at the adhesion site.
Although the mechanism(s) by which Mbc-activated Rac1 accomplishes this role have yet to be elucidated, Rac1 is known to interact with components of the WAVE/SCAR pathway in Drosophila and mammalian cells (Derivery and Gautreau, 2010; Schenck et al., 2003). In mammals, SCAR exists as part of a multiprotein complex composed of HEM/Kette, Abi, Sra1 and HSPC300. These subunits control SCAR stability and localization at the membrane (Derivery and Gautreau, 2010). Moreover, the pentameric SCAR complex can be activated by GTP-bound Rac (Ismail et al., 2009; Lebensohn and Kirschner, 2009) to promote actin polymerization by Arp2/3 (Derivery and Gautreau, 2010). HEM/Kette and SCAR play crucial roles in Drosophila myoblast fusion (Gildor et al., 2009; Richardson et al., 2007; Schroter et al., 2004), and Rac1 has been shown to synergize with SCAR in the myoblast/myotube. Moreover, SCAR is absent from sites of fusion in rac1 mutant embryos (Gildor et al., 2009). Notably, recent studies have shown that SCAR is required in both cell types (Sens et al., 2010), though it remains to be determined whether it plays similar roles in each. In summary, however, our studies support a mechanism in which Mbc/Elmo mediates the cell-type specific activation of Rac1 and, in turn, activation of WAVE/SCAR to promote an invasive actin-associated structure in the FCMs.
Supplementary Material
Acknowledgements
We thank M. Frasch for Eve antisera, J. Reinitz for Kr antisera, R. Palmer for Alk antisera and D. Kiehart for anti-MHC antibody. We thank S. D. Menon for UAS-Rols, rols1728-20L, rP298-lacZ and rP298-Gal4; S. Bogdan for UAS-Actin-mCherry; P. Schedl for sxl-GFP; and M. Ruiz-Gomez for UAS-kirre. We thank the Stowers Institute Cytometry and Imaging core facilities for assistance. This work was supported by the Stowers Institute for Medical Research and NIH award RO1 AR44274 to S.M.A. Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.057653/-/DC1
References
- Abmayr S. M., Balagopalan L., Galletta B. J., Hong S. J. (2003). Cell and molecular biology of myoblast fusion. Int. Rev. Cytol. 225, 33-89 [DOI] [PubMed] [Google Scholar]
- Abmayr S. M., Balagopalan L., Galletta B. J., Hong S. J. (2005). Myogenesis and muscle development. In Comprehensive Molecular Insect Science (ed. Gilbert L. I., Iatrou K., Gill S. S.), pp. 1-43 Amsterdam: Elsevier; [Google Scholar]
- Albert M. L., Kim J. I., Birge R. B. (2000). alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat. Cell Biol. 2, 899-905 [DOI] [PubMed] [Google Scholar]
- Artero R. D., Castanon I., Baylies M. K. (2001). The immunoglobulin-like protein Hibris functions as a dose-dependent regulator of myoblast fusion and is differentially controlled by Ras and Notch signaling. Development 128, 4251-4264 [DOI] [PubMed] [Google Scholar]
- Bai J., Binari R., Ni J. Q., Vijayakanthan M., Li H. S., Perrimon N. (2008). RNA interference screening in Drosophila primary cells for genes involved in muscle assembly and maintenance. Development 135, 1439-1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J., Sepp K. J., Perrimon N. (2009). Culture of Drosophila primary cells dissociated from gastrula embryos and their use in RNAi screening. Nat. Protoc. 4, 1502-1512 [DOI] [PubMed] [Google Scholar]
- Balagopalan L., Chen M. H., Geisbrecht E. R., Abmayr S. M. (2006). The CDM superfamily protein MBC directs myoblast fusion through a mechanism that requires phosphatidylinositol 3,4,5-triphosphate binding but is independent of direct interaction with DCrk. Mol. Cell. Biol. 26, 9442-9455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barolo S., Carver L. A., Posakony J. W. (2000). GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. Biotechniques 29, 726-732 [DOI] [PubMed] [Google Scholar]
- Baylies M. K., Bate M., Ruiz-Gomez M. (1998). Myogenesis: a view from Drosophila. Cell 93, 921-927 [DOI] [PubMed] [Google Scholar]
- Beckett K., Baylies M. K. (2007). 3D analysis of founder cell and fusion competent myoblast arrangements outlines a new model of myoblast fusion. Dev. Biol. 309, 113-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S., Schafer G., Kesper D. A., Holz A., Eriksson T., Palmer R. H., Beck L., Klambt C., Renkawitz-Pohl R., Onel S. F. (2008). WASP and SCAR have distinct roles in activating the Arp2/3 complex during myoblast fusion. J. Cell Sci. 121, 1303-1313 [DOI] [PubMed] [Google Scholar]
- Bernstein S. I., Fyrberg E. A., Donady J. J. (1978). Isolation and partial characterization of Drosophila myoblasts from primary cultures of embryonic cells. J. Cell Biol. 78, 856-865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco E. E., Mulloy J. C., Zheng Y. (2009). Rac1 GTPase: a “Rac” of all trades. Cell. Mol. Life Sci. 66, 370-374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour B. A., Chakravarti M., West J. M., Abmayr S. M. (2000). Drosophila SNS, a member of the Immunoglobulin Superfamily that is essential for myoblast fusion. Genes Dev. 14, 1498-1511 [PMC free article] [PubMed] [Google Scholar]
- Bulchand S., Menon S. D., George S. E., Chia W. (2010). The intracellular domain of Dumbfounded affects myoblast fusion efficiency and interacts with Rolling pebbles and Loner. PLoS One 5, e9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldieri G., Ayala I., Attanasio F., Buccione R. (2009a). Cell and molecular biology of invadopodia. Int. Rev. Cell Mol. Biol. 275, 1-34 [DOI] [PubMed] [Google Scholar]
- Caldieri G., Giacchetti G., Beznoussenko G., Attanasio F., Ayala I., Buccione R. (2009b). Invadopodia biogenesis is regulated by caveolin-mediated modulation of membrane cholesterol levels. J. Cell. Mol. Med. 13, 1728-1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E. H., Olson E. N. (2001). Antisocial, an intracellular adaptor protein, is required for myoblast fusion in Drosophila. Dev. Cell 1, 705-715 [DOI] [PubMed] [Google Scholar]
- Cote J. F., Vuori K. (2007). GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 17, 383-393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote J. F., Motoyama A. B., Bush J. A., Vuori K. (2005). A novel and evolutionarily conserved Ptdlns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat. Cell Biol. 7, 797-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E., Gautreau A. (2010). Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. BioEssays 32, 119-131 [DOI] [PubMed] [Google Scholar]
- Dworak H. A., Charles M. A., Pellerano L. B., Sink H. (2001). Characterization of Drosophila hibris, a gene related to human nephrin. Development 128, 4265-4276 [DOI] [PubMed] [Google Scholar]
- Erickson M. R. S., Galletta B. J., Abmayr S. M. (1997). Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure and cytoskeletal organization. J. Cell Biol. 138, 589-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch M., Hoey T., Rushlow C., Doyle H., Levine M. (1987). Characterization and localization of the even-skipped protein of Drosophila. EMBO J. 6, 749-759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke R., Gohl C., Dharmalingam E., Grevelhorster A., Zahedi B., Harden N., Kessels M., Qualmann B., Bogdan S. (2009). Drosophila Cip4/Toca-1 integrates membrane trafficking and actin dynamics through WASP and SCAR/WAVE. Curr. Biol. 19, 1429-1437 [DOI] [PubMed] [Google Scholar]
- Galletta B. J., Chakravarti M., Banerjee R., Abmayr S. M. (2004). SNS: adhesive properties, localization requirements and ectodomain dependence in S2 cells and embryonic myoblasts. Mech. Dev. 121, 1455-1468 [DOI] [PubMed] [Google Scholar]
- Gawden-Bone C., Zhou Z., King E., Prescott A., Watts C., Lucocq J. (2010). Dendritic cell podosomes are protrusive and invade the extracellular matrix using metalloproteinase MMP-14. J. Cell Sci. 123, 1427-1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbrecht E. R., Haralalka S., Swanson S. K., Florens L., Washburn M. P., Abmayr S. M. (2008). Drosophila ELMO/CED-12 interacts with Myoblast city to direct myoblast fusion and ommatidial organization. Dev. Biol. 314, 137-149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildor B., Massarwa R., Shilo B. Z., Schejter E. D. (2009). The SCAR and WASp nucleation-promoting factors act sequentially to mediate Drosophila myoblast fusion. EMBO Rep. 10, 1043-1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L. (1999). The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221-227 [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S., Ng J., Tzu J., Dietzl G., Sun Y., Harms M., Nardine T., Luo L., Dickson B. J. (2002). Rac function and regulation during Drosophila development. Nature 416, 438-442 [DOI] [PubMed] [Google Scholar]
- Haralalka S., Abmayr S. M. (2010). Myoblast fusion in Drosophila. Exp. Cell Res. 316, 3007-3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman S. J., Ridley A. J. (2008). Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690-701 [DOI] [PubMed] [Google Scholar]
- Ishimaru S., Ueda R., Hinohara Y., Ohtani M., Hanafusa H. (2004). PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. EMBO J. 23, 3984-3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail A. M., Padrick S. B., Chen B., Umetani J., Rosen M. K. (2009). The WAVE regulatory complex is inhibited. Nat. Struct. Mol. Biol. 16, 561-563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesper D. A., Stute C., Buttgereit D., Kreiskother N., Vishnu S., Fischbach K. F., Renkawitz-Pohl R. (2007). Myoblast fusion in Drosophila melanogaster is mediated through a fusion-restricted myogenic-adhesive structure (FuRMAS). Dev. Dyn. 236, 404-415 [DOI] [PubMed] [Google Scholar]
- Kim S., Shilagardi K., Zhang S., Hong S. N., Sens K. L., Bo J., Gonzalez G. A., Chen E. H. (2007). A critical function for the actin cytoskeleton in targeted exocytosis of prefusion vesicles during myoblast fusion. Dev. Cell 12, 571-586 [DOI] [PubMed] [Google Scholar]
- Kocherlakota K. S., Wu J. M., McDermott J., Abmayr S. M. (2008). Analysis of the cell adhesion molecule sticks-and-stones reveals multiple redundant functional domains, protein-interaction motifs and phosphorylated tyrosines that direct myoblast fusion in Drosophila melanogaster. Genetics 178, 1371-1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosman D., Small S., Reinitz J. (1998). Rapid preparation of a panel of polyclonal antibodies to Drosophila segmentation proteins. Dev. Genes Evol. 208, 290-294 [DOI] [PubMed] [Google Scholar]
- Lebensohn A. M., Kirschner M. W. (2009). Activation of the WAVE complex by coincident signals controls actin assembly. Mol. Cell 36, 512-524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S. (2007). The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17, 107-117 [DOI] [PubMed] [Google Scholar]
- Linder S., Aepfelbacher M. (2003). Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 13, 376-385 [DOI] [PubMed] [Google Scholar]
- Lu M., Ravichandran K. S. (2006). Dock180-ELMO cooperation in Rac activation. Methods Enzymol. 406, 388-402 [DOI] [PubMed] [Google Scholar]
- Luo L., Liao Y. J., Jan L. Y., Jan Y. N. (1994). Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 8, 1787-1802 [DOI] [PubMed] [Google Scholar]
- Meller N., Merlot S., Guda C. (2005). CZH proteins: a new family of Rho-GEFs. J. Cell Sci. 118, 4937-4946 [DOI] [PubMed] [Google Scholar]
- Menon S. D., Chia W. (2001). Drosophila rolling pebbles: a multidomain protein required for myoblast fusion that recruits D-Titin in response to the myoblast attractant Dumbfounded. Dev. Cell 1, 691-703 [DOI] [PubMed] [Google Scholar]
- Menon S. D., Osman Z., Chenchill K., Chia W. (2005). A positive feedback loop between Dumbfounded and Rolling pebbles leads to myotube enlargement in Drosophila. J. Cell Biol. 169, 909-920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose A., Isshiki T., Takeichi M. (1998). Regional specification of muscle progenitors in Drosophila: the role of the msh homeobox gene. Development 125, 215-223 [DOI] [PubMed] [Google Scholar]
- Onel S. F., Renkawitz-Pohl R. (2009). FuRMAS: triggering myoblast fusion in Drosophila. Dev. Dyn. 238, 1513-1525 [DOI] [PubMed] [Google Scholar]
- Postner M. A., Miller K. G., Wieschaus E. F. (1992). Maternal effect mutations of the sponge locus affect actin cytoskeletal rearrangements in Drosophila melanogaster embryos. J. Cell Biol. 119, 1205-1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau A., Buttgereit D., Holz A., Fetter R., Doberstein S. K., Paululat A., Staudt N., Skeath J., Michelson A. M., Renkawitz-Pohl R. (2001). rolling pebbles (rols) is required in Drosophila muscle precursors for recruitment of myoblasts for fusion. Development 128, 5061-5073 [DOI] [PubMed] [Google Scholar]
- Richardson B. E., Beckett K., Nowak S. J., Baylies M. K. (2007). SCAR/WAVE and Arp2/3 are crucial for cytoskeletal remodeling at the site of myoblast fusion. Development 134, 4357-4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochlin K. M., Yu S., Roy S., Baylies M. K. (2010). Myoblast fusion: When it takes more to make one. Dev. Biol. 341, 66-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Gomez M., Coutts N., Price A., Taylor M. V., Bate M. (2000). Drosophila dumbfounded: a myoblast attractant essential for fusion. Cell 102, 189-198 [DOI] [PubMed] [Google Scholar]
- Rushton E., Drysdale R., Abmayr S. M., Michelson A. M., Bate M. (1995). Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development. Development 121, 1979-1988 [DOI] [PubMed] [Google Scholar]
- Schenck A., Bardoni B., Langmann C., Harden N., Mandel J. L., Giangrande A. (2003). CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38, 887-898 [DOI] [PubMed] [Google Scholar]
- Schroter R. H., Lier S., Holz A., Bogdan S., Klambt C., Beck L., Renkawitz-Pohl R. (2004). kette and blown fuse interact genetically during the second fusion step of myogenesis in Drosophila. Development 131, 4501-4509 [DOI] [PubMed] [Google Scholar]
- Sens K. L., Zhang S., Jin P., Duan R., Zhang G., Luo F., Parachini L., Chen E. H. (2010). An invasive podosome-like structure promotes fusion pore formation during myoblast fusion. J. Cell Biol. 191, 1013-1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton C., Kocherlakota K. S., Zhuang S., Abmayr S. M. (2009). The immunoglobulin superfamily member Hbs functions redundantly with Sns in interactions between founder and fusion-competent myoblasts. Development 136, 1159-1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunkelnberg M., Bonengel B., Moda L. M., Hertenstein A., de Couet H. G., Ramos R. G., Fischbach K. F. (2001). rst and its paralogue kirre act redundantly during embryonic muscle development in Drosophila. Development 128, 4229-4239 [DOI] [PubMed] [Google Scholar]
- Stute C., Schimmelpfeng K., Renkawitz-Pohl R., Palmer R. H., Holz A. (2004). Myoblast determination in the somatic and visceral mesoderm depends on Notch signalling as well as on milliways(miliAlk) as receptor for Jeb signalling. Development 131, 743-754 [DOI] [PubMed] [Google Scholar]
- Vasyutina E., Martarelli B., Brakebusch C., Wende H., Birchmeier C. (2009). The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc. Natl. Acad. Sci. USA 106, 8935-8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignjevic D., Montagnac G. (2008). Reorganisation of the dendritic actin network during cancer cell migration and invasion. Semin. Cancer Biol. 18, 12-22 [DOI] [PubMed] [Google Scholar]
- Wernimont S. A., Cortesio C. L., Simonson W. T., Huttenlocher A. (2008). Adhesions ring: a structural comparison between podosomes and the immune synapse. Eur. J. Cell Biol. 87, 507-515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Chen E. H. (2008). Ultrastructural analysis of myoblast fusion in Drosophila. Methods Mol. Biol. 475, 275-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}