

Abstract
Resistance to apoptosis is a hallmark of many solid tumors, including pancreatic cancers, and may be the underlying basis for the suboptimal response to chemo-radiation therapies. Overexpression of a family of inhibitor of apoptosis proteins (IAP) is commonly observed in pancreatic malignancies. We determined the therapeutic efficacy of recently described small-molecule antagonists of the X-linked IAP (XIAP) in preclinical models of pancreatic cancer. Primary pancreatic cancers were assessed for XIAP expression by immunohistochemistry, using a pancreatic cancer tissue microarray. XIAP small-molecule antagonists (“XAntag”; compounds 1396-11 and 1396-12) and the related compound 1396-28 were tested in vitro in a panel of human pancreatic cancer cell lines (Panc1, Capan1, and BxPC3) and in vivo in s.c. xenograft models for their ability to induce apoptosis and impede neoplastic growth. In addition, pancreatic cancer cell lines were treated with XAntags in conjunction with either tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) or with radiation to determine potential synergy for such dual targeting of the apoptotic machinery. XIAP was overexpressed in 14 of 18 (77%) of primary pancreatic cancers. The XAntags 1396-11 and 1396-12, but not the inactive isomer 1396-28, induced profound apoptosis in multiple pancreatic cancer cell lines tested in vitro, with a IC50 in the range of 2 to 5 μmol/L. Mechanistic specificity of the XAntags for the baculoviral IAP repeat-2 domain of XIAP was shown by preferential activation of downstream “effector” caspases (caspase-3 and caspase-7) versus the upstream “initiator” caspase-9. S.c. BxPC3 xenograft growth in athymic mice was significantly inhibited by monotherapy with XAntags; treated xenografts showed marked apoptosis and increased cleavage of caspase-3. Notably, striking synergy was demonstrable when XAntags were combined with either TRAIL or radiation therapy, as measured by growth inhibition in vitro and reduced colony formation in soft agar of pancreatic cancer cell lines, at dosages where these therapeutic modalities had minimal to modest effects when used alone. Finally, XAntags in combination with the standard-of-care agent for advanced pancreatic cancer, gemcitabine, resulted in significantly greater inhibition of in vitro growth than gemcitabine alone. Our results confirm that pharmacologic inhibition of XIAP is a potent therapeutic modality in pancreatic cancers. These antagonists are independently capable of inducing pancreatic cancer cell death and also show synergy when combined with proapoptotic ligands (TRAIL), with radiation, and with a conventional antimetabolite, gemcitabine. These preclinical results suggest that targeting of the apoptotic machinery in pancreatic cancers with XAntags is a promising therapeutic option that warrants further evaluation.
Introduction
Pancreatic cancer is the fourth most common cause of cancer-related mortality in the United States, with approximately 32,000 deaths annually from this neoplasm (1). The overwhelming majority of patients present with advanced, inoperable disease and systemic chemoradiation therapy remains as the only treatment recourse for these individuals. Unfortunately, conventional therapeutic approaches have had minimal success in ameliorating the dismal prognosis of pancreatic cancer, and for the most part therefore, pancreatic cancer remains a disease of near uniform lethality (2).
Resistance to apoptosis is a commonly observed phenomenon in many cancers (3). Neoplastic cells overcome the apoptotic machinery and, hence, the propensity to be naturally eliminated, through a variety of mechanisms, including the overexpression of antiapoptotic proteins (e.g., Bcl-2) or the inactivation of proapoptotic molecules (e.g., epigenetic silencing of caspase-8; refs. 4, 5). Because many therapeutic modalities principally act by promoting apoptosis, alterations in this intracellular cascade can render neoplastic cancer cells resistant to therapy (6). A family of endogenous antiapoptotic proteins known as inhibitors of apoptosis proteins (IAP), which bind and repress proapoptotic caspases in their quiescent `zymogen' state, is frequently overexpressed in both solid and hematologic malignancies (7–12), including pancreatic cancer (13, 14). It is postulated that IAPs may be a major cause of the resistance to chemoradiation therapy- induced apoptosis observed in neoplastic cells; therefore, blockade of IAP function while simultaneously initiating cellular apoptosis would have the effect of overcoming this resistance state (15, 16).
Eight IAP family members have been identified in humans, and they share a variable number of the so-called baculoviral IAP repeat (BIR) domain (17). Of these, the X-linked IAP (XIAP) protein has been extensively studied for its role in human neoplasia and is known to inhibit caspase-3, caspase-7, and caspase-9 (18). Further, studies have revealed that of the three BIR domains of XIAP, BIR-2 inhibits the downstream caspase-3 and caspase-7, whereas BIR-3 inhibits the upstream caspase-9 (19–21). In light of its frequent overexpression in human cancers and its known function as a roadblock to apoptosis, XIAP also represents a candidate therapeutic target in cancer cells (22).
Recently, small-molecule phenylurea-based chemical inhibitors of XIAP (“XAntags”) were identified by large-scale combinatorial library screening (23). This and subsequent studies have confirmed that the active XAntags, but not their inactive structural analogues, could induce apoptosis in a variety of human cancer cell lines and xenografts (24–26). Furthermore, it was determined that these XAntags act by binding to its BIR-2 domain, resulting in elevated activity of the downstream caspase-3 and caspase-7 (the “executioner” caspases; ref. 23). Thus, the action of these exogenous XAntags was found to be mechanistically distinct from that of the endogenous inhibitor second modulator of apoptotic proteases, which predominantly binds to the BIR-3 domain (27).
We explored the role of XAntags in pancreatic cancer, not only as an independent therapeutic modality but also as an “apoptosis sensitizer,” wherein we combined the small-molecule XAntags with upstream proapoptotic stimuli [e.g., ligand-mediated death receptor activation using the tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)], radiation, and conventional antimetabolite, gemcitabine. Our results show that inhibition of XIAP activity potently sensitizes pancreatic cancer cells to apoptosis and indicates the potential of XAntags as a novel therapeutic strategy for pancreatic cancer.
Materials and Methods
Human Pancreatic Cancer Cell Lines
Human pancreatic cancer cell lines Panc1, Capan1, and BxPC3 were obtained from the American Type Culture Collection (Manassas, VA). Panc1 was maintained in DMEM, whereas Capan1 and BxPC3 were maintained in RPMI 1640, all medium being supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin.
XIAP Small-Molecule Inhibitors,TRAIL, and Radiation
XIAP small-molecule inhibitors 1396-11 and 1396-12 and the inactive structural analogue 1396-28 were synthesized and purified as described (23); the structures of these compounds have been described previously. The compounds were dissolved in 1% DMSO for in vitro assays. TRAIL was obtained from Alexis Biochemicals (San Diego, CA) and administered for in vitro dosing as described below for 3-(4-5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) and soft agar assays. Radiation to pancreatic cancer cells plated in 60-mm dishes was administered using a constant voltage, 225 kVp X-ray source. Aluminum (4 mm thick) was placed in the beam to filter out the lowest energy X-rays. The dishes were placed on a wooden surface f59 cm from the source. At this distance, the radiation field covered a surface area of 17.6 × 17.6 cm2. A dose rate of 126 cGy/min was measured using radiochromic film exposed under the same conditions. Dose uniformity across cell dish (perpendicular to the beam axis) was within ±5%.
Cell Viability (MTS) Assay
Cell viability was measured using the CellTiter 96 Aqueous Cell Proliferation Assay (Promega, Madison, WI), which relies on the conversion of a tetrazolium compound (MTS) to a colored formazan product by the activity of living cells (28). Briefly, pancreatic cancer cells were plated in 96-well plates and treated with varying concentrations of the XAntags (1396-11, 1396-12, and 1396-34 and the inactive control 1396-28) for 96 h, at which point the assay was terminated, and relative growth inhibition compared with vehicle-treated cells was measured using the CellTiter 96 reagent as described in the manufacturer's protocol. Viability assays were also done using a combination of XAntag 1396-11 (2 μmol/L) and TRAIL (100 ng), as above, to determine therapeutic synergy. Combination efficacy with gemcitabine was assessed using a range of gemcitabine dosages (0.4, 08, 1, and 2 μmol/L), with or without a constant dose of 1396-11 (2 μmol/L), and growth inhibition was measured by MTS assay. All assays were done in triplicate, and means + SEs were calculated.
Western Blot Assay for Cleaved Caspase-3
Western blot analysis was done for detecting cleaved caspase-3 (Cell Signaling, Danvers, MA). Briefly, cells were treated with 1396-11 and 1396-12, or vehicle treated for 48 h, and protein lysates were made from cell pellets. Radioimmunoprecipitation assay lysis buffer [20 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1% NP40, 0.5% sodium deoxycholate, 1 mmol/L EDTA, 0.1% SDS] was used; protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) and phenylmethylsulfonyl fluoride were added before lysing the cells. Cells were lysed on ice for 30 min with occasional gentle agitation of the tubes. Cell debris was separated by centrifugation at 13,200 rpm for 30 min at 4°C. Protein lysates were resolved by electrophoresis on 12% Tris-glycine gel (Invitrogen, Carlsbad, CA) and electrotransferred on nitrocellulose membranes (LC 2000, Invitrogen). Standard immunoblotting procedures were followed with slight modification: nitrocellulose membranes were blocked overnight at room temperature and incubated in primary cleaved caspase-3 antibody (1:1,000) overnight at 4°C. Anti-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used as a loading control.
Measurement of Caspase Activity
Activity of cellular caspases was measured using the Caspase-Glo-9 and Caspase-Glo-3/7 assay reagents (Promega), according to the manufacturer's instructions. The assays provide a luminogenic caspase-3/caspase-7 or caspase-9 substrate, respectively, in a buffer system optimized for caspase activity measurement in cell lysates. The luciferase signal generated is proportional to the amount of caspase activity present. Briefly, cells were plated in opaque 96-well plates in triplicate and treated with the XIAP inhibitors. After 24 h, the assay reagents were added to the plates at room temperature and incubated for 60 min, and the luminescence was measured using a plate reader. All assays were done in triplicate, and means + SEs were calculated for each condition.
Colony-Forming Assay in Soft Agar
Colony formation in soft agar was assessed for the two combination therapies, 1396-11 and TRAIL and 1396-11 and radiation. Briefly, 2 mL of mixture of serum supplemented medium and 0.5% agar containing 100 nmol/L 1396-11 and 20 ng TRAIL at 40°C were added in a 35-mm culture dish and allowed to solidify (base agar). Next, on top of the base layer was added a mixture of serum supplemented medium and 0.35% agar (total of 2 mL) containing 5,000 cells in the presence of the 100 nmol/L 1396-11 and 20 ng TRAIL at 40°C and allowed to solidify (top agar). For the combination experiments with radiation and XAntag, cells were treated with 2 μmol/L 1396-11 overnight and then irradiated as described above for a total dose of either 2 or 6 Gy; the irradiated cells were then used for the soft agar assay. Subsequently, the dishes were kept in tissue culture incubator maintained at 37°C and 5% CO2 for 14 days to allow for colony growth. All assays were done in triplicate. The colony assay was terminated at day 14, when plates were stained, and colonies were counted on ChemiDoc XRS instrument (Bio-Rad, Hercules, CA).
Immunohistochemical XIAP Labeling in Pancreatic Cancer Tissues
Immunohistochemistry for measuring XIAP protein levels in resected human primary pancreatic cancer specimens was done on a pancreatic cancer tissue microarray (29). A 1:100 dilution of mouse polyclonal anti-XIAP antibody (BD Biosciences, Franklin Lakes, NJ) was used. Briefly, following deparaffinization of tissue microarray sections, antigen retrieval was done in EDTA buffer (pH 9.1) for 30 min, with primary antibody incubation at 37°C for 2 h. After secondary antibody incubation (30 min), chromogenic detection was done using 3,3′-diaminobenzi-dine, and slides were counterstained with Mayer's hematoxylin. XIAP labeling was assessed using previously described criteria for cytoplasmic labeling (29).
In vivo S.c. Xenograft Studies Using XIAP Antagonists
BxPC3 cells (5 × 106) were suspended in phosphate buffer saline (PBS, pH 7.4) and Matrigel matrix (BD Biosciences). The cell suspension was injected s.c. into the flank of CD1 nu/nu male mice (Charles River Laboratories, Wilmington, MA). Xenografts were allowed to grow for 14 days, and baseline xenograft size was measured using an external electronic digital caliper (Fisher Scientific, Pittsburgh, PA); the average tumor size for the entire cohort was 130 mm3 before treatment. Mice were randomized to three arms, with five mice per arm, and administered either 1396-11 or 1396-12 at a dose of 30 mg/kg/d i.p., for 5 consecutive days, with the control arm receiving vehicle (DMSO) only. Mice were weighed once weekly over the course of the next 3 weeks, to exclude any potential systemic toxicities from the XAntags. The xenografted tumors were measured weekly for 3 weeks posttherapy, with mean tumor volumes calculated for each arm and normalized to the tumor volume at the initiation of the 3-week observation period. Mice were sacrificed subsequently and tissues were fixed in formalin and prepared for immunohistochemical analysis.
Immunohistochemistry for Phospho-Histone H3 and Cleaved Caspase-3 in Pancreatic Cancer Xenografts
Immunohistochemical analysis was done on harvested xenografts from XIAP antagonist- and vehicle-treated mice using antibodies to the mitotic protein phospho-histone H3 (dilution 1:500; Upstate, Charlottesville, VA) and to cleaved caspase-3, as described above.
Results
XIAP Is Frequently Overexpressed in Pancreatic Cancers
Cytoplasmic XIAP overexpression was seen in 14 of 18 (77%) of primary pancreatic cancers on the tissue micro-array. In Fig. 1, we show representative examples of XIAP labeling in two primary pancreatic cancers, with the staining being homogeneous and comparable across the 14 cases with XIAP overexpression. In contrast, minimal to absent labeling was seen in nonneoplastic pancreatic ductules.
Figure 1.

Tissue microarrays stained with anti-XIAP antibody confirm cytoplasmic overexpression of XIAP protein in human primary pancreatic cancers. A tissue microarray with 18 archival pancreatic cancers was stained with anti-XIAP antibody, and two representative cases are shown at ×20 magnification. Note complete absence of labeling in stromal and inflammatory components.
XAntags Induce Cell Death and Caspase Activation in Cultured Pancreatic Cancer Cell Lines
The active XAntags (1396-11 and 1396-12), but not the related compound 1396-28, caused dose-dependent growth inhibition of all three pancreatic cancer cell lines, as measured by MTS assay (Fig. 2). The IC50 for both active compounds was in the range of 2 to 5 μmol/L. Western blot analysis for cleaved caspase-3 following exposure to XAntags showed increased cellular processing of this apoptotic protease in Capan1 and Panc1 cells (Fig. 3), consistent with an apoptotic mechanism. To further clarify the mechanism of apoptosis induced by the XAntags, Panc1 and BxPC3 cells were treated with 1396-11 and 1396-12, and relative levels of activity of the upstream (“initiator”) caspase-9 and the downstream (“effector”) caspase-3/caspase-7 were measured using a luminometric assay. Treatment with the XIAP antagonists resulted in marked increases in the levels of caspase-3/caspase-7 activity, whereas the level of caspase-9 activity was essentially unchanged (Fig. 4). These results confirm that the XIAP antagonists used in this study function through the activation of downstream caspases and are consistent with their stated mechanism of action by binding the BIR-2 domain of XIAP (21, 23).
Figure 2.

XAntags induce cell death in cultured pancreatic cancer cell lines. Viability assays for three pancreatic cancer cell lines, Capan1, Panc1, and BxPC3, were done using two active XAntags 1396-11 and 1396-12 and inactive control 1396-28. Cells were cultured for 96 h before doing MTS assays. Dosages are color coded and annotated for all three compounds; assays were done in triplicate. Columns, mean; bars, SE.
Figure 3.
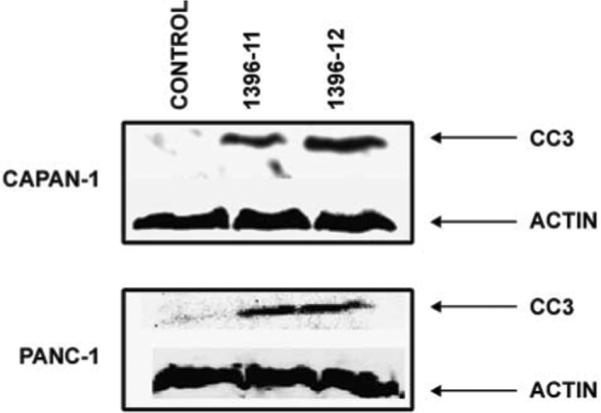
Western blot analysis shows up-regulation of cleaved caspase-3 levels in response to XAntag exposure in Capan1 and Panc1 cell lines. Pancreatic cancer cells were treated with 1396-11 or 1396-12 for 48 h, before preparing lysates for Western blot analysis. Blots were probed with antibodies specific for cleaved caspase-3 or β-actin.
Figure 4.

XAntags induce increases in effector caspase activity in cultured pancreatic cancer cell lines. Panc1 (top) and BxPC3 (bottom) cells were cultured for 24 h with 2 μmol/L 1396-12, or vehicle control, and then cell lysates were prepared and assessed for caspase activity using luminometric assays. The fold change reflects relative increase in luminometric emission from caspase substrates (separate substrates are used for caspase-3/caspase-7 and for caspase-9) compared with vehicle-treated controls. Assays were done in triplicate. Columns, mean; bars, SE.
XAntags Display In vivo Antitumor Activity on Pancreatic Cancer Xenografts
S.c. xenografts were generated from BxPC3 cell lines and treated independently with the XAntags or with vehicle DMSO (five mice per arm), as described above. There was no significant difference in body weights between treated and control mice (data not shown); further, no gross organ toxicities were observed at the time of necropsy. Mono-therapy with XAntags 1396-11 or 1396-12 resulted in growth inhibition at 3 weeks compared with vehicle-treated xenografts (Fig. 5A), with 1396-12 showing a somewhat greater efficacy than 1396-11. The xenografts were harvested at 3 weeks, and morphologic examination confirmed “lakes” of apoptotic cells in the XAntag-treated tumors compared with the vehicle-treated xenografts (Fig. 5B). The induction of marked increases in processing of caspase-3 was confirmed by immunohistochemical labeling for caspase-3 in serial histologic sections obtained from the XAntag-treated tumors.
Figure 5.

XAntags exhibit in vivo antitumor activity in a pancreatic cancer xenograft model. A, s.c. xenografts were generated using BxPC3 pancreatic cancer cells and randomized for treatment with XAntag monotherapy or with vehicle only as described in the text (n = 5 mice per arm). Following 5 five consecutive days of therapy, the mice were followed for 3 wks, with serial weekly measurements of xenograft volume. Y-axis, mean tumor volume for each arm, normalized to the mean tumor volume (100%) at the end of 5 days of therapy and beginning of the 3-wk observation period. Columns, mean; bars, SE. B, xenografts were harvested at the end of 3 wks from animals treated with vehicle only (control) or with 1396-11 and 1396-12 and examined for morphologic alterations by H&E staining (left), as well as evidence of apoptotic markers (cleaved caspase-3) by immunohistochemistry (right). “Lakes” of apoptotic cells are seen in xenografts administered with XAntags 1396-11 or 1396-12, as evidenced by nuclear pyknosis and karyorrhexis. The induction of apoptosis is mirrored by marked up-regulation of cleaved caspase-3 in both treatment arms. In contrast, minimal apoptosis and cleaved capsase-3 expression is seen in the control xenografts.
XAntags Exhibit Apparent Synergy withTRAIL, Radiation, and Gemcitabine
We then examined the effects of combining XAntags with a variety of therapeutic modalities that independently induce apoptosis, such as TRAIL, to explore potential therapeutic synergy. As seen in Fig. 6, the combination of the XAntag 1396-11 with TRAIL resulted in significant growth inhibition measured by MTS assay compared with either therapeutic modality alone, for Capan1 and Panc1 cell lines (all P values <0.01). The results were particularly striking for Capan1, which is completely resistant to TRAIL-mediated apoptosis, where this resistance was circumvented by the addition of XAntag 1396-11 (P = 0.0001).
Figure 6.

XAntag 1396-11 shows apparent synergy with TRAIL against cultured pancreatic cancer cells. Viability assays were done for cells cultured for 96 h with XAntag 1396-11 alone (2 μmol/L), TRAIL alone (100 ng), or the combination (COMBO) using two pancreatic cancer cell lines, Panc1 and Capan1. Growth inhibition was compared relative to vehicle control. All experiments were done in triplicate. Columns, mean; bars, SE.
Colony assays in soft agar were done to determine whether the combination of XAntag and TRAIL can affect clonogenic growth. Greater inhibition of colony formation was observed at 2 weeks in Capan1 cells when exposed to the combination therapy versus either modality alone (Fig. 7). Whereas neither therapeutic modality showed independent efficacy at the dosages used, the combination resulted in dramatic and significant inhibition of colony formation (P < 0.0001 and 0.0006, respectively). The results were less striking for Panc1 cells, which showed intrinsic TRAIL sensitivity (data not shown). However, even in Panc1, we found a significant decrease in colony growth with the combination therapy (P = 0.0135).
Figure 7.

XAntag 1396-11 shows apparent synergy with TRAIL against cultured pancreatic cancer cells. A, colony assays in soft agar were done using Capan1 cells exposed to XAntag 1396-11 alone (100 ng), TRAIL alone (20 ng), and the combination exposure. Representative results, showing photographs of stained soft agar plates at 2 wks. B, colonies were counted. Columns, mean; bars, SE.
Next, experiments were done to determine whether XAntags can radiosensitize pancreatic cancer cells. Accordingly, colony assays in soft agar were done for cells pretreated with XAntag alone, radiation alone, and the combination. For both Capan1 and Panc1 cells, pretreatment with the XAntag 1396-11 significantly increased the sensitivity to radiation at the lower dose of 2 Gy (Fig. 8A shows soft agar plates for Panc1, whereas Fig. 8B enumerates the colony counts in both cell lines). The results were particularly striking for Panc1 cells, where nearly complete abrogation of colony formation was observed with the combination of 2 Gy irradiation and 2 μmol/L 1396-11 (P = 0.003). Significant enhancement of radiation effect was also observed at 6 Gy with Capan1 cells (P = 0.04). Panc1 colony formation was abrogated by radiation itself at the higher dose; therefore, an effect of combination therapy could not be discerned.
Figure 8.

XAntag 1396-11 shows apparent synergy with radiation against cultured pancreatic cancer cells. Colony assays in soft agar were done to evaluate the effects of XAntag on radiosensitization of pancreatic cancer cell lines. In addition to the control arm, five treatment arms were generated for Panc1 cells before plating for colony assay: equal numbers of cells were pretreated overnight with XAntag 1396-11 (2 μmol/L) without exposure to radiation, exposed to two escalating dosages of radiation (2 and 6 Gy) without pretreatment with XAntag, or exposed to radiation (2 and 6 Gy) following an overnight incubation with XAntag 1396-11. The assays were terminated at 2 wks. A, representative plates. B, colonies were counted. Columns, mean; bars, SE.
Finally, we examined whether XAntags may apparently synergize with gemcitabine, which is the standard of care for advanced pancreatic cancers. In vitro experiments confirmed that the combination of XAntag and gemcitabine results in enhanced growth inhibition compared with gemcitabine alone, and these effects on cell growth are observed even at the lowest range of gemcitabine dose (0.4 μmol/L) used (Fig. 9).
Figure 9.

XAntags sensitize pancreatic cancer cell lines to gemcitabine. Panc1 and BxPC3 cells were treated with escalating dosages of gemcitabine (0.4, 0.8, 1, and 2 μmol/L) with or without a constant dose of XAntag 1396–11 (2 μmol/L). Cell viability assays were done at 96 h. Cell viability assays were normalized relative to control (vehicle treated) cells. Columns, mean of triplicates; bars, SE.
Discussion
The IAP family of antiapoptotic proteins is composed of eight members in humans, including XIAP (30). Various members of this family are overexpressed in many human cancers (4, 5). Considerable experimental evidence has been presented to suggest that IAP proteins contribute to chemoradiation resistance in solid tumors and that down-regulation of IAP proteins increases the sensitivity of neoplastic cells to a broad range of therapeutic agents (15, 16). Thus, inhibition of XIAP function in a variety of human cancer cell lines has been shown to positively affect their response to proapoptotic death receptor ligands, such as TRAIL, as well as to conventional cytotoxic anticancer drugs and to radiation (31–38). XIAP exerts it antiapoptotic effects by sequestration of essential “executioner” caspases (e.g., caspase-3 and caspase-7), and targeted deletion of XIAP by homologous recombination has shown that this molecule is a nonredundant mediator of TRAIL sensitivity in cancer cells (39).
Given the seminal importance of XIAP and other IAP proteins in mediating chemoresistance, it is not surprising that there has been a concerted effort to therapeutically target this protein family in human cancer. Multiple investigators have used RNA interference to down-regulate XIAP transcripts in cancer cell lines and have shown the consequent restoration of sensitivity to TRAIL, as well as to other chemotherapeutic agents, such as paclitaxel (31, 32, 34, 40). Although encouraging, these studies are currently limited to In vitro assays, primarily due to the absence of reliable delivery systems for synthetic small interfering RNA in vivo. A second approach for targeting XIAP, which is currently in early-phase clinical trials, involves the use of antisense oligonucleotides (33, 38, 41–43). Specifically, AEG35156 is a second-generation XIAP antisense oligonucleotide that is currently undergoing evaluation as monotherapy, as well as in combination with docetaxel (Taxotere) and other agents, in solid cancers and in leukemia (44). Unlike small interfering RNAs, the antisense oligonucleotide approach seems to have had greater in vivo promise to date, and the results of these trials are eagerly awaited. Overall, these reports underscore the potential clinical relevance of XIAP inhibition in human cancer and have provided the motivation to explore a third alternative (i.e., synthetic small molecules for targeting XIAP In vitro and in vivo). In 2004, Schimmer et al. (23) identified multiple small molecules showing potent XIAP inhibitory activity from a large scale combinatorial library screen. Since that time, the preclinical efficacy of these chemical XAntags has been confirmed for several types of solid and hematologic malignancies (reviewed in ref. 22). Unlike antisense oligonucleotides, which depend on absolute reduction of cellular XIAP levels for their effects, the XAntags are functional inhibitors of this protein that disrupt the interaction of XIAP with downstream executioner caspases (21).
Our motivation to explore XIAP inhibition as a therapeutic strategy in pancreatic cancers arose from the dismal prognosis of this malignancy (<5% 5-year survival rates) and the lack of significant efficacy for most conventional chemotherapeutic modalities (1). We were also encouraged by previous reports showing the radiosensitization of cancer cells by XIAP down-regulation (35, 38), a finding with clinical implications for a modestly radiosensitive malignancy, such as pancreatic cancer (2). We show here that XIAP is overexpressed in the majority of human pancreatic cancers, as assessed by immunohistochemistry, confirming for the first time data that have been reported previously only in cell line models (13, 14). Although the numbers of cases analyzed are relatively small, the trend clearly underscores that XIAP up-regulation is a common phenomenon in pancreatic cancer. The mechanism of XIAP overexpression in pancreatic cancer is unknown, although recently, the ras oncogene has been reported to up-regulate XIAP levels independent of epidermal growth factor receptor signaling in intestinal epithelial cells (45); given that activated KRAS is a signature mutation of pancreatic cancer (present in 90% of cases; ref. 46), it is tempting to speculate that the observed XIAP up-regulation in this malignancy might be oncogenic ras dependent.
Analogous to other solid cancers, the potential clinical effect of XIAP down-regulation has been shown recently in pancreatic cancers. These studies used RNA interference in pancreatic cancer cell lines to improve their responsiveness to TRAIL (47) or to conventional metabolites, such as 5-fluorouracil and gemcitabine (40). Although these data are encouraging and support our major findings, the ability to induce RNA interference in vivo in the clinical setting has not yet been shown. In contrast, the small-molecule antagonists described in this study are readily suited for systemic administration. In our limited studies, as well as previous reports (23, 25, 26), XAntags do not seem to cause incidental adverse effects in xenograft-bearing mice. We were able to achieve impressive down-regulation of pancreatic cancer xenograft growth in vivo with XAntag monotherapy, further validating XIAP as a target in pancreatic cancer. Our experiments also show that the effects of XAntags on pancreatic cancer cells are associated with activation of downstream caspases (caspase-3 and caspase-7) In vitro and with striking increases in intratumoral apoptosis and cleaved caspase-3 levels in treated tumor xenografts.
Our study also shows the potential for synergy between XAntags and other therapeutic modalities commonly used for pancreatic cancer (e.g., radiation and gemcitabine). We show that both In vitro growth (as measured by MTS viability assays) and clonogenic survival (as measured by colony growth in soft agar) are impeded by combination therapy with XAntag, often at dosages where each individual modality has only modest or minimal effects. The apparent synergy with radiation is particularly relevant as “neoadjuvant” therapies for borderline resectable pancreatic cancer are being increasingly adapted (48, 49). Thus, the addition of systemic XAntag to these protocols has the potential for achieving greater locoregional disease control. Our results also confirm that XAntags can overcome resistance to TRAIL in cultured pancreatic cancers, thus offering the possibility for combination therapy with death receptor ligands and XAntags. The recent availability of monoclonal death receptor agonist antibodies provides an avenue for such protocols in advanced disease states, where the disease is surgically inoperable (50, 51).
In summary, our report shows that XIAP protein is overexpressed in the majority of human pancreatic cancers and suggests that targeting of this protein with small-molecule antagonists is a valid therapeutic strategy for pancreatic cancer. Future studies are warranted in preclinical models of pancreatic cancer, examining the in vivo efficacy of XAntag in combination with death receptor agonists, radiation and/or antimetabolites, to determine therapeutic efficacy and toxicity.
Acknowledgments
Grant support: R01CA113669, R21DK072532, AACR-PanCAN Career Development Award, the Sol Goldman Pancreatic Cancer Research Center and the Lustgarten Foundation for Pancreatic Cancer Research (A. Maitra). U19 CA113318 and the California Tobacco-Related Disease Program (J.C. Reed).
References
- 1.Yeo TP, Hruban RH, Leach SD, et al. Pancreatic cancer. Curr Probl Cancer. 2002;26:176–275. doi: 10.1067/mcn.2002.129579. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Debatin KM. Apoptosis pathways in cancer and cancer therapy. Cancer Immunol Immunother. 2004;53:153–9. doi: 10.1007/s00262-003-0474-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 6.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–10. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira CG, van der Valk P, Span SW, et al. Assessment of IAP (inhibitor of apoptosis) proteins as predictors of response to chemotherapy in advanced non-small-cell lung cancer patients. Ann Oncol. 2001;12:799–805. doi: 10.1023/a:1011167113067. [DOI] [PubMed] [Google Scholar]
- 8.Liu SS, Tsang BK, Cheung AN, et al. Anti-apoptotic proteins, apoptotic and proliferative parameters, and their prognostic significance in cervical carcinoma. Eur J Cancer. 2001;37:1104–10. doi: 10.1016/s0959-8049(01)00085-5. [DOI] [PubMed] [Google Scholar]
- 9.McEleny KR, Watson RW, Coffey RN, O'Neill AJ, Fitzpatrick JM. Inhibitors of apoptosis proteins in prostate cancer cell lines. Prostate. 2002;51:133–40. doi: 10.1002/pros.10061. [DOI] [PubMed] [Google Scholar]
- 10.Shiraki K, Sugimoto K, Yamanaka Y, et al. Overexpression of X-linked inhibitor of apoptosis in human hepatocellular carcinoma. Int J Mol Med. 2003;12:705–8. [PubMed] [Google Scholar]
- 11.Endo T, Abe S, Seidlar HB, et al. Expression of IAP family proteins in colon cancers from patients with different age groups. Cancer Immunol Immunother. 2004;53:770–6. doi: 10.1007/s00262-004-0534-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakagawa Y, Hasegawa M, Kurata M, et al. Expression of IAP-family proteins in adult acute mixed lineage leukemia (AMLL) Am J Hematol. 2005;78:173–80. doi: 10.1002/ajh.20285. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Cao Z, Yan H, Wood WC. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res. 2003;63:6815–24. [PubMed] [Google Scholar]
- 14.Trauzold A, Schmiedel S, Roder C, et al. Multiple and synergistic deregulations of apoptosis-controlling genes in pancreatic carcinoma cells. Br J Cancer. 2003;89:1714–21. doi: 10.1038/sj.bjc.6601330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–85. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 16.Reed JC. Apoptosis-targeted therapies for cancer. Cancer cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- 17.Verhagen AM, Coulson EJ, Vaux DL. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-7-reviews3009. REVIEWS3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–4. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 19.Shiozaki EN, Chai J, Rigotti DJ, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11:519–27. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 20.Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24:645–55. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Cuddy M, Samuel T, et al. Cellular, biochemical, and genetic analysis of mechanism of small molecule IAP inhibitors. J Biol Chem. 2004;279:48168–76. doi: 10.1074/jbc.M405022200. [DOI] [PubMed] [Google Scholar]
- 22.Schimmer AD, Dalili S, Batey RA, Riedl SJ. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 2006;13:179–88. doi: 10.1038/sj.cdd.4401826. [DOI] [PubMed] [Google Scholar]
- 23.Schimmer AD, Welsh K, Pinilla C, et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 24.Berezovskaya O, Schimmer AD, Glinskii AB, et al. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005;65:2378–86. doi: 10.1158/0008-5472.CAN-04-2649. [DOI] [PubMed] [Google Scholar]
- 25.Carter BZ, Gronda M, Wang Z, et al. Small-molecule XIAP inhibitors derepress downstream effector caspases and induce apoptosis of acute myeloid leukemia cells. Blood. 2005;105:4043–50. doi: 10.1182/blood-2004-08-3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kater AP, Dicker F, Mangiola M, et al. Inhibitors of XIAP sensitize CD40-activated chronic lymphocytic leukemia cells to CD95-mediated apoptosis. Blood. 2005;106:1742–8. doi: 10.1182/blood-2005-02-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Sun C, Olejniczak ET, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–8. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 28.Karikari CA, Mullendore M, Eshleman JR, et al. Homozygous deletions of methylthioadenosine phosphorylase in human biliary tract cancers. Mol Cancer Ther. 2005;4:1860–6. doi: 10.1158/1535-7163.MCT-05-0103. [DOI] [PubMed] [Google Scholar]
- 29.Hustinx SR, Hruban RH, Leoni LM, et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: a potential new target for therapy. Cancer Biol Ther. 2005;4:83–6. doi: 10.4161/cbt.4.1.1380. [DOI] [PubMed] [Google Scholar]
- 30.Reed JC, Doctor KS, Godzik A. The domains of apoptosis: a genomics perspective. Sci STKE. 2004;2004:re9. doi: 10.1126/stke.2392004re9. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi Y, Shiraki K, Fuke H, et al. Targeting of X-linked inhibitor of apoptosis protein or survivin by short interfering RNAs sensitize hepatoma cells to TNF-related apoptosis-inducing ligand- and chemotherapeutic agent-induced cell death. Oncol Rep. 2005;14:1311–6. [PubMed] [Google Scholar]
- 32.McManus DC, Lefebvre CA, Cherton-Horvat G, et al. Loss of XIAP protein expression by RNAi and antisense approaches sensitizes cancer cells to functionally diverse chemotherapeutics. Oncogene. 2004;23:8105–17. doi: 10.1038/sj.onc.1207967. [DOI] [PubMed] [Google Scholar]
- 33.Amantana A, London CA, Iversen PL, Devi GR. X-linked inhibitor of apoptosis protein inhibition induces apoptosis and enhances chemotherapy sensitivity in human prostate cancer cells. Mol Cancer Ther. 2004;3:699–707. [PubMed] [Google Scholar]
- 34.Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs BS, Lindner DJ, Borden EC. Downregulation of Bcl-2, FLIP, or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2004;11:915–23. doi: 10.1038/sj.cdd.4401416. [DOI] [PubMed] [Google Scholar]
- 35.Roa WH, Chen H, Fulton D, et al. X-linked inhibitor regulating TRAIL-induced apoptosis in chemoresistant human primary glioblastoma cells. Clin Invest Med. 2003;26:231–42. [PubMed] [Google Scholar]
- 36.Ng CP, Zisman A, Bonavida B. Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: role of XIAP in resistance. Prostate. 2002;53:286–99. doi: 10.1002/pros.10155. [DOI] [PubMed] [Google Scholar]
- 37.Ng CP, Bonavida B. X-linked inhibitor of apoptosis (XIAP) blocks Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis of prostate cancer cells in the presence of mitochondrial activation: sensitization by overexpression of second mitochondria-derived activator of caspase/direct IAP-binding protein with low pl (Smac/DIABLO) MolCancer Ther. 2002;1:1051–8. [PubMed] [Google Scholar]
- 38.Cao C, Mu Y, Hallahan DE, Lu B. XIAP and survivin as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene. 2004;23:7047–52. doi: 10.1038/sj.onc.1207929. [DOI] [PubMed] [Google Scholar]
- 39.Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 2004;64:3006–8. doi: 10.1158/0008-5472.can-04-0046. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Jian Z, Xia K, et al. XIAP is related to the chemoresistance and inhibited its expression by RNA interference sensitize pancreatic carcinoma cells to chemotherapeutics. Pancreas. 2006;32:288–96. doi: 10.1097/01.mpa.0000218314.67111.fb. [DOI] [PubMed] [Google Scholar]
- 41.Tong QS, Zheng LD, Wang L, et al. Downregulation of XIAP expression induces apoptosis and enhances chemotherapeutic sensitivity in human gastric cancer cells. Cancer Gene Ther. 2005;12:509–14. doi: 10.1038/sj.cgt.7700813. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y, Cherton-Horvat G, Dragowska V, et al. Antisense oligonucleotides targeting XIAP induce apoptosis and enhance chemotherapeutic activity against human lung cancer cells in vitro and in vivo. Clin Cancer Res. 2003;9:2826–36. [PubMed] [Google Scholar]
- 43.Bilim V, Kasahara T, Hara N, Takahashi K, Tomita Y. Role of XIAP in the malignant phenotype of transitional cell cancer (TCC) and therapeutic activity of XIAP antisense oligonucleotides against multidrug-resistant TCC in vitro. Int J Cancer. 2003;103:29–37. doi: 10.1002/ijc.10776. [DOI] [PubMed] [Google Scholar]
- 44.Lacasse EC, Cherton-Horvat GG, Hewitt KE, et al. Preclinical characterization of AEG35156/GEM 640, a second-generation antisense oligonucleotide targeting X-linked inhibitor of apoptosis. Clin Cancer Res. 2006;12:5231–41. doi: 10.1158/1078-0432.CCR-06-0608. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z, Li H, Derouet M, et al. ras Oncogene triggers up-regulation of cIAP2 and XIAP in intestinal epithelial cells: epidermal growth factor receptor-dependent and -independent mechanisms of ras-induced transformation. J BiolChem. 2005;280:37383–92. doi: 10.1074/jbc.M503724200. [DOI] [PubMed] [Google Scholar]
- 46.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- 47.Vogler M, Durr K, Jovanovic M, Debatin KM, Fulda S. Regulation of TRAIL-induced apoptosis by XIAP in pancreatic carcinoma cells. Oncogene. 2007;26:248–57. doi: 10.1038/sj.onc.1209776. [DOI] [PubMed] [Google Scholar]
- 48.Raut CP, Evans DB, Crane CH, Pisters PW, Wolff RA. Neoadjuvant therapy for resectable pancreatic cancer. Surg Oncol Clin N Am. 2004;13:639–61. ix. doi: 10.1016/j.soc.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Pisters PW, Wolff RA, Crane CH, Evans DB. Combined-modality treatment for operable pancreatic adenocarcinoma. Oncology (Huntingt) 2005;19:393–404. 9; discussion 9 – 10, 12-6. [PubMed] [Google Scholar]
- 50.Griffith TS, Rauch CT, Smolak PJ, et al. Functional analysis of TRAIL receptors using monoclonal antibodies. J Immunol. 1999;162:2597–605. [PubMed] [Google Scholar]
- 51.Merchant MS, Yang X, Melchionda F, et al. Interferon γ enhances the effectiveness of tumor necrosis factor-related apoptosis-inducing ligand receptor agonists in a xenograft modelof Ewing's sarcoma. Cancer Res. 2004;64:8349–56. doi: 10.1158/0008-5472.CAN-04-1705. [DOI] [PubMed] [Google Scholar]