

Abstract
This critical review will focus on the application of shape-persistent receptors for anions that derive their rigidity and optoelectronic properties from the inclusion of arylethynyl linkages. It will highlight a few of the design strategies involved in engineering selective and sensitive fluorescent probes and how arylacetylenes can offer a design pathway to some of the more desirable properties of a selective sensor. Additionally, knowledge gained in the study of these receptors in organic media often leads to improved receptor design and the production of chromogenic and fluorogenic probes capable of detecting specific substrates among the multitude of ions present in biological systems. In this ocean of potential targets exists a large number of geometrically distinct anions, which present their own problems to the design of receptors with complementary binding for each preferred coordination geometry. Our interest in targeting charged substrates, specifically how previous work on receptors for cations or neutral guests can be adapted to anions, will be addressed. Additionally, we will focus on the design and development of supramolecular arylethynyl systems, their shape-persistence and fluorogenic or chromogenic optoelectronic responses to complexation. We will also examine briefly how the “chemistry in the cuvet” translates into biological media.
Introduction
This critical review highlights the role functionalized ethynylarenes play in supramolecular sensor design. We begin with a non-comprehensive survey of representative examples of ethynyl- and butadiynyl-based receptors for neutral and biologically relevant guests and the translation of these systems into sensors for anionic species. We conclude with an overview of related work in our lab and provide a brief survey of the burgeoning field of arylethynyl probe development for biologically relevant anions.
1.1 Arylethynyl receptors
Many synthetic receptors rely on some degree of preorganization in their approach to achieving high affinities and selectivities. Flexible receptors can achieve increased size-selectivity and preorganization through macrocyclization, and numerous, in-depth reviews have been written which cover the usefulness of the macrocyclic effect in increasing binding affinities.1-4 However, macrocyclic receptors tend to exhibit slow binding kinetics1,2 and indeed many sensor applications rely more upon kinetic selectivity rather than a large contribution from preorganization of the receptor. In addition it can be the case that the guest bound most strongly is not the guest that gives rise to the largest colorimetric or fluorometric response, which is difficult to predict. This gives rise to a delicate balance in designing probes capable of binding a target selectively, while also exhibiting a response for that specific analyte.
There exist many examples of conformationally rigid receptors,5-7 most of which exploit the inherent rigidity in conjugated π-systems. Expanded porphyrins,8,9 calixpyrroles or related calixarenes,10-12 and innumerable other nitrogen13,14 or oxygen containing heterocycles15 have been synthesized and their affinities for both ionic and neutral guests studied. In much the same fashion phenylacetylenes have provided structure and optoelectronic handles to a multitude of coordination and host-guest complexes.16 Macrocyclic host molecules as well as shape-persistent acyclic ligands for metal ions have benefited from their linear, rigid geometries and relatively simple derivatization.17-20
In some cases the acetylenes themselves have served as the binding site for transition metal guests.21 The synthesis and characterization of a Ni0 complex of dehydroannulene 1 was reported in 1985.22 More often, however, the rigid acetylenic linker serves to reinforce a desired binding conformation. The synthesis and characterization of phenylacetylenic macrocycles capable of differentiating transition metals as well as serving as rudimentary proton sensors has also been achieved.23-25 Twistophane 2 was shown to signal PdII and HgII by a distinct fluorescence quenching response, and to signal H+ by bathochromic shifting and quenching of the fluorescence emission.23
Phenylacetylenes have well-studied fluorescence emission properties,26, 27 and the optoelectronic response to perturbation of their ground-state conformations can be a useful spectroscopic handle. The conjugation of donor and acceptor groups via alkyne linkages28 is much studied and this charge transfer process has been used as a fluorescent handle for the visualization of binding events.29 Dibutylamine-functionalized dehydrobenzoannulene 3 was found to shift fluorescence emission based upon H+ concentration (as trifluoroacetic acid). Interestingly, it was found that emission shifting was dependent upon stepwise protonation of the dibutylaniline moieties, which indicated independent manipulation of the frontier molecular orbital energies and thus tunable charge transfer pathways.

In 2002 ethynyl-linked pyrrole-naphthyridine compound 4 was found to selectively bind glucopyranoside.30 The free receptor was found to adopt a slightly twisted conformation that exhibited a fluorescence emission maximum at 475 nm (τf ≈ 1.25 ns), which decreased upon addition of octyl β-D-glucopyranoside (OGU). A new emission band at 535 nm (τf ≈ 0.95 ns) grew in intensity as a 1:1 complex was formed. The association constants for this complex determined by both fluorescence and UV-Vis absorbance measurements were in good agreement (Ka = 5.3 × 103 M−1 and Ka = 4.8 × 103 M−1 in CH2Cl2, respectively). Interestingly, the association constant for octyl β-D-galactopyranoside was found to be only 1800 M−1, which is impressive considering these saccharides differ only in the orientation of the 4-hydroxyl group. The optoelectronic response in this system was attributed to a rigidification and planarization of the pyrrole-naphthyridine moieties, which served to enhance the charge transfer in this D-π-A-A-π-D system.
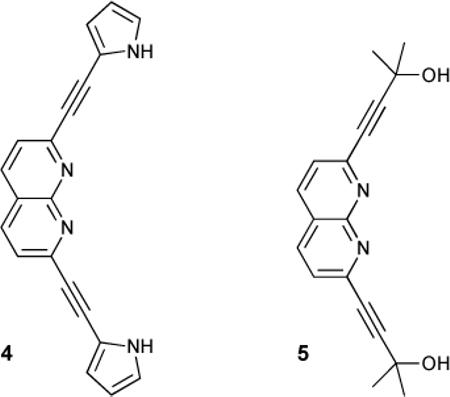
To test this an analogous receptor 5 was synthesized in which the binding sites were not conjugated through the core. Binding of OGU to this receptor was weaker due to the relative differences in acidity at the binding sites, but fluorescence emission intensity still increased, albeit not bathochromically shifted as could be expected by rigidification in this system. Additionally, modification of the pyrrolyl moieties of 4 to indolyl units gave the normally CD-silent receptor strong fluorescence-detected CD spectra upon binding, due to chirality transfer from the substrates.31
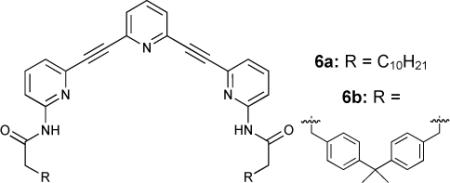
A simple illustration of how flexible macrocyclization of rigid arylethynyl scaffolds can increase binding constants is found in two pyridine based systems 6a and 6b.32 Acyclic 6a bound ribofuranosides poorly in CDCl3 (Ka = 30 M−1) but macrocyclization of this cleft-like receptor increased binding constants two orders of magnitude (Ka = 2400 M−1). Related poly(ethyleneglycol) linked derivatives of this receptor motif allowed the expanded terpyridine core to adopt a slightly wider conformation while maintaining preorganization.32a Due to this longer, more flexible linker these receptors exhibited still higher affinities for larger monosaccharides.
A number of phosphorylated binol-based macrocycles have been investigated as receptors for neutral guests, such as saccharides.33-35 The polyanionic cavities in receptors 7a and 7b were targeted at the hydroxyl moieties of the sugars, a strategy which proved effective even in slightly competitive media. It was found that macrocyclization can be a hindrance in these very rigid systems; cyclotrimer 7a was found to be incapable of binding monosaccharides in the cavity, but was still capable of binding OGU (Ka = 3500 M−1 in CD3CN) ostensibly through a face-to-face interaction.34 However, cyclotetramer 7b was large enough to bind pyranose in its cavity, with a corresponding increase in association constant (Ka = 4500 M−1 for OGU), while extended cyclotetramer 8 was selective for disaccharides over monosaccharides even in very competitive media (Ka's of 10,700 – 12,500 M−1 in 88:12 CD3CN-CD3OD).35 The rigidity of the macrocyles in these cases helped to reinforce their size selectivity between guests.
This size regulation can also be extended to polymers of phenylacetylenic subunits. Polymers of 9 were found to exhibit saccharide-dependent induction of chirality.36

Hydrogen bonding with saccharide guests was reinforced by the rigidity and relative angle of the alkyne linkers between pyridine units, which necessarily directed all of the hydrogen-bond accepting lone pairs to the interior of the cavity. This reinforcement also biased the polymer for the 2,3,4,6-OH groups of β-glucoside from other monosaccharides or their derivatives. The above examples, while by no means exhaustive, illustrate a few of the design strategies involved in engineering a selective and sensitive fluorescent probe, and how phenylacetylenes offer one such viable design pathway.
1.2 Approaches to receptor design
A viable non-covalent receptor must strike a balance between selectivity, solubility, robustness and, in the case of sensors, signalling or response.1-3 There are a few distinct strategies for designing effective non-covalent, fluorionophores. These can be classified simply as either “FSR” (fluorophore-spacer-receptor) or displacement.37-39
In the FSR system, an independently tunable binding site is linked covalently to a signalling unit. The role of the signalling unit is simply to transduce the chemical information upon coordination from the binding site into a specific fluorescent response. This historically has been the most common approach to engineering fluorogenic hosts to complex ionic targets, and many reviews exist.37,39 There has been considerable success with what could be called a noncovalent version of FSR.38 This dye-displacement method relies upon careful modulation of a bound fluorophore or chromophore that is designed to have a weaker affinity for the host than the target analyte.
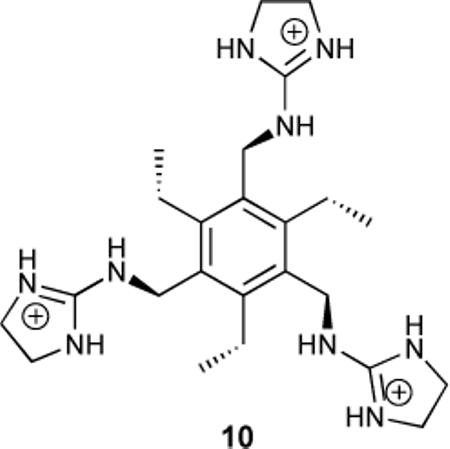
In 1998 there was reported the synthesis of the hexa-substituted benzene-based tripodal receptor 10 that used three guanidinium groups as binding sites.40 Carboxyfluorescein was found to bind with a Ka = 4.7 × 103 M−1 in aqueous buffer, while citrate bound with Ka = 2.9 × 105 M−1. Modulation of the pKa of the phenolic proton of carboxyfluorescein was implicated in the decreased fluorescence of free dye versus the host-dye complex.
Alternatively, replacement of a non-conjugated spacer with a conjugated linker has proven an interesting approach to receptor design. It allows for discrete modulation of the optoelectronic response of a fluorogenic receptor via conformation rather than just electronic effects from guest inclusion, as well as rigidifying the receptor. This provides an element of size-based recognition and can be exploited to modulate the fluorescent response, either through twisting of the π-system41-43 or extension of excitation/emission into the near-IR for use in biological systems.44 Regardless of the desired properties, this approach is closely related to traditional FSR receptors, although in this instance the spacer plays a more active role in enforcing receptor conformation. Integration of these components can access smaller molecular sensing systems as well as provide additional information about the binding geometry via modulation of optoelectronic response.

1.3 Modularity
Convergent syntheses are a common route in molecule assembly. The well-studied techniques of arene-acetylene cross-coupling can allow the supramolecular chemist to synthesize and derivatize a wide array of functional subunits with spacers that transduce signal themselves, rather than separating these into independent moieties in the receptor.45 These can then be easily linked together in a convergent fashion through a multitude of cross-coupling reactions, most of which are characterized by their relatively benign reaction conditions.46 This modular approach to synthesis allows for quick and effective screening of candidates by allowing quick and subtle changes to each building block. Most of the phenylacetylene work mentioned thus far has made use of sequential cross-couplings of independently synthesized subunits, and the budding field of alkyne metathesis may open up another efficient and modular strategy in the synthesis of ethynyl or butadiynyl-containing small molecules.47,48 In tuning binding parameters such as cavity size, bite angle or the number or relative position of binding sites, secondary properties (i.e., optoelectronic response, photostability or solubility) of the receptor scaffold can be adjusted as well, and simplicity when modulating all of these parameters is key to quick and efficient discovery of “Goldilocks” candidates for viable receptors.
1.4 Switchability in Sensitivity
Controlling the affinity of a host for a specific analyte via allosteric modulation (e.g., pH or ionic environment) is an interesting tool for modifying the selectivity of a binding site.49,50 This allosteric behaviour is relatively rare in synthetic receptors, but has particular implications for designing biomimetic receptors that will operate within the diminished pH windows in biological systems, where either “turn-on” or “turn-off” binding may be desired in response to pH changes in cellular compartments. Notably, switchable conformational control has been shown in rotaxanes51 and in “FSR” type hydrogen-bonding receptors.52,53 As an example, the switchable complexation of uracil in two amine-triazine-crown ether receptors has been recently reported.52 Protonation of the amine initiated an intramolecular hydrogen-bonding interaction between the ammonium moiety and the crown ether that sterically blocked the triazine binding site.
Switchability in a sensor can also come from conformational change in induced-fit sensors. In these cases clever design can limit the conformational degrees of freedom such that only a single analyte gives the most intense response. For example, this can be accomplished by appending fluorophores such that conformational change upon binding brings them into proximity for excimer formation (e.g., FRET)54 or redox active groups can have their environments changed in such a way as to give rise to a known modulation in potential for non-optical sensing.55,56 Phenylacetylenic systems can be exploited when engineering optically responsive receptors in much the same way as discussed below.
Extended bis-ethynylpyridine compounds 11 and 12 were reported to bind deoxyribosides in CDCl3 (Ka = 690 M−1 for 11 and Ka = 19000 M−1 for 12).57 The modification of the hydrogen bond accepting pyridine lone pair to a hydrogen bond donating pyridinone was responsible for this significant increase in affinity. This switchability, although engineered into the receptor during the synthesis, has interesting implications for the design of receptors capable of binding anionic guests.
As an example, pyridylthiourea 13a exhibits switchable affinities for either acetate or halide anions.58 Protonation as a switch in this case provides an additional hydrogen bonding contact which further stabilizes the spherical halides. Treating the protonated receptor with excess acetate led to deprotonation and binding of the residual acetate anion. Replacing the arene C-H with a methyl group yielded 13b, which had no affinity for acetate or hydrogen halides, thus highlighting the role arene C-H hydrogen bonding can play in designing receptors.
2.1 Tuning Receptors for Anionic Targets
As anions have become the focus of more and more research efforts, the modification and application of known cation receptor design criteria for anionic targets has grown as well. For many years the importance of anions in the natural world was overlooked. Relatively benign chloride, carbonate and sulfate, as well as toxic arsenate anions, are found naturally in water the world over,61 or in runoff and acid rain in the case of industrial pollutants.62 Anthropogenic anions can be expanded to include phosphate and nitrates from agriculture,63 or pertechnetate and perchlorate from industrial waste streams.64,65 Their ubiquity in the natural world has lead to an increase of research in this field with modest advances made in the characterization and sequestration of electron-rich wastewater contaminants. This has led to elegant designer nanomaterials that offer selectivity for some of these myriad problematic anionic pollutants present in both developing and developed countries.66,67b
In spite of their prevalence, targeting anionic substrates has its own inherent challenges. Their low charge to radius ratio, high solvation energies, polarizability and the large range of preferred geometries (or lack thereof) make them difficult targets in competitive media.1-3 Protic solvents tend to form extremely stable hydrogen bonds with most anions, which mandates an extremely strong host-guest interaction if any binding is to be accomplished. On the other hand, relatively nonpolar solvents give rise to ion-pairing, which can have a significant negative influence on binding ability. In addition, many biological anions exist only within narrow pH windows. Nature has many strategies to overcome these difficulties, a fact illustrated beautifully by the complexity of many of the natural receptors whose function depends upon their selectivity for specific anions. A particularly elegant example was the elucidation of the StCIC and EcCIC chloride ion channels via X-ray crystallography in 2002.68 These CIC ion channels are selective for Cl− and Br− through partial positive charges within the channels, rather than a fully electrostatic interaction with the nearby lysine and arginine residues which would bind too strongly and inhibit the function of the channel. Interestingly, the gating mechanism necessary for the channel to function is hypothesized to depend on two chloride binding events, and that even small changes in the anionic substrate, i.e., from chloride to bromide anion, alter the operational efficiency of the channel.69 In spite of insights such as these into the complexity of Nature's use of anion coordination chemistry in cell regulation, our understanding of how the properties of specific anions affect changes in these systems is severely limited.
2.2 Common functionalities for anion receptors and probes
This review focuses on receptors with linked sp-carbon atoms to which are append a variety of functional groups: arenes such as pyrrole, indole, carbazole, or carbonyl-containing amide, urea, thiourea or sulfonamide functionalities.78 This combination can translate into well-defined binding sites, whether they be based strictly on polycyclic arenes or involve extension via acetylenic linkers, as well as impart distinct optoelectronic properties to these receptors. While the inclusion of metal centers is an important design motif,59 contributions from organometallic receptors for anions have been recently reviewed60 and are beyond the scope of this review.
An early example of an arene-alkyne based receptor attempted to reinforce face-to-face interactions between the host and guest via multiple butadiyne linkages that were not in conjugation. Investigation of the binding affinities for heterotricyclic receptors 14a-d found that these bound large, flat substrates well with the highest binding constants observed for the largest substrates (e.g., terephthalate2− > 2,6-naphthalenedicarboxylate2− > 2,6-anthraquinonedisulfonate2i) as these fit the large, reinforced binding pocket better.70
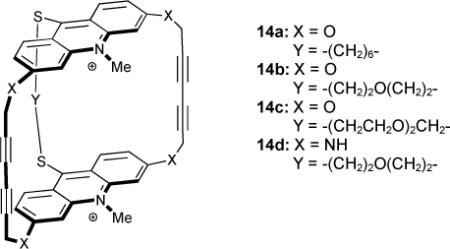
The binding constants with neutral adenosine were measured in an aqueous buffer (log Ka = 4.00, 3.87, 3.86 and 4.00, 14a-d respectively) and followed the same trend with doubly charged AMP (log Ka = 4.08, 3.79, 3.92 and 4.08).
Alkyne-linked macrocyclic indole and indolocarbazole receptors 15 and 16 were synthesized and their anion affinities probed in acetonitrile.71 It was found that further rigidification of receptor 15 to 16 very modestly increased binding constants (e.g. Cl− Ka = 1.5 × 106 M−1 to 2.1 × 106 M−1, N3− Ka = 8.8 × 105 M−1 to 9.1 × 105 M−1, H2PO4− Ka = 2.1 × 106 M−1 to 3.2 × 106 M−1). A crystal structure of the 16•Cl− complex revealed that the binding pocket was slightly too small to incorporate the Cl− anion completely. Both of these receptors exhibited increased affinities over previously studied acyclic receptors 17 and 18.72 Rigidification in this system had the same effect as in the macrocyclic versions (17: Ka = 5100 M−1 to 18: 110,000 M−1 for Cl−). In more recent work, 19 was used to study the preferred binding geometry of azide, halide and oxoanionic guests.73
Changing the ethynyl linker in 16 to butadiynyl in 19 decreased the affinity for Cl− anion over four fold, although the affinity for Br− and I− anions was greater for the larger cavity of 19. There was no affinity for acetate, but other polyoxo-anions (H2PO4−, NO3− and HSO4−) exhibited increased affinities of one to two orders of magnitude. Azide bound in an orthogonal fashion (normal to the macrocycle plane) in ethynyl linked 16 with one N atom in the cavity, but butadiynyl 19 had a large enough binding pocket to fully accommodate azide in a linear fashion, with a concomitant increase in binding constant (Ka = 2300 M−1 to 81,000 M−1). The binding events in these receptors were followed by UV-Vis or 1H-NMR spectroscopy; the change in their fluorescence emission was not reported. The cleft-like derivative 20 was studied as well and found to strongly and selectively bind H2PO4− (Ka = 1.1 × 105 M−1) due to the inclusion of two additional hydrogen bond acceptors in the ethynyl “arms”.74 Although the binding constant for this system is smaller than for their previously reported macrocyclic receptor 16, the selectivity increased, highlighting the well-designed binding pocket for the geometry of the desired guest. As expected, when binding guests with hydrogen bond donating capability the pyridine nitrogens were pointed into the cavity (e.g., 20•H2PO4−). The pyridine nitrogens rotated out when binding guests lacking hydrogen bond donating ability (e.g., 20•Cl−), and this was accompanied by a downfield shift of the C-H protons at the 3 position in the arene rings indicating C-H…anion hydrogen bonding. This underscores the need for some flexibility in the conformation of the binding site for selective receptors, as well as the contribution of an increased degree of flexibility in the scaffold.


Invariably receptor systems must contain some signalling unit to function as a sensor, as showcased in the phenylacetylene-based tripodal receptor for heparin in serum 21.75 In 21 the three ammonium-bearing arms were extended from the fluorophore body by ethynyl linkages, which proved to be the structural criterion necessary for effective binding of anionic heparin. In contrast, 22, with its closely packed tripodal arms, proved insufficiently sensitive to be viable in biological media such as serum.76 This was attributed to non-specific binding of proteins in serum which displaced the dye bound in the receptor. Additionally, this provided elegant precedent for the ability of small-molecule synthetic receptors to be viable probes for complex biological substrates in competitive media.

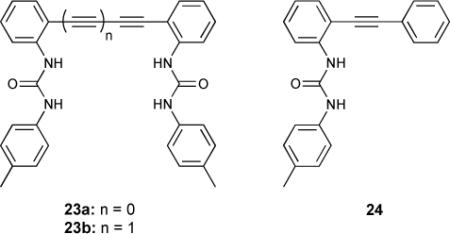
Recent work has made use of fluorescent “turn-off” anion sensing modes77 and phenylacetylene subunits for both “turn-on” signalling and binding site size enforcement.78 The chloride binding properties of diphenylacetylene-based anion receptors 23a,b exhibited “turn-off” fluorescence in the presence of a suitable guest. Bisurea 23a exhibited quenched fluorescence emission in its unbound “open” state. The DFT minimized conformation had the typical low fluorescence observed in diphenylacetylenes due to the dark πy*← πx (1A1u) transition being close in energy to the emissive πx*← πx (1B1u) transition. Fitting a suitably sized guest in the receptor cleft (in this case Cl− anion) induced planarity by rotation around the alkynyl bond. This increased the energy of the 1A1u transition, and the bound receptor thus became emissive. Binding constants for model systems 23b and 24 were also measured against halide and oxoanions. Receptor 23a had the highest affinity for Cl− (log ß = 2.557 versus 1.831 for 23b and <1 for 24). The length of the phenylacetylenic linker influenced size selectivity, as Br− was bound by 23a and not by 23b. None of the receptors exhibited any affinity for NO3−.
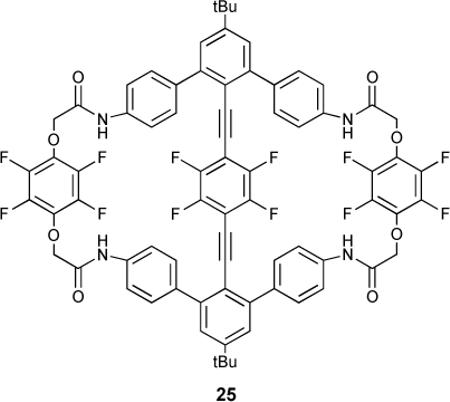
The allosteric binding of 25, a macrocyclic receptor with a central tetrafluorophenyl “turnstile” suspended in the cavity by ethynyl bridges, was studied by NMR spectroscopy.79 The turnstile inhibited both amide binding sites in the free receptor. Upon binding one equivalent of a guest the tetrafluorophenyl gate was opened and allowed access to the second binding site. Acetate, Br−, and phosphate bound cooperatively (Hill coefficients of 1.4, 1.5 and 1.4, respectively). Interestingly, Br− anion was large enough (1.82Å in an octahedral environment) to exhibit cooperative binding, but Cl− (1.67Å) ostensibly did not, although chloride had the higher Ka1 with approximately equivalent second binding constants. This was reflected in the Hill coefficient for Cl− anion (1.1).
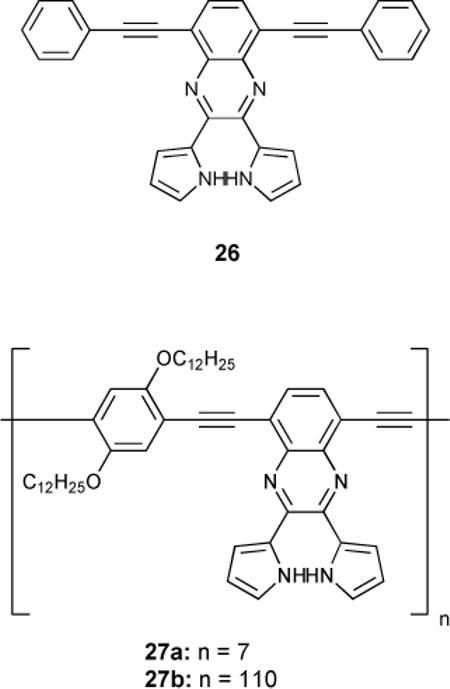
Based on earlier work,80 dipyrrolylquinoxaline derivative 26 was reported to bind anions with both a chromogenic and fluorogenic response.81 This monomer exhibited affinity for anions (F− > HP2O7−3 > CN− > OAc− > H2PO4− ≈ Cl− ≈ Br− ≈ I− ≈ NO−3) correlating to the relative basicity of the guest, although fluoride and pyrophosphate were found to deprotonate the receptor. Polymerization to 27a and 27b was accompanied by a 34-fold increase in optoelectronic response, which translates into increased sensitivity with increased repeat units.
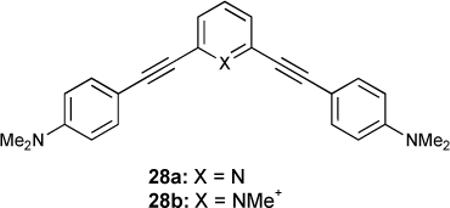
Although beyond the scope of this review, it is worth noting the two-photon absorption (TPA) properties that some phenylacetylenes can lend to a probe. Cross-conjugated D-πA-π-D bis(anilinoethynyl)-functionalized pyridine 28 was synthesized and its properties as a two-photon induced polymerization (TPIP) initiator were studied.82 While free-base 28a was too poor an acceptor to provide a good TPA cross section, cationic 28b was assumed to have a much higher value, though this ultimately could not be verified due to the poor solubility of 28b. While applications for a target specific polymerization initiator may exist, the application of these to two-photon imaging techniques also warrants interest. It has also been reported that TPA fluorophores of the A-π-A type can be modulated via their planarity, which has also been a demonstrated handle for supramolecular sensors.83 A combination of selective targeting and low energy excitation and emission is a promising application for phenylacetylene-based sensors.
3.1 2,6-bis(arylethynyl)pyridine-based receptors
2,6-Bis(arylethynyl)pyridines have found utility in numerous areas of chemistry. The inherent properties of these ethynylpyridines (conjugation, absorption/emission, pH dependence, metal binding capability, rigidity etc.) have been exploited for applications in: liquid crystals, light-emitting materials, rotaxane-type structures, molecular magnets, antiangiogenic activity, polymer composites and coordination complexes.84-87 In contrast, the supramolecular chemistry of 2,6-bis(arylethynyl)pyridines has received little attention, perhaps in part due to the scarcity of uniting the fields of molecule/ion recognition with the synthesis of highly-conjugated, carbon-rich materials. Most of the host/guest studies reported have focused on exploiting the pyridine lone pair to bind metal ion,88 or organoiodides.85 Alternatively, we hypothesized that the unique absorption/emission properties of arylethynylpyridines in tandem with their structural rigidity aptly positions them to function as small molecule or ion receptors. Moreover, we envisioned that the 2,6-bis(arylethynyl)pyridine scaffold and its derivatives would serve as a versatile building block for the development of receptor molecules that target a variety of guests depending on the protonation state of the pyridine and the type of functional group appended to the arylethynyl unit.
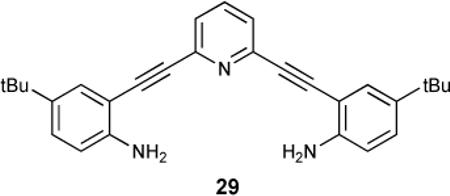
Our work thus far has focused on derivatives of the 2,6-bis(anilinoethynyl)pyridine 29.89,90 Recent investigations in the Haley lab of the structural and electronic properties of pyridine-based cyclynes27 and metallacycles91 prompted our consideration of their use in molecular recognition applications. A highly conjugated, structurally rigid phenylacetylene scaffold incorporating the inward-directed and tunable (protonated or free base) pyridine functionality affords a pre-organized receptor capable of an optoelectronic response (e.g., change in emission) upon guest complexation. By opening up the macrocycle to provide a binding pocket for guest molecules, our acyclic design is now capable of adapting to a range of guest sizes. This is achieved via a three-point binding motif where the amide hydrogens offer tunable hydrogen-bond donating sites along with protonation of the pyridine lone-pair as a switchable hydrogen-bond accepting or donating site. Additionally, exploiting the easily derivatized aniniline functionality as a synthetic handle makes the modular approach to receptor design feasible.
As an example, acetyl-protected mercaptoamide 30 crystallizes in a polymeric chain as the neutral compound.92 Protonation of the receptor with HCl gas bubbled through solution provides a racemic mixture of crystals with induced helicity from the bound anion. Low temperature 1H-NMR spectroscopy indicated rapid interconversion between the two possible helicities by significant broadening of the methylene proton signals.
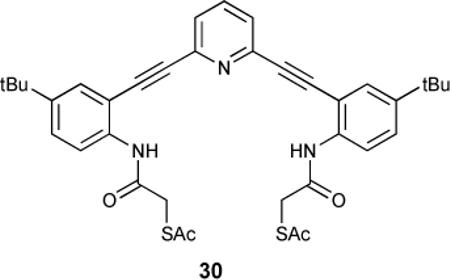
3.2 Sulfonamide and urea as hydrogen bond-donating binding sites
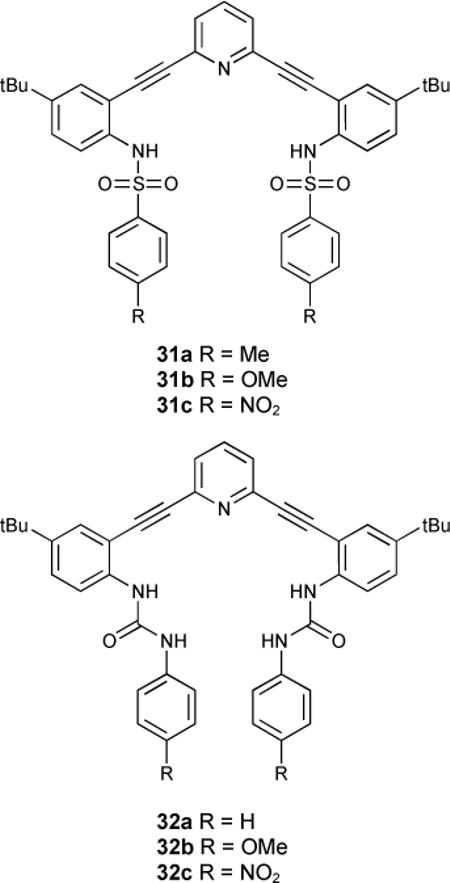
Both urea and sulfonamide derivatives 31 and 32 were investigated as anion receptors by UV-Vis, 1H-NMR and fluorescence spectroscopic studies.89
Sulfonamide receptors yielded [2 + 2] complexes from numerous solvents. Crystallization by either diffusion or evaporation yielded complexes with two water guests or two halide guests (if the receptors were protonated). Surprisingly, a heterodimer (H31a+•Cl−)•(31a•H2O) was obtained from treatment of the receptor with concentrated HCl, where one chloride and one water guest filled the binding pocket between the two receptor units (Figure 1). This dimeric species contained one protonated receptor whose pyridinium hydrogen shared a strong hydrogen bond with the chloride guest, which itself shared a hydrogen bond to a water guest. A final hydrogen bond from the water to the lone pair of the freebase receptor stitched the heterodimeric complex together. An additional four hydrogen bonds from the sulfonamide units of each receptor formed a network of helical hydrogen bonds running between the two receptors. Both the water and the halide guest freely exchanged in the system, and hence the heterodimeric structure exhibited properties intermediate to either of the homogenous dimers. In addition to the stabilization of the seven total hydrogen bonds, π-stacking interactions between the two receptors also contributed to the stabilization of the complex. Both hydrogen halide and H-OH shared the same strucural role in this self-assembled system, a synergistic effect that has recently become more widely appreciated as an important structural role in protein folding.
Fig. 1.

Crystal structure representations (both stick and CPK) of (H31a+•Cl−)•(31a•H2O)
These studies have prompted us to exploit the modular nature of our synthetic strategy to vary the linking arms, functional groups, cores and binding units in the multitopic receptors.
In contrast to the dimeric structures obtained in the sulfonamide analogues, urea receptors 32a,b crystallized in [4 + 4] tetrameric fashion with solvent molecules in the binding pockets (Figure 2).90 Crystals obtained from DMSO/MeOH/toluene mixture or pure ethanol were found in either an “S” or “W” conformation, and these stacked in an “SWWS” repeat unit. Protonation of receptor 32a with HCl yielded single crystals of a [1 + 1] complex (Figure 3).
Fig. 2.

Crystal structure representation of one half of 32a (“S” in blue and “W” in orange) with bound MeOH solvent90
Fig. 3.

ORTEP representation (50% probability level) of H32a+•Cl− (t-butyl groups are omitted for clarity)
Binding constants in water saturated chloroform with NBu4+ salts followed the Hofmeister series in the neutral receptor (treated as [1 + 1] for fitting): 2100 M−1 for Cl−, 400 M−1 for Br−, iodide had no measurable binding. In contrast trifluoroacetic acid protonated receptor bound halides in water saturated chloroform with the opposite affinity: 41,700 M−1 for Cl−, 61,700 M−1 for Br− and 83,200 M−1 for iodide.
Geminate “turn on” and “turn off” fluorescence emission was observed for these receptors. In both sulfonamide and urea derivatives, protonation of the receptors in solution led to either quenching or enhancement of fluorescence based upon the pendant functionality. Electron-rich sulfonamide 31b was fluorescent in the neutral state, but protonation by oxoacids (TFA or acetic acid), or hydrogen halides (HCl or HBr) quenched fluorescence emission and bathochromically shifted the residual emission band. In electron-poor 31c, the opposite behaviour was observed: non-fluorescent neutral receptors fluoresced with an emission maximum at 515 nm (Figure 4).93
Fig. 4.

Normalized fluorescence emission of neutral and TFA-protonated bis-sulfonamides 31b,c
This trend also held true for the urea receptors 32b,c which were investigated for their signalling ability in vitro. Methoxy-substituted phenylurea 32b fluoresced in NIH3t3 murine embryo fibroblasts treated with a high Cl− buffer, while NO2-substituted phenylurea 32c did not behave in the “OFF-ON” fashion expected, most likely due to solubility issues. Control experiments with nitrate buffers (no Cl−) exhibited minimal fluorescence (Figure 5).
Fig. 5.

Epifluorescence microscope images of NIH3T3 murine embryo fibroblasts incubated in 10% aqueous DMSO with 32b (a) Cl− containing buffer, (b) Normarski phase image of same and (c) NO3− buffer (low Cl−)93
These results underscore the need for a full understanding of how a promising receptor in the cuvet can be successfully translated to biological media.
4.1 Biological applications of phenylacetylene-based sensors
There are several biochemical factors to consider in the design of new intracellular ion indicators. These include the loading of the indicator(s) into target cells,94-98 maximizing optimal detection parameters99 with minimal toxicity100 and the ability to selectively quantitate the ion concentration in question within the cells or tissues being examined.101 Within these areas are several additional factors that affect utility including compartmentalization or partitioning within the cell into specific organelles,102 matching optical excitation/emission wavelengths to match common instrumentation in use103 and the use of ratiometric determination methods to help quantitate binding and ion levels.104,105 Finally, factors such as specificity versus other intracellular ions,106 water solubility,107 relative binding constants for the intracellular ion or ions108 and the effect of pH on binding99,109 and fluorescence emission101 are also important factors to be optimized for biochemical probe optimization. Of course, it is impossible to optimize all parameters, but of ultimate importance are ion selectivity and the ability to quantitate intracellular concentration.
Anions in cells are present at roughly 70% of all enzymatic sites and are responsible for everything from the activation of signal transduction pathways to maintaining osmotic pressure and cell volume.1,110 They are involved in many disease pathways, from cystic fibrosis to osteoporosis.111-113 In mitochondria, at least 14 different anion transport pathways have been identified114 and are responsible for the regulation and trafficking of ADP, ATP, citrate, maleate and halide anions, among others.115,116 The ubiquity of these molecules in living organisms, and their importance in regulating life systems can best be summed up in the realization of the polyanionic character of both DNA and RNA. The acidity of intracellular compartments has been implicated in some disease pathways, notably in the cellular breach of anthrax lethal toxin and edema toxin.117
This breach has been correlated with decreased endosomal pH, which can be achieved either by concomittant movement of K+, or an influx of Cl−.
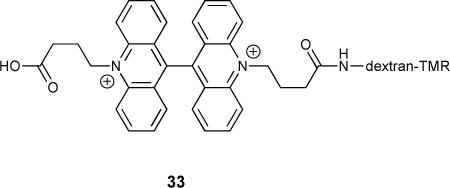
One recently reported example of a ratioable fluorescent Cl− probe was dextran-tethered acridinium system 33, used for charting endosomal Cl− concentration in tandem with endosomal pH.109 The biaccridinium subunit of the receptor was found to be quenched by Cl− anion with a Stern-Volmer constant of 36 M−1, and to be insensitive to non-halide anions (nitrate, phosphate, bicarbonate and sulfate). By tethering this system to tetramethylrhodamine, which is insensitive to Cl−, they developed a ratioable sensor for in vivo application that allowed them to observe the change in Cl− concentration in endosomes with decreasing pH. Using this same technique, it was also shown that the intracellular CIC-3 Cl− channel regulated the vacuolar H+ pump during endosomal acidification.118 The ease with which a well designed small molecule sensor can visualize these complex biological responses demonstrates the import role these receptors will play in furthering our understanding of cellular processes.
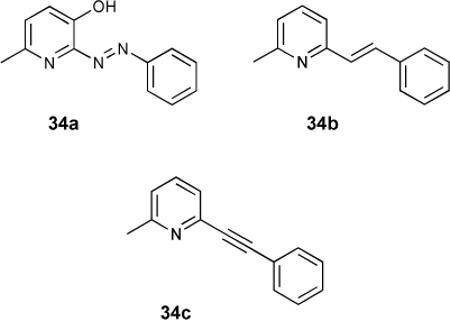

A series of linked pyridylarenes 34 were used as selective antagonists for the metabotropic glutamate receptor subtype 5 (mGlu5).119 It was found that phenylethynyl derivative 34c (MPEP) was the most active antagonist, and bound the receptor much more effectively than the non-ethynylated receptors. This work was continued with radiolabelled derivatives 35a120 and 35b-d.121 These radiolabelled compounds were thought to be appropriate for binding and following the distribution of mGluR5 receptors. Radiolabelled 35a possessed a high affinity (Kd = 2 nM) and was over 90% specific for mGlu5 receptor over the 1-10 nM range.
Expanding upon 35a, ligands 35b-d were also utilized as PET imaging agents in Sprague-Dawley rats.121 Bioaccumulation of the radiolabelled agents were tracked by in vivo microPET imaging (Figure 6), and found to have the highest binding in the olfactory bulb, followed by striatum, hippocampus and cortex localization. It was hypothesized that the glutamate receptor mGluR5 might have major physiological function in the oflactory area as olfactory damage in neurodegenerative diseases is consistent and severe.122 This would also suggest that early diagnosis of Parkinson's or Alzheimer's could be achieved via the olfactory bulb.
Fig. 6.

Coronal, axial and sagittal PET images of derivatives of 35 in anesthetized rat brain at 8-10 min after administration of radiolabelled ligand. The coronal level 15.2 corresponds to the olfactory area; 11.2 the cingulate level; 10.0 the striatal level; 4.2 the hippocampal level; and −2.8 the cerebellar level. The axial and sagittal views illustrate the distribution of the radioactivity at the mid-striatal level. (Reprinted from ref. 121 with permission from Elsevier)
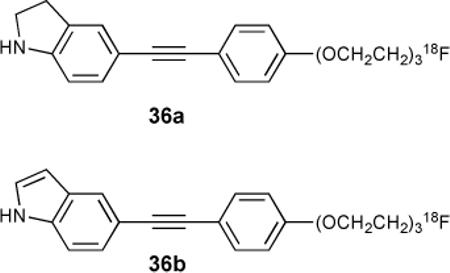
Related work with ß-amyloid plaques (Aß plaques) as pathological features of Alzheimer's onset made use of indolinyl- and indolylphenylacetylenes as PET imaging agents.123 Both 36a and 36b were found to bind Aß plaques, with 36b being slightly more selective in early trials (via autoradiography). No binding constants were reported, although relative binding was inferred from washout rates.
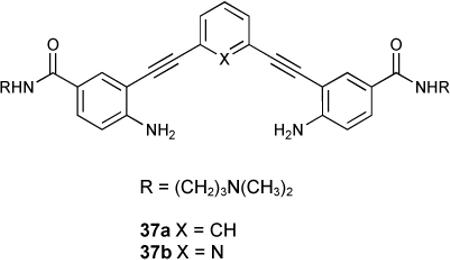
Phenylethynylamides 37a,b were found to be potent Gquadruplex binders.124 Binding was studied in this system by FRET melting assays, surface plasmon resonance and CD spectroscopy. Moderate stabilization with G-quadruplexes were observed for both ligands. No discernible duplex DNA stabilization was reported. As there is evidence that small molecules can bind G-quadruplexes and modulate transcription, selectivity like this is promising for future application of these acyclic compounds in this area. Additionally, the 1H-NMR experiments reported were carried out in aqueous buffer, notable due to the low solubility for related analogues (no amide functionality) and water-solubility for many arylethynyl probes is a barrier to their use as bioimaging agents.
Conclusions
Phenylacetylenes have emerged as important components in the design of an increasingly diverse array of exciting supramolecular host-guest complexes. Their unique combination of optoelectronic properties and shape persistence has played an important role in the development of many novel coordination compounds, fluorogenic and chromogenic probes and biological imaging agents. The relative insolubility of many of these compounds in aqueous media still remains a challenge in the development of this class of probe architectures. While metal-containing complexes have been extensively reviewed, host-guest complexes that make use of only simple organic phenylacetylenes have only recently received increasing attention. Their contribution to the rational and effective design of receptors able to target both neutral and ionic guests with concomitant optoelectronic response upon binding is at the core of this advancement. Additionally, commonly used synthetic techniques allow for facile and efficient preparation of a variety of novel candidates for study, which further drives innovation in this field, suggesting potential applications in molecular probe development, nanotechnology, and even recognition of biologically-relevant molecules and ions.125
Our contribution to the work highlighted in this review was funded by the National Science Foundation (NSF-0718242), the National Institutes of Health (GM087398-01A1) and the University of Oregon (UO). C.N.C. acknowledges the NSF for an Integrative Graduate Education and Research Traineeship (DGE-0549503). D.W.J. is a Cottrell Scholar of Research Corporation for Science Advancement and gratefully acknowledges the NSF for a CAREER award.
Biographies
 Calden N. Carroll is a native of Flagstaff, Arizona and received his extended B.S. in Chemistry in 2004 from Northern Arizona University. He is currently a Ph. D. candidate in the laboratories of Profs. Michael M. Haley and Darren W. Johnson at the University of Oregon where he studies the binding affinities of small molecule receptors for anionic guests when he is not on the river. He spends a lot of time rafting.
Calden N. Carroll is a native of Flagstaff, Arizona and received his extended B.S. in Chemistry in 2004 from Northern Arizona University. He is currently a Ph. D. candidate in the laboratories of Profs. Michael M. Haley and Darren W. Johnson at the University of Oregon where he studies the binding affinities of small molecule receptors for anionic guests when he is not on the river. He spends a lot of time rafting.
 John J. Naleway received his Ph. D. in 1981 from Marquette University under the direction of Prof. Norman Hoffman. After completing postdoctoral research at The University of Alberta under Prof. Raymond Lemieux and at the University of Wisconsin under Prof. Laurens Anderson, he joined the staff at Monsanto Healthcare - Searle Pharmaceuticals, and was later the Laboratory Director and Director of Substrates Research at Molecular Probes, Inc. He is currently an Adjunct Facutly member in the Department of Chemistry at the University of Oregon and President of Marker Gene Technologies, Inc. in Eugene, Oregon where his research is centered around live cell assays for drug discovery and fluorescent chemosensors.
John J. Naleway received his Ph. D. in 1981 from Marquette University under the direction of Prof. Norman Hoffman. After completing postdoctoral research at The University of Alberta under Prof. Raymond Lemieux and at the University of Wisconsin under Prof. Laurens Anderson, he joined the staff at Monsanto Healthcare - Searle Pharmaceuticals, and was later the Laboratory Director and Director of Substrates Research at Molecular Probes, Inc. He is currently an Adjunct Facutly member in the Department of Chemistry at the University of Oregon and President of Marker Gene Technologies, Inc. in Eugene, Oregon where his research is centered around live cell assays for drug discovery and fluorescent chemosensors.
 Michael M. Haley studied cyclopropene and cycloproparene chemistry with Prof. Ed Billups at Rice University where he received both his B.A. (1987) and Ph.D. degrees (1991). In 1991 he received a National Science Foundation Postdoctoral Fellowship to work with Prof. Peter Vollhardt on [N]phenylene chemistry at the University of California, Berkeley. In 1993 he joined the faculty at the University of Oregon where he is currently a Professor and Head of the Chemistry Department, as well as a member of the Materials Science Institute. Among the awards he has received are a National Science Foundation CAREER Award (1995), a Camille Dreyfus Teacher-Scholar Award (1998), an Alexander von Humboldt Research Fellowship (2000), Thomas F. Herman Distinguished Teaching Award (2002), and University of Oregon Fund for Faculty Members Excellence Award (2007). His current research focuses on the chemistry of dehydrobenzoannulenes, indenofluorenes, molecules based on phenyl-acetylene scaffolding and other novel carbon-rich systems.
Michael M. Haley studied cyclopropene and cycloproparene chemistry with Prof. Ed Billups at Rice University where he received both his B.A. (1987) and Ph.D. degrees (1991). In 1991 he received a National Science Foundation Postdoctoral Fellowship to work with Prof. Peter Vollhardt on [N]phenylene chemistry at the University of California, Berkeley. In 1993 he joined the faculty at the University of Oregon where he is currently a Professor and Head of the Chemistry Department, as well as a member of the Materials Science Institute. Among the awards he has received are a National Science Foundation CAREER Award (1995), a Camille Dreyfus Teacher-Scholar Award (1998), an Alexander von Humboldt Research Fellowship (2000), Thomas F. Herman Distinguished Teaching Award (2002), and University of Oregon Fund for Faculty Members Excellence Award (2007). His current research focuses on the chemistry of dehydrobenzoannulenes, indenofluorenes, molecules based on phenyl-acetylene scaffolding and other novel carbon-rich systems.
 Darren W. Johnson received his B.S. in Chemistry at the University of Texas at Austin in 1996, where he performed undergraduate research under the direction of Jonathan L. Sessler. He earned his Ph. D. in Chemistry in 2000 from the University of California at Berkeley working with Kenneth N. Raymond, and he then spent two years at the Scripps Research Institute as a National Institutes of Health post-doctoral fellow with Julius Rebek, Jr. He joined the chemistry faculty at the University of Oregon in 2003, where he is currently an Associate Professor. He is a Cottrell Scholar of Research Corporation for Science Advancement and a National Science Foundation CAREER awardee. Research in his group uses supramolecular chemistry as a tool to explore a variety of problems in coordination chemistry, molecule/ion recognition and inorganic cluster synthesis.
Darren W. Johnson received his B.S. in Chemistry at the University of Texas at Austin in 1996, where he performed undergraduate research under the direction of Jonathan L. Sessler. He earned his Ph. D. in Chemistry in 2000 from the University of California at Berkeley working with Kenneth N. Raymond, and he then spent two years at the Scripps Research Institute as a National Institutes of Health post-doctoral fellow with Julius Rebek, Jr. He joined the chemistry faculty at the University of Oregon in 2003, where he is currently an Associate Professor. He is a Cottrell Scholar of Research Corporation for Science Advancement and a National Science Foundation CAREER awardee. Research in his group uses supramolecular chemistry as a tool to explore a variety of problems in coordination chemistry, molecule/ion recognition and inorganic cluster synthesis.
Footnotes
Part of a themed issue on the supramolecular chemistry of anionic species.
Notes and references
- 1.Sessler JL, Gale PA, Cho W-S. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]
- 2.Cho W-S, Sessler JL. In: Functional Synthetic Receptors. Schrader T, Hamilton AD, editors. Wiley-VCH; Weinheim: 2005. pp. 165–256. [Google Scholar]
- 3.Bianchi A, Bowman-James K, Garcia-España E, editors. Supramolecular Chemistry of Anions. Wiley-VCH; New York: 1997. [Google Scholar]
- 4.Houssain A. Curr. Org. Chem. 2008;12:1231–1256. [Google Scholar]
- 5.Campbell K, Tykwinski RR. In: Carbon-rich Compounds: From Molecules to Materials. Haley MM, Tykwinski RR, editors. Wiley-VCH; New York: 2006. pp. 229–294. [Google Scholar]
- 6.Welti R, Diederich F. Helv. Chim. Acta. 2003;86:494. [Google Scholar]
- 7.Huang C-Y, Cabell LA, Anslyn EV. J. Am. Chem. Soc. 1994;116:2778. [Google Scholar]
- 8.Sessler JL, Vivian AE, Seidel D, Burrell AK, Hoehner M, Mody TD, Gebauer A, Weghorn SJ, Lynch V. Coord. Chem. Rev. 2000;216-217:411–434. [Google Scholar]
- 9.Jasat A, Dolphin D. Chem. Rev. 1997;97:2267–2340. doi: 10.1021/cr950078b. [DOI] [PubMed] [Google Scholar]
- 10.Gale PA, Anzenbacher P, Jr., Sessler JL. Coord. Chem. Rev. 2001;222:57–102. [Google Scholar]
- 11.Anzenbacher P, Jr., Nishiyabu R, Palacios MA. Coord. Chem. Rev. 2006;250:2929–2938. [Google Scholar]
- 12.Atwood JL, Holman KT, Steed JW. J. Chem. Soc., Chem. Commun. 1996:1401–1407. [Google Scholar]
- 13.Sessler JL, Rubin BL, Camiolo S, Cho W-S, Pantos GD, Lynch VM. Supramolecular Chem. 2006;18:103–109. [Google Scholar]
- 14.Gale PA. Chem. Commun. 2005:3761–3772. doi: 10.1039/b504596g. [DOI] [PubMed] [Google Scholar]
- 15.Evan-Salem T, Baruch I, Avram L, Cohen Y, Palmer LC, Rebek J., Jr. Proc. Nat. Acad. Sci. 2006;103:12296–12300. doi: 10.1073/pnas.0604757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leininger S, Olenyuk B, Stang PJ. Chem. Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 17.Anderson HL, Sanders JKM. Chem. Commun. 1996:946–947. [Google Scholar]
- 18.Ferrara JD, Tessier-Youngs C, Youngs WJ. Organometallics. 1987;6:676–678. [Google Scholar]
- 19.Iyoda M, Sirinintasak S, Nishiyama Y, Vorasingha A, Sultana F, Nakao K, Kuwatani Y, Matsuyama H, Yoshida M, Miyake Y. Synthesis. 2004:1527–1531. [Google Scholar]
- 20.Ferrara JD, Tanaka AA, Fierro C, Tessier-Youngs C, Youngs WJ. Organometallics. 1989;8:2089–2098. [Google Scholar]
- 21.Ferrara JD, Djebli A, Tessier-Youngs C, Youngs WJ. J. Am. Chem. Soc. 1988;110:647–649. [Google Scholar]
- 22.Ferrara JD, Tessier-Youngs C, Youngs WJ. J. Am. Chem. Soc. 1985;107:6719. [Google Scholar]
- 23.Baxter PNW. Chem. Eur. J. 2003;9:2531–2541. [Google Scholar]
- 24.Baxter PNW. Chem. Eur. J. 2002;8:5250–5264. doi: 10.1002/1521-3765(20021115)8:22<5250::AID-CHEM5250>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 25.Baxter PNW, Dali-Youcef R. J. Org. Chem. 2005;70:4935–4953. doi: 10.1021/jo040276f. [DOI] [PubMed] [Google Scholar]
- 26.Samori S, Tojo S, Fujitsuka M, Spitler EL, Haley MM, Majima T. J. Org. Chem. 2007;72:2785–2793. doi: 10.1021/jo062326h. [DOI] [PubMed] [Google Scholar]
- 27.Spitler EL, McClintock SP, Haley MM. J. Org. Chem. 2007;72:6692–6699. doi: 10.1021/jo070827c. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Wan X, Xue X, Li Y, Yu A, Chen Y. Eur. J. Org. Chem. 2010:1681–1687. [Google Scholar]
- 29.Spitler EL, Haley MM. Tetrahedron. 2008;64:11469–11474. [Google Scholar]
- 30.Liao J-H, Chen C-T, Chou H-C, Cheng C-C, Chou P-T, Fang J-M, Slanina Z, Chow TJ. Org. Lett. 2002;4:3107–3110. doi: 10.1021/ol0264096. [DOI] [PubMed] [Google Scholar]
- 31.Fang JM, Selvi S, Liao JH, Slanina Z, Chen CT, Chou PT. J. Am. Chem. Soc. 2004;126:3559. doi: 10.1021/ja039237w. [DOI] [PubMed] [Google Scholar]
- 32.(a) Inouye M, Miyake T, Furusyo M, Nakazumi H. J. Am. Chem. Soc. 1995;117:12416. [Google Scholar]; (b) Inouye M, Chiba J, Nakazumi H. J. Org. Chem. 1999;64:8170–8176. doi: 10.1021/jo9911138. [DOI] [PubMed] [Google Scholar]
- 33.Anderson S, Neidlein U, Gramlich V, Diederich F. Angew. Chem. Int. Ed. Engl. 1995;34:1596. [Google Scholar]
- 34.Neidlein U, Diederich F. Chem. Commun. 1996:1493. [Google Scholar]
- 35.Droz AS, Neidlein U, Anderson S, Seiler P, Diederich F. Helv. Chim. Acta. 2001;84:2243. [Google Scholar]
- 36.Inouye M, Waki M, Abe H. J. Am. Chem. Soc. 2004;126:2022. doi: 10.1021/ja039371g. [DOI] [PubMed] [Google Scholar]
- 37.Martínez-Máñez R, Sancenón F. Chem. Rev. 2003;103:4419–4476. doi: 10.1021/cr010421e. [DOI] [PubMed] [Google Scholar]
- 38.Anslyn E. J. Org. Chem. 2006;72:687–699. doi: 10.1021/jo0617971. [DOI] [PubMed] [Google Scholar]
- 39.Gunnlaugsson T, Glynn M, Tocci GM, Kruger PE, Pfeffer FM. Coord. Chem. Rev. 2006;250:3094–3117. [Google Scholar]
- 40.Metzger A, Anslyn EV. Angew. Chem. Int. Ed. 1998;37:649–651. doi: 10.1002/(SICI)1521-3773(19980316)37:5<649::AID-ANIE649>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 41.Malashikhin SA, Baldridge KK, Finney NS. Org. Lett. 2010;12:940–943. doi: 10.1021/ol902902m. [DOI] [PubMed] [Google Scholar]
- 42.Kondo S, Sato M. Tetrahedron. 2006;62:4844. [Google Scholar]
- 43.Anthony JE, Khan SI, Rubin Y. Tetrahedron. 1997;38:3499–3502. [Google Scholar]
- 44.Taniguchi M, Cramer DL, Bhise AD, Kee HL, Bocian DF, Holten D, Lindsey JS. New. J. Chem. 2008;32:947–958. [Google Scholar]
- 45.Droz AS, Diederich F. J. Chem. Soc., Perkin Trans. 1. 2000:4224–4226. [Google Scholar]
- 46.Young JK, Moore JS. In: Modern Acetylene Chemistry. Stang PJ, Diederich F, editors. Wiley-VCH; New York: 1995. pp. 415–442. [Google Scholar]
- 47.Beer S, Hrib CG, Jones PG, Brandhorst K, Grunenberg J, Tamm M. Angew. Chem. Int. Ed. 2007;46:8890–8894. doi: 10.1002/anie.200703184. [DOI] [PubMed] [Google Scholar]
- 48.Zhang W, Moore JS. Adv. Syn. Catal. 2007;349:93–120. [Google Scholar]
- 49.Scrimgeour KG. Chemistry and Control of Enzymatic Reactions. Academic Press, Inc.; New York: 1977. [Google Scholar]
- 50.Schetz JA, Sibley DR. J. Pharmacol Exp. Ther. 2001;296:359. [PubMed] [Google Scholar]
- 51.(a) Martinez-Diaz M-V, Spencer N, Stoddart F. Angew. Chem. Int. Ed. Engl. 1997;36:1904. [Google Scholar]; (b) Huang Y-L, Hung W-C, Lai C-C, Liu Y-H, Peng SM, Chiu S-H. Angew. Chem., Int. Ed. 2007;46:6629–6633. doi: 10.1002/anie.200702197. [DOI] [PubMed] [Google Scholar]
- 52.Al-Sayah MH, Branda NR. Org. Lett. 2002;4:881. doi: 10.1021/ol0170915. [DOI] [PubMed] [Google Scholar]
- 53.Al-Sayah MH, Branda NR. Angew. Chem., Int. Ed. Engl. 2000;39:945. [PubMed] [Google Scholar]
- 54.Filby MH, Dickson SJ, Zaccheroni N, Prodi L, Bonacchi S, Montalti M, Paterson MJ, Humphries TD, Chiorboli C, Steed JW. J. Am. Chem. Soc. 2008;130:4105–4113. doi: 10.1021/ja711012d. [DOI] [PubMed] [Google Scholar]
- 55.Bucher C, Devillers CH, Moutet J-C, Royal G, Saint-Aman E. New J. Chem. 2004;28:1584–1589. [Google Scholar]
- 56.Miyaji H, Gasser G, Green SJ, Molard Y, Strawbridge SM, Tucker JHR. Chem. Commun. 2005:5355–5357. doi: 10.1039/b510513g. [DOI] [PubMed] [Google Scholar]
- 57.Inouye M, Takahashi K, Nakazumi H. J. Am. Chem. Soc. 1999;121:341. [Google Scholar]
- 58.Rashadan S, Light ME, Kilburn JD. Chem. Commun. 2006:4578–4580. doi: 10.1039/b611138f. [DOI] [PubMed] [Google Scholar]
- 59.Steed JW. Chem. Soc. Rev. 2008;38:506–519. doi: 10.1039/b810364j. [DOI] [PubMed] [Google Scholar]
- 60.Amendola V, Bonizzoni M, Esteban-Goméz D, Fabrizzi L, Licchelli M, Sancenón F, Taglietti A. Coord. Chem. Rev. 2006;250:1451–1470. [Google Scholar]
- 61.Cercla Priority List of Hazardous Substances. 2007 http://www.atsdr.cdc.gov/cercla.
- 62.US EPA Case Study – Arsenic Treatment Technologies, Tucson, Arizona. 2003 http://www.epa.gov/safewater/arsenic/publications.html.
- 63.Liu C-Q, Li S-L, Lang Y-C, Xiao H-Y. Env. Sci. Technol. 2006;40:6928. doi: 10.1021/es0610129. [DOI] [PubMed] [Google Scholar]
- 64.Katayev EA, Kolesnikov GV, Sessler JL. Chem. Soc. Rev. 2009;38:1572–1586. doi: 10.1039/b806468g. [DOI] [PubMed] [Google Scholar]
- 65.Cherimisinoff NP. Pollution Engineering. 2001;33:38. [Google Scholar]
- 66.Maji SK, Pal A, Pal T. J. Haz. Mat. 2008;151:811–820. doi: 10.1016/j.jhazmat.2007.06.060. [DOI] [PubMed] [Google Scholar]
- 67.(a) Gu B, Brown GM, Bonnesen PV, Liang L, Moyer BA, Ober R, Alexandratos SD. Env. Sci. Technol. 2000;34:1075. [Google Scholar]; (b) Chuoyyok W, Wiacek RJ, Pattamakomsan K, Sangvanich T, Grudzien RM, Fryxell GE, Yantasee W. Env. Sci. Technol. 2010;44:3073. doi: 10.1021/es100787m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dutzler R, Campbell EB, Cadene M, Chait BT, Mackinnon R. Nature. 2002;415:287. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 69.Rychkov GY, Pusch M, Roberts ML, Jentsch TJ, Bretag AH. J. Gen. Physiol. 2001;530:379–393. [Google Scholar]
- 70.Cudic P, Zinic M, Tomisic V, Simeon V, Vigneron J-P, Lehn J-M. J. Chem. Soc., Chem. Commun. 1995:1073. [Google Scholar]
- 71.Chang K-J, Moon D, Lah MS, Jeong K-S. Angew. Chem., Int. Ed. 2005;44:7926. doi: 10.1002/anie.200503121. [DOI] [PubMed] [Google Scholar]
- 72.Kim N-K, Chang K-J, Moon D, Lah MS, Jeong K-S. Chem. Commun. 2007:3401–3403. doi: 10.1039/b707032b. [DOI] [PubMed] [Google Scholar]
- 73.Chang K-J, Chae MK, Lee C, Lee J-Y, Jeong K-S. Tetrahedron Lett. 2006;47:6385. [Google Scholar]
- 74.Kwon TH, Jeong K-S. Tetrahedron Lett. 2006;47:8539. [Google Scholar]
- 75.Wright AT, Zhong Z, Anslyn EV. Angew. Chem. Int Ed. 2005;44:5679–5682. doi: 10.1002/anie.200501437. [DOI] [PubMed] [Google Scholar]
- 76.Zhong Z, Anslyn EV. J. Am. Chem. Soc. 2002;124:9014–9015. doi: 10.1021/ja020505k. [DOI] [PubMed] [Google Scholar]
- 77.Swinburne AN, Paterson MJ, Beeby A, Steed JW. Org. Biomol. Chem. 2010;8:1010–1016. doi: 10.1039/b919821k. [DOI] [PubMed] [Google Scholar]
- 78.Swinburne AN, Paterson MJ, Beeby A, Steed JW. Chem. Eur. J. 2010;16:2714–2718. doi: 10.1002/chem.200903293. [DOI] [PubMed] [Google Scholar]
- 79.Hirata O, Takeuchi M, Shinkai S. Chem. Commun. 2005:3805–3807. doi: 10.1039/b506883e. [DOI] [PubMed] [Google Scholar]
- 80.Black CB, Andrioletti B, Try AC, Ruiperez C, Sessler JL. J. Am. Chem. Soc. 1999;121:10438–10439. [Google Scholar]
- 81.Wu C-Y, Chen M-S, Lin C-A, Lin S-C, Sun S-S. Chem. Eur. J. 2006;12:2263–2269. doi: 10.1002/chem.200500804. [DOI] [PubMed] [Google Scholar]
- 82.Pucher N, Rosspeinter A, Satzinger V, Schmidt V, Gescheidt G, Stampfl J, Liska R. Macromolecules. 2009;42:6519–6528. [Google Scholar]
- 83.Porres L, Charlot M, Entwistle CD, Beeby A, Marder TB, Blanchard-Desce M. Proc. SPIE. 2005;5943:559340F. doi: 10.1039/b416605a. [DOI] [PubMed] [Google Scholar]
- 84.Ahn CM, Shin W-S, Woo HB, Lee S, Lee H-W. Bio. Org. Med. Chem. Lett. 2004;14:3893. doi: 10.1016/j.bmcl.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 85.Holmes BT, Deb P, Pennington WT, Hanks TW. J. Polym. Res. 2006:133. [Google Scholar]
- 86.Rajadurai C, Ivanova A, Enkelmann V, Baumgarten M. J. Org. Chem. 2003;68:9907. doi: 10.1021/jo034597n. [DOI] [PubMed] [Google Scholar]
- 87.Yamaguchi Y, Kobayashi S, Wakamiya T, Matsubara Y, Yoshida Z-I. Angew. Chem. Int. Ed. 2005;44:7040. doi: 10.1002/anie.200502214. [DOI] [PubMed] [Google Scholar]
- 88.Phelps D, Crihfield A, Hartwell J, Hanks TW, Pennington WT, Bailey RD. Mol. Cryst. Liq. Cryst. 2000;354:1111. [Google Scholar]
- 89.Berryman OB, Johnson CA, Zakharov LN, Haley MM, Johnson DW. Angew. Chem. Int. Ed. 2008;47:117. doi: 10.1002/anie.200703971. [DOI] [PubMed] [Google Scholar]
- 90.Carroll CN, Berryman OB, Johnson CA, Zakharov LN, Haley MM, Johnson DW. Chem. Commun. 2009:2520. doi: 10.1039/b901643k. [DOI] [PubMed] [Google Scholar]
- 91.Johnson CA, Baker BA, Berryman OB, Zakharov LN, O'Connor MJ, Haley MM. J. Organomet. Chem. 2006;691:413. [Google Scholar]
- 92.Johnson CA, Berryman OB, Sather AC, Zakharov LN, Haley MM, Johnson DW. Cryst. Growth Des. 2009;9:4247. doi: 10.1021/cg5018856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carroll CN, Coombs BA, Johnson CA, McClintock SP, Naleway JJ, Haley MM, Johnson DW. unpublished results. [Google Scholar]
- 94.Tsien RY. Nature. 1981;290:527. doi: 10.1038/290527a0. [DOI] [PubMed] [Google Scholar]
- 95.Bush DS, Jones RL. Plant Physiol. 1990;93:841. doi: 10.1104/pp.93.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Steinberg TH, Newman AS, Swanson JA, Silverstein SC. J. Biol. Chem. 1987;262:8884. [PubMed] [Google Scholar]
- 97.Pilas B, Durack G. Cytometry. 1997;28:316. [PubMed] [Google Scholar]
- 98.Doyle AD, Lee J. Biotechniques. 2002;33:358. doi: 10.2144/02332rr02. [DOI] [PubMed] [Google Scholar]
- 99.Martínez-Zaguilán R, Parnami G, Lynch RM. Cell Calcium. 1996;19:337. doi: 10.1016/s0143-4160(96)90074-3. [DOI] [PubMed] [Google Scholar]
- 100.Katerinopoulos HE. Curr. Pharm. Design. 2004;10:3835. doi: 10.2174/1381612043382666. [DOI] [PubMed] [Google Scholar]
- 101.Lattanzio FA. Biochem. Biophys. Res. Commun. 1991;177:184. doi: 10.1016/0006-291x(91)91966-g. [DOI] [PubMed] [Google Scholar]
- 102.Di Virgilio F, Steinberg TH, Silverstein SC. Cell Calcium. 1990;11:57. doi: 10.1016/0143-4160(90)90059-4. [DOI] [PubMed] [Google Scholar]
- 103.Clark HA, Kopelman R, Tjalkens R, Philbert MA. Anal. Chem. 1999;71:4837. doi: 10.1021/ac990630n. [DOI] [PubMed] [Google Scholar]
- 104.Silver RB. Methods Cell Biol. 1998;56:237. doi: 10.1016/s0091-679x(08)60429-x. [DOI] [PubMed] [Google Scholar]
- 105.Bright GR, Fisher GW, Rogowska J, Taylor DL. Methods Cell Biol. 1989;30:157. doi: 10.1016/s0091-679x(08)60979-6. [DOI] [PubMed] [Google Scholar]
- 106.Carpenter RD, Verkman AS. Org. Lett. 2010;12:1160. doi: 10.1021/ol902836c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Biwersi J, Farah N, Wang YX, Ketchum R, Verkman AS. Am. J. Physiol. 1992;262-1:C242. doi: 10.1152/ajpcell.1992.262.1.C243. [DOI] [PubMed] [Google Scholar]
- 108.Grynkiewicz G, Poenie M, Tsien RY. J. Biol. Chem. 1985;260:3440. [PubMed] [Google Scholar]
- 109.Sonawane ND, Thiagarajah JR, Verkman AS. J. Biol. Chem. 2002;277:5506. doi: 10.1074/jbc.M110818200. [DOI] [PubMed] [Google Scholar]
- 110.Schroeder JI. Plant Mol. Biol. 1995;28:353. doi: 10.1007/BF00020385. [DOI] [PubMed] [Google Scholar]
- 111.Renkawek K, Bosman GJ. NeuroReport. 1995;6:929. doi: 10.1097/00001756-199504190-00026. [DOI] [PubMed] [Google Scholar]
- 112.Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Science. 1991;253:202. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 113.Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friederich W, Delling G, Jentsch TJ. Cell. 2001;104:205. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 114.Kaplan RS. J. Membr. Biol. 2001;179:165. doi: 10.1007/s002320010046. [DOI] [PubMed] [Google Scholar]
- 115.Thompson RJ, Akana HCSR, Finnigan C, Howell KE, Caldwell JH. Am. J. Physiol. Cell Physiol. 2006;290:C499. doi: 10.1152/ajpcell.00585.2004. [DOI] [PubMed] [Google Scholar]
- 116.Okada SF, O'Neal WK, Huang P, Nicholas RA, Ostrowski LE, Craigen WJ, Lazarowski ER, Boucher RC. J. Gen. Phys. 2004;124:513. doi: 10.1085/jgp.200409154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang S, Finkelstein A, Collier RJ. Proc. Natl. Acad. Sci. 2004;101:16756. doi: 10.1073/pnas.0405754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S, Verkman AS. J. Biol. Chem. 2005;280:1241. doi: 10.1074/jbc.M407030200. [DOI] [PubMed] [Google Scholar]
- 119.Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Veliçelebi G, Kuhn R. Neuropharmacology. 1999;38:1493. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 120.Gasparini F, Andres H, Flor PJ, Heinrich M, Inderbitzin W, Lingenhöhl K, Müller H, Munk VC, Omilusik K, Stierlin C, Stoehr N, Vranesic I, Kuhn R. Bioorg. Med. Chem. Lett. 2002;12:407. doi: 10.1016/s0960-894x(01)00767-3. [DOI] [PubMed] [Google Scholar]
- 121.Yu M, Tueckmantel W, Wang X, Zhu A, Kozikowski AP, Brownell A-L. Nucl. Med. Biol. 2005;32:631. doi: 10.1016/j.nucmedbio.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 122.Nores JM, Biacabe B, Bonfils P. Ann. Med. Interne (Paris) 2000;151:97. [PubMed] [Google Scholar]
- 123.Qu W, Choi S-R, Hou C, Zhuang Z, Oya S, Zhang W, Kung M-P, Manchandra R, Skovronsky DM, Kung HF. Bioorg. Med. Chem. Lett. 2008;18:4823. doi: 10.1016/j.bmcl.2008.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dash J, Shirude PS, Hsu S-TD, Balasubramanian S. J. Am. Chem. Soc. 2008;130:15950. doi: 10.1021/ja8046552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gale PA. Chem. Commun. 2010 ASAP: DOI:10.1039/C0CC00656D. [Google Scholar]