

Abstract
Using whole cell patch clamp in thin brain stem slices, we tested the effects of cholecystokinin (CCK) on identified gastric-projecting neurons of the rat dorsal motor nucleus of the vagus (DMV). Perfusion with the sulfated form of CCK octapeptide (CCK8s, 30 pM–300 nM, EC50 ~4 nM) induced a concentration-dependent inward current in 35 and 41% of corpus- and antrum/ pylorus-projecting DMV neurons, respectively. Conversely, none of the fundus-projecting DMV neurons responded to perfusion with CCK8s. The CCK8s-induced inward current was accompanied by a 65 ± 17% increase in membrane input resistance and reversed at 90 ± 4 mV, indicating that the excitatory effects of CCK8s were mediated by the closure of a potassium conductance. Pretreatment with the synaptic blocker TTX (0.3–1 μM) reduced the CCK8s-induced current, suggesting that a portion of the CCK8s-induced current was mediated indirectly via an action on presynaptic neurons apposing the DMV membrane. Pretreatment with the selective CCK-A receptor antagonist lorglumide (0.3–3 μM) attenuated the CCK8s-induced inward current in a concentration-dependent manner, with a maximum inhibition of 69 ± 12% obtained with 3 μM lorglumide. Conversely, pretreatment with the selective CCK-B antagonist triglumide did not attenuate the CCK8s-induced inward current; pretreatment with triglumide (3 μM) and lorglumide (1 μM) attenuated the CCK8s-induced current to the same extent as pretreatment with lorglumide alone. Immunohistochemical experiments showed that CCK-A receptors were localized on the membrane of 34, 65, and 60% of fundus-, corpus-, and antrum/pylorus-projecting DMV neurons, respectively. Our data indicate that CCK-A receptors are present on a subpopulation of gastric-projecting neurons and that their activation leads to excitation of the DMV membrane.
Keywords: vagovagal reflex, gastrointestinal motility
Cholecystokinin (CCK) is released from small intestinal enteroendocrine cells in response to nutrients entering the duodenum (for review see Refs. 22, 27, 39). CCK then induces multiple effects along the gastrointestinal tract, such as gastric relaxation, decreased gastric acid secretion, and increased pancreatic exocrine secretion, as well as reduction of food intake (21, 25, 32–34, 39, 45). The actions of CCK are mediated by activation of two G protein-coupled receptors: the CCK-A (or CCK-1) and the CCK-B (or CCK-2) receptor (28).
The most publicized site of action of CCK is at the level of vagal sensory neurons, where CCK is proposed to act in a paracrine fashion. Such a mechanism of action is supported by a large amount of data indicating that 1) CCK-containing cells are present on the serosal surface of the small intestine; 2) CCK receptors are present on, and transported within, vagal afferents; 3) a large percentage of vagal sensory neurons contain CCK and transcribe CCK-A receptor mRNA; 4) CCK activates intestinal vagal afferent neurons, and this activation is mimicked by intestinal stimuli and attenuated by CCK-A receptor antagonists; and 5) perivagal capsaicin treatment, by destroying sensory C-fibers, significantly attenuates the effects of systemic administration of CCK (2, 3, 12, 18, 31, 33).
However, several lines of evidence indicate that the effects of CCK, especially the satiety effect, might also be mediated via actions at sites other than the peripheral vagal sensory afferents. In fact, systemic administration of the selective CCK-A receptor antagonist devazepide, which crosses the blood-brain barrier, increases food intake in capsaicin-treated, as well as vagotomized, rats (35), and CCK antagonists also attenuate food intake after administration of nutrients that do not increase plasma CCK (7). Furthermore, food intake induces the release of hypothalamic CCK, and microinjections of CCK into the hypothalamus inhibit food intake (4, 13, 41). Similarly, microinjections of CCK into the dorsal vagal complex [DVC; i.e., the nucleus tractus solitarius (NTS) and the dorsal motor nucleus of the vagus (DMV)] inhibit food intake and induce gastric relaxation, and systemic administration of CCK induces c-Fos expression in the DMV and NTS, where CCK receptors have been shown to be present (4, 24, 36, 37).
Electrophysiological analysis of the effects of CCK on DVC cells produced mixed results with respect to the actual effects of CCK as well as the receptor subtypes involved. Plata-Salaman et al. (29) reported that CCK depolarized a large subset of DMV neurons but also hyperpolarized a small fraction of them; both effects were mediated via activation of CCK-A receptors. Conversely, Branchereau et al. (5, 6) reported that application of CCK induced an excitation, an inhibition, or a biphasic excitatory-inhibitory effect on solitary complex neurons. They also concluded that the effects of the sulfated form of CCK octapeptide (CCK8s) were determined by activation of CCK-A and CCK-B receptors; however, their recordings did not distinguish between NTS and DMV neurons.
The aims of the present study, thus, were to investigate and analyze the pharmacological responses of identified gastric-projecting DMV neurons to perfusion with CCK8s.
MATERIALS AND METHODS
Retrograde tracing
Rats were purchased from Charles River Laboratories (Wilmington, MA). Animal care and experimental procedures were performed with the approval of the Animal Care and Utilization Committee.
Gastrointestinal (GI)-projecting DMV neurons were labeled as described previously (9). Briefly, 10- to 12-day-old Sprague-Dawley rat pups of either gender were anesthetized (indicated by abolition of the foot-pinch withdrawal reflex) with a 3% solution of isoflurane with air (400–600 ml/min). To maintain anesthesia, the head of the rat was placed in a custom-made anesthetic chamber through which the isoflurane mixture was perfused. A laparotomy was performed, during which crystals of the retrograde tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate [DiI, DiIC18(3)] were applied to the serosal surface of the stomach fundus, corpus, or antrum/ pylorus, and the application site (which covered an ~3- to 5-mm2 area along the greater curvature) was embedded in a fast-hardening epoxy resin, which was allowed to dry for several minutes before the entire surgical area was washed with warm saline (9). The wound was closed with 5-0 suture, and the animal was allowed to recover for 10–15 days.
Electrophysiology
The brain stems were removed as described previously (9). Briefly, the rats were anesthetized with halothane; when a deep level of anesthesia was induced, we killed the rats by severing the major blood vessels in the chest. The brain stem was removed and placed in oxygenated Krebs solution at 4°C (see Solution composition). The site of DiI labeling in the stomach was confirmed by visual inspection of the organ. The brain stem was used only from those animals in which the glue covering the site of DiI application was still in place at the time of the experiment. A Vibratome was used to cut six to eight 2- to 300-μm-thick coronal sections containing the DVC, and the sections were stored in oxygenated Krebs solution at 30°C for ≥1 h before use. A single slice was transferred to a custom-made 500-μl perfusion chamber and kept in place using a nylon mesh. The chamber was maintained at 35 ± 1°C by perfusion with warmed, oxygenated Krebs solution at a rate of 2.5–3.0 ml/min.
Before electrophysiological recording, GI-projecting DMV neurons were identified using a microscope (model E600-FN, Nikon) equipped with epifluorescent filters suitable for visualizing DiI. Once the identity of a labeled neuron was confirmed, whole cell recordings were made under bright-field illumination using differential interference contrast (Nomarski) optics.
Whole cell recordings were made with patch pipettes (3- to 8-MΩ resistance) filled with a potassium gluconate solution (see Solution composition) using an Axoclamp 2B single-electrode voltage-clamp amplifier (Axon Instruments, Union City, CA). Recordings were made only from neurons unequivocally labeled with DiI. Data were sampled every 100 μs and filtered at 2 kHz, digitized via a Digidata 1200C interface (Axon Instruments), and acquired, stored, and analyzed on an IBM personal computer utilizing pClamp 8 software (Axon Instruments). Recordings were accepted only if the series resistance was <15 MΩ. In all voltage-clamp experiments, the cells were held at −50 mV. Current-voltage relation curves were constructed by stepping the membrane from −50 to −120 mV in −10-mV increments for 0.5–1 s. The input resistance was calculated by measuring the instantaneous current displacement obtained by stepping the membrane from −50 to −70 mV. To assess the effects of drugs, each neuron served as its own control (i.e., the results obtained after administration of a drug were compared with those obtained before administration using Student’s paired t-test). Cells were classified as responders if CCK8s (30–100 nM) induced a current of at least ± 20 pA. Values are means ± SE. Significance was set at P <0.05.
Concentration-response curves were constructed from neurons in which at least three concentrations of CCK8s were tested. At least 5 min were allowed between successive drug applications. Antagonists were applied for 10 min before the agonist was reapplied.
Immunohistochemistry
Rats were injected with fluorogold (20 μg/1 ml saline ip per rat) to label vagal preganglionic neurons innervating the subdiaphragmatic viscera, allowing delineation of the boundaries of the DMV (17, 30, 46).
With the use of a custom-made anesthetic chamber, rats were anesthetized deeply (abolition of foot-pinch withdrawal reflex) with isoflurane, and the abdominal and thoracic areas were shaved and cleaned with 70% ethanol. After an abdominal laparotomy, the stomach was freed from the liver and reflected gently to one side to facilitate access to the gastric wall. Gastric-projecting DMV neurons were retrogradely labeled via microinjections of rhodamine beads (17, 46) or apposition of DiI crystals on the fundus, corpus, or antrum/ pylorus (9). As previously described, care was taken to restrict the dyes within the stomach wall and to reduce the possibility of perforating the mucosa. The laparotomy was closed with 5-0 sutures, and the rats were allowed to recover for 5–15 days to allow retrograde transport of the fluorescent marker to the brain stem. On the day of the experiments, rats were killed with an overdose of halothane and perfused transcardially with 200 ml of chilled saline followed by 200 ml of chilled Zamboni’s solution (see Solution composition). In another series of experiments, rats were anesthetized with thiobutabarbital (Inactin, 120 mg/kg ip) and injected with CCK8s (2–8 μg/kg ip). After 30–90 min, the animals were fixed as described above. After extraction, brain stems were immersed in chilled Zamboni’s fixative overnight, washed with PBS, and cut into 40-μm-thick coronal sections with a cryostat. The brain stem slices were then mounted on gelatin-coated coverslips and incubated with rabbit anti-CCK-A receptor antibodies (CURE, University of California Los Angeles) diluted 1:200 with 0.1% PBS-BSA for 24 h at 4°C or with rabbit anti-CCK-A phosphorylated receptor diluted 1:250 (R & D Systems, Minneapolis, MN). Sections were then washed in 1% PBS-BSA (3 times over a 2-h period) at room temperature and incubated with secondary goat anti-rabbit antibodies conjugated to Alexa 488 (1:500) for 2 h at 37°C. The brain stem slices were washed in PBS containing 0.1% goat antiserum for 2 h at room temperature before being coverslipped with Fluoromount-G (Southern Biotechnology, Birmingham, AL).
Specimens were examined using a Nikon E400 microscope fitted with epifluorescent filters for tetramethylrhodamine isothiocyanate, FITC, and UV. Cells labeled with rhodamine beads or DiI and/or CCK-A visualized with Alexa 488 were counted on alternate brain stem slices by two independent investigators who were unaware of the treatment. If the cell count differed by >10%, a third investigator analyzed the brain stem slices. The final cell count was the mathematical average of the independent cell counts.
To minimize errors in the counting of DMV somata, we counted only those neurons in which the nucleus was clearly visible. Despite this precaution, we have to consider cell counts as best proportional estimates only, rather than absolute values, when comparing labeled subpopulations. This is of paramount importance, because the surface of the stomach wall covered by application of the retrograde tracers was limited to ~3–5 mm2. Therefore, this provided only a small, but representative, fraction of labeled DMV neurons projecting to the gastric areas of interest.
Sections of brain stem were analyzed from ~2 mm caudal to the posterior tip to ~3 mm rostral to the anterior tip of the area postrema. Cell count values are means ± SE.
Photographs were taken using a SPOT digital camera (Sterling Heights, MI) mounted on a Nikon E400 fluorescent microscope. Digital images were enhanced using Adobe Photoshop.
Solution composition
Krebs solution contained (in mM) 126 NaCl, 25 NaHCO3, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, and 11 dextrose, with pH maintained at 7.4 by bubbling with 95% O2-5% CO2. Intracellular solution consisted of (in mM) 128 potassium gluconate, 10 KCl, 0.3 CaCl2, 1 MgCl2, 10 HEPES, 1 EGTA, 2 ATP, and 0.25 GTP, with pH adjusted to 7.35 with KOH. PBS consisted of (in mM) 115 NaCl, 75 Na2HPO4 · 7H2O, 7.5 KH2PO4, and 0.15% Triton X. Zamboni’s fixative contained 1.6% (wt/vol) paraformaldehyde, 19 mM KH2PO4, and 100 mM Na2HPO4 · 7H2O in 240 ml of saturated picric acid and 1,600 ml of H2O, with pH adjusted to 7.4 with HCl.
Statistical analysis
Values are means ± SE. Intergroup comparisons were analyzed with one-way ANOVA followed by the conservative Bonferroni test for individual post hoc comparisons, Student’s paired t-test, or χ2 test. Significance was defined as P <0.05.
Drugs and chemicals
Drugs were made fresh immediately before use and applied to the bath via a series of manually operated valves. Nonsulfated CCK (CCKns) was purchased from Bachem (King of Prussia, PA), DiI from Molecular Probes (Eugene, OR), and fluorogold from Fluorochrome (Denver, CO). Phosphorylated CCK-A receptor antibody was a gift from Dr. A. E. Kalyuzhny (R & D); CCK-A receptor antibody (nonphosphorylated) was a gift from CURE. All other chemicals, including CCK8s, were purchased from Sigma (St. Louis, MO).
RESULTS
Whole cell patch-clamp studies were conducted on 267 neurons identified as 21 fundus-, 87 corpus-, and 159 antrum/ pylorus-projecting neurons.
CCK8s induced an inward current in subpopulations of DMV neurons
Two minutes of perfusion with CCK8s (30–100 nM) induced an inward current in 34% of corpus-projecting neurons (i.e., 30 of 87) and 41% of antrum/pylorus-projecting neurons (i.e., 66 of 159). The remaining 168 (i.e., 64%) neurons, including all the fundus neurons, did not respond to CCK8s. Three more neurons responded to CCK8s with a 50 ± 13 pA outward current. The low occurrence of this type of response to CCK8s prevented us from conducting any type of pharmacological analysis.
Because the response of corpus- and antrum/pylorus-projecting neurons to CCK8s did not differ between the groups, the data obtained from DMV neurons projecting to these two areas were pooled. Furthermore, there was no apparent preferential distribution of cells, whether responsive or unresponsive to CCK8s, along the rostrocaudal extent of the DMV.
The response to perfusion with CCK8s was concentration dependent (30 pM–300 nM), the EC50 was ~4 nM, the maximum response was reached with 30 nM, and the maximum current amplitude was 42.3 ± 4.7 pA (Fig. 1). Six cells responsive to CCK8s in voltage-clamp configuration were also tested in current-clamp configuration; the spontaneous firing rate was 1.15 ± 0.26 and 2.01 ± 0.26 pulses per second in control and after perfusion with 30 nM CCK8s, respectively (i.e., 104 ± 27% of control, P <0.05). Twenty-four additional neurons that did not respond to 30 nM CCK8s in the voltage-clamp configuration were also unresponsive to 30 nM CCK8s perfusion when tested in the current-clamp configuration.
Fig. 1.

Sulfated cholecystokinin octapeptide (CCK8s) induced a concentration-dependent inward current. A: representative voltage-clamp traces illustrating the concentration-dependent inward current induced by superfusion of CCK8. A recovery period of ≥5 min was allowed between successive applications, during which membrane potential returned to baseline values. Parallel oblique bars indicate a 2- to 5-min interval. Holding potential −50 mV. B: concentration-response curve for the CCK8-induced inward current.
When two applications of 30 nM CCK8s were conducted within 5 min of each other, the CCK-induced inward current did not show tachyphylaxis. The first superfusion of CCK8s induced a 43.5 ± 5.5 pA inward current. After washout of CCK8s, the membrane returned to its initial values. A second superfusion of CCK8s induced a 38.0 ± 3.6 pA inward current (i.e., 93 ± 5.9% of the 1st application, P >0.05, n = 13; Fig. 2, A and B).
Fig. 2.

CCK8-induced inward current does not show tachyphylaxis or desensitization. A: representative voltage-clamp traces comparing amplitude of the current obtained by 2 applications (1 and 2) of 30 nM CCK8s 5 min apart. Holding potential −50 mV. B: amplitudes of inward current induced by CCK8s in 2 successive applications 5 min apart. Maximal current amplitude is not significantly reduced between the 2 applications of CCK8. C: representative voltage-clamp trace. CCK8s (30 nM)-induced response does not desensitize even in the continued presence of CCK8. Holding potential −50 mV.
When the application of 30 nM CCK8s was continued for 5 min, the amplitude of the peak inward current (49.4 ± 2.5 pA) did not decay throughout the period of superfusion; i.e., the CCK8s-induced current did not desensitize. The cells recovered to baseline after discontinuation of CCK8s superfusion (Fig. 2C).
To detect whether the CCK8s-induced current was due to a direct effect of CCK8s on the DMV cell, we compared the amplitude of the CCK8s (30–100 nM)-induced inward current in the absence and presence of the synaptic transmission blocker tetrodotoxin (TTX, 0.3 μM). In six neurons, CCK8s induced a 48.0 ± 5.57 pA inward current that recovered to baseline on washout. After 10 min of perfusion with TTX, reapplication of CCK8s in the presence of TTX induced a 37.8 ± 5.71 pA inward current (i.e., 80 ± 9.7% of control, P <0.05 vs. CCK8s alone; Fig. 3).
Fig. 3.

A large proportion of the CCK8-induced current is via direct activation of the membrane of the dorsal motor nucleus of the vagus (DMV). A: representative voltage-clamp traces showing CCK8s (100 nM)-induced inward current in control and after 10 min of perfusion with the synaptic transmission blocker TTX (0.3 μM). Parallel oblique bars indicate a 2-min interval. Holding potential −50 mV. B: amplitudes of inward current induced by CCK8s in the absence and presence of TTX. *P <0.05 vs. CCK8s alone.
The reversal potential of the CCK8s-induced inward current was measured by stepping the membrane from −50 to −120 mV in 10-mV increments (Fig. 4A). In 11 cells responsive to CCK8s, the reversal potential of the CCK8s-induced current was −89 ± 4.5 mV (Fig. 4B). In 8 of the 11 cells, CCK8s increased the input resistance, measured from −50 to −70 mV, from 356 ± 52 MΩ in control to 560 ± 70 MΩ in CCK8s (i.e., 65 ± 17% of control, P <0.05; Fig. 4C). In the three remaining cells, perfusion with CCK8s did not affect the input resistance.
Fig. 4.

CCK8-induced inward current has a reversal potential close to that for potassium. DMV neurons were voltage clamped at −50 mV and stepped to −120 mV for 800 ms every 5 s in −10-mV increments in control conditions and after perfusion with 30 nM CCK8s. B: current-voltage plot. Reversal potential for CCK8-induced current is close to that for potassium. Each data point represents average of 6–24 neurons. C: input resistance in control and after perfusion with 30 nM CCK8s. CCK8s perfusion induced a 65 ± 17% increase in input resistance. *P <0.05 vs. control.
CCKns (30 nM–3 μM) was tested on eight CCK8s-responsive DMV neurons. Perfusion with 30 nM CCK8s induced a 57 ± 6.40 pA inward current; after washout of CCK8s and return of the membrane to baseline, perfusion with 30 nM–1 μM CCKns did not induce any measurable current, but perfusion with 3 μM CCKns induced a 25 ± 5.2 pA inward current.
CCK8s-induced inward current is mediated by CCK-A receptors
In 18 DMV neurons, perfusion with 30 nM CCK8s induced a 68.5 ± 5.96 pA inward current. After washout of CCK8s and 10 min of superfusion with the selective CCK-A receptor antagonist lorglumide (0.3–3 μM), which per se had no effect on the holding current, the CCK8s-induced inward current was significantly attenuated in a concentration-dependent manner (Fig. 5). In contrast, 10 min of pretreatment with the selective CCK-B receptor antagonist triglumide (0.3–3 μM, n = 12) did not attenuate significantly the CCK8s-induced inward current. Even at the highest concentration tested (3 μM), pretreatment with triglumide did not significantly reduce the CCK8s-induced current (Fig. 5). In six cells in which 3 μM triglumide was first tested against CCK8s, 10 min of perfusion with 1 μM lorglumide and 3 μM triglumide reduced the CCK8s-induced current to the same extent as CCK8s + lorglumide alone (P <0.05 vs. CCK8s alone, P >0.05 vs. CCK8s + lorglumide alone; Fig. 5).
Fig. 5.
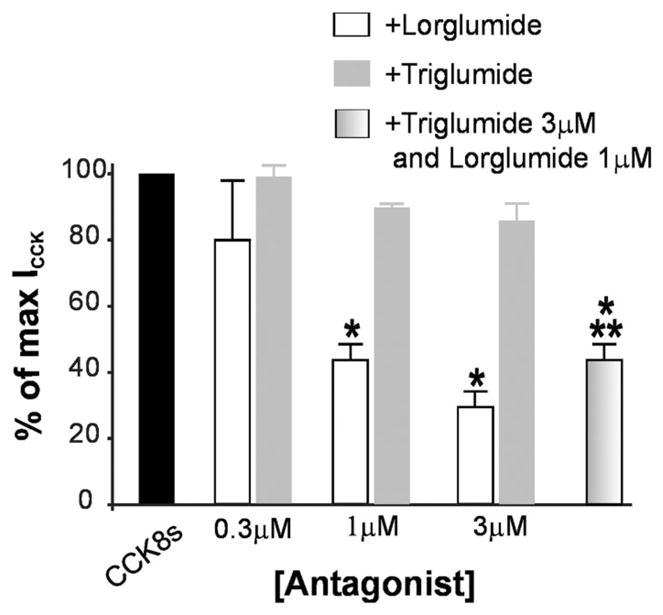
CCK8-induced inward current (ICCK) is mediated by CCK-A receptors. Normalized data show that pretreatment with the selective CCK-A receptor antagonist lorglumide, but not the selective CCK-B receptor antagonist triglumide, attenuated the maximal response to CCK in a concentration-related manner. Pretreatment with lorglumide + triglumide did not increase the antagonism induced by lorglumide alone. *P <0.05 vs. CCK8s alone; **P >0.05 vs. lorglumide alone.
CCK-A receptors are present on the membrane of a sub-population of DMV neurons
A total of 26 rats were used: 9 were labeled with apposition of dyes on the fundus, 11 on the corpus, and 6 on the serosal surface of the antrum/pylorus.
DiI- or rhodamine bead-filled neurons were seen throughout the rostrocaudal extent of the DMV. We counted an average of 40.2 ± 8.16 dye-labeled DMV cells in each animal.
Two different antibodies from CCK-A receptors were tested: the first antibody was raised against the nonphosphorylated receptor and the second against the phosphorylated, i.e., activated (15), receptor. Immunoreactivity to the phosphorylated CCK-A receptor was not observed in four additional rats that did not receive intraperitoneal injection of CCK8s (see MATERIALS AND METHODS), confirming that the antibody was sufficiently specific to recognize the activated, i.e., phosphorylated, form of CCK-A receptor only.
Results obtained from rats pretreated with intraperitoneal injection of CCK8s and tested with the phosphorylated form of the CCK-A receptor or from rats tested with the nonphosphorylated form of the CCK-A receptors were qualitatively and quantitatively similar; thus the data were pooled.
CCK-A receptor immunoreactivity was evenly distributed throughout the rostrocaudal extent of the DMV. The immunoreactivity was restricted to the perinuclear cytoplasm and was not detectable in adjacent processes. CCK-A receptor immunoreactivity colocalized with 65 ± 4.8% of corpus-projecting, 60 ± 3.7% of antrum/pylorus-projecting, and 34 ± 6.8% of fundus-projecting neurons (P <0.05 vs. corpus and antrum/ pylorus; Fig. 6).
Fig. 6.

Immunohistochemical localization of CCK-A receptors. Top: low-magnification micrograph depicting localization of CCK-A receptors (green, FITC filters) and corpus-projecting DMV neurons (red; tetramethylrhodamine isothiocyanate filters). a: Higher magnification of encircled area on the left showing a corpus-projecting DMV neuron (arrow) that did not contain CCK-A receptors and a unidentified DMV neuron (double-headed arrow) that expressed CCK-A receptors. b: Higher magnification of encircled area on the right showing a corpus-projecting DMV neuron (arrow) and an unidentified DMV neuron (double-headed arrow) that expressed CCK-A receptors. B: percentage of identified gastric-projecting [fundus, corpus, and antrum/pylorus (A/P)] DMV neurons expressing CCK-A receptors. CC, central canal; XII, hypoglossus nucleus. *P <0.05 vs. fundus-projecting neurons.
DISCUSSION
In this study, we have shown that 1) CCK8s induces a concentration-dependent inward current in ~40% of identified corpus- and antrum/pylorus-projecting DMV neurons but has no effect on fundus-projecting neurons, 2) the CCK8s-induced inward current is likely to be mediated by closure of a potassium conductance, 3) the CCK8s-induced inward current is mediated by activation of CCK-A receptors, and 4) CCK-A receptors can be identified on a subpopulation of gastric-projecting DMV neurons.
These data suggest that functional CCK-A, but not CCK-B, receptors are present on subgroups of gastric-projecting DMV neurons. It is possible that these CCK-A receptors are physiologically relevant in the mechanism of action of CCK in the central nervous system (CNS). Given the low circulating concentrations of gut-derived CCK, however, we cannot exclude the possibility that the CCK acting on gastric-projecting DMV neurons is derived from some other source. Thus the full physiological relevance may remain to be established.
Our conclusions are based on the following evidence. Approximately 40% of corpus- and antrum/pylorus-projecting DMV neurons respond to perfusion with CCK8s with an inward current. Conversely, none of the fundus-projecting neurons responded to perfusion with CCK8s. Our data confirm and extend the initial observation by Plata-Salaman and colleagues (29), who showed that ~43% of unidentified DMV neurons were depolarized by CCK8s. Contrary to Plata-Salaman and colleagues, who reported that 11% of DMV neurons were hyperpolarized by CCK8s, we observed an outward current (i.e., hyperpolarization) in only 3 of 267 neurons. A possible reason for this discrepancy might be that Plata-Salaman and colleagues recorded from DMV neurons projecting to areas other than those studied in our investigation, which was restricted to gastric-projecting cells. Our data, showing that CCK8s induces an inward current (i.e., an excitatory effect) in gastric-projecting DMV neurons, are in agreement with data showing an excitatory effect of CCK8s in other CNS areas (10, 11, 28, 43).
The CCK8s-induced inward current is attenuated in a concentration-dependent manner by pretreatment with the selective CCK-A receptor antagonist lorglumide. Conversely, the CCK-B-preferring receptor agonist CCK8ns does not induce any current when perfused onto DMV neurons at concentrations as high as 1 μM; a small inward current was induced, however, on administration of 3 μM CCKns, strongly suggesting a nonselective effect. Similarly, pretreatment with the selective CCK-B receptor antagonist triglumide does not attenuate the CCK8s-induced inward current. Furthermore, perfusion with a combination of lorglumide and triglumide attenuates the CCK8s-induced inward current to the same extent as lorglumide alone. Our in vitro data, showing that the effects of CCK8s on DMV neurons are mediated via interaction with CCK-A receptors, are in agreement with biochemical and in vivo results showing that, overall, the satiety and GI effects of CCK8s are mediated via CCK-A receptors (16, 23, 26, 34, 35). Of particular relevance are the data showing that the CCK-mediated effects at the level of the brain stem are mediated via activation of CCK-A receptors (4, 16, 35).
The inward current induced by perfusion with CCK8s is accompanied by a significantly increased input resistance. The reversal potential of the current is close to −90 mV, i.e., close to the reversal potential for potassium, suggesting that the excitatory effects of CCK8s are mediated via closure of a potassium conductance. These results are similar to data reported in other CNS regions, where the excitatory effects of CCK8s were shown to be mediated by closure of potassium conductance (5, 10, 29), although Dun et al. (14) suggested that the effects of CCK in nodose ganglion cells were mediated by an increase in a nonselective cationic conductance.
Our data also show that, after blockade of synaptic transmission with TTX, the response to CCK8s is significantly reduced, suggesting that a portion of the excitatory response to CCK8s is mediated by inputs impinging on DMV neurons, similar to that observed for the parabrachial nucleus and hippocampus (8, 40). Indeed, several authors indicated an excitatory effect of CCK on areas, such as the NTS and area postrema, that have robust projections to DMV and, most importantly, would be preserved in our experimental conditions (1, 4–6, 16, 36, 38).
Finally, our electrophysiological results are supported by our immunohistochemical data. With the use of two different types of CCK-A receptor antibodies, one recognizing the CCK-A receptor in its phosphorylated state and the other recognizing the nonphosphorylated CCK-A receptor, we observed that CCK-A receptor immunoreactivity is present in a significantly larger proportion of corpus- or antrum/pylorus- than fundus-projecting neurons. CCK-A receptor labeling observed in this study was restricted to cell bodies, as previously reported with autoradiographic and immunochemical methods (19, 24). Interestingly, the phosphorylated CCK-A receptor immunoreactivity was detectable only after intraperitoneal administration of CCK8s, indicating that CCK-A receptors were activated/ phosphorylated by circulating CCK8s (15). These data further support the possible role of CCK8s in the central regulation of satiety or gastric functions.
Physiological significance
The CCK-induced gastric relaxation is thought to be one of the mechanisms that mediate the satiety effects of CCK. The DMV comprises the preganglionic parasympathetic neurons that finely modulate gastric tone and motility. Cholinergic neurons in the DMV, in fact, impinge on postganglionic cells that comprise the excitatory cholinergic or the inhibitory nonadrenergic noncholinergic (NANC) pathways that regulate gastric functions (44). CNS-mediated gastric relaxation can thus be obtained via inhibition of DMV neurons that comprise the cholinergic excitatory pathway or via excitation of DMV cells that comprise the inhibitory NANC pathway. Our results, showing that CCK8s has an excitatory effect on DMV neurons, seem to support the hypothesis that the CCK8s-mediated gastric relaxation occurs via an increase in the activity of NANC pathways to the corpus wall, although this mechanism of action is still controversial (42). Under physiological conditions, it is not clear how CCK would reach the brain stem to exert its actions. In fact, the postprandial plasma concentration of CCK is in the low picomolar range, which would not be sufficient to excite DMV neurons. Furthermore, CCK does not readily cross the blood-brain barrier, although portions of the DVC have a “leaky” blood-brain barrier. It is possible, however, that the effects of CCK on DMV neurons are determined by CCK acting as a neurotransmitter or neuromodulator. Indeed, CCK-containing neural projections originating from the NTS or the hypothalamus are present throughout the rostrocaudal extent of the DMV (20).
The hypothesis that the effects of CCK are mediated via activation of an NANC pathway falls short of explaining the lack of effects of CCK8s on fundus-projecting DMV neurons. Because most of the gastric relaxation is mediated by the proximal stomach, which is also the area that supposedly receives most of the NANC innervation, it is possible that the effects of CCK8s are indirect, i.e., mediated by an effect of CCK8s on local inputs impinging on gastric-projecting DMV neurons. Indeed, our data showing that some of the effects of CCK8s are abolished by TTX treatment might indicate this possibility. The lack of CCK8s effects on fundus-projecting neurons might be ascribed to subthreshold (i.e., below resolution) effects on the DMV membrane.
In summary, in the present study, we have shown that CCK-A receptors mediate the excitatory effects of CCK8s on subsets of corpus- and antrum/pylorus-projecting DMV neurons, an action mediated by the closure of a potassium conductance. We have also shown that CCK-A receptor immunoreactivity is present on the membrane of subgroups of gastric-projecting DMV neurons. Taken together, these data would suggest that the CCK8s-mediated gastric relaxation occurs via activation of NANC pathways.
Acknowledgments
We acknowledge F. Holly Coleman for technical support, Drs. K. N. Browning, R. C. Rogers, G. E. Hermann, and H. R. Berthoud for comments on previous versions of the manuscript, and Cesare M. Travagli for support and encouragement.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-55530.
References
- 1.Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. CCK activates pro-opiomelanocortin neurons and visceral afferent synaptic transmission in nucleus tractus solitarius (Abstract) Proc 33rd Annu Meeting Soc Neurosci. 2003 [Google Scholar]
- 2.Berthoud HR, Patterson LM. Anatomical relationship between vagal afferent fibers and CCK-immunoreactive entero-endocrine cells in the rat small intestinal mucosa. Acta Anat (Basel) 1996;156:123–131. doi: 10.1159/000147837. [DOI] [PubMed] [Google Scholar]
- 3.Blackshaw LA, Grundy D. Effects of cholecystokinin (CCK-8) on two classes of gastroduodenal vagal afferent fibre. J Auton Nerv Syst. 1990;31:191–202. doi: 10.1016/0165-1838(90)90185-l. [DOI] [PubMed] [Google Scholar]
- 4.Blevins JE, Stanley BG, Reidelberger RD. Brain regions where cholecystokinin suppresses feeding in rats. Brain Res. 2000;860:1–10. doi: 10.1016/s0006-8993(99)02477-4. [DOI] [PubMed] [Google Scholar]
- 5.Branchereau P, Bohme GA, Champagnat J, Morin-Surun MP, Durieux C, Blanchard JC, Roques BP, Denavit-Saubie M. CholecystokininA and cholecystokininB receptors in neurons of the brainstem solitary complex of the rat: pharmacological identification. J Pharmacol Exp Ther. 1992;260:1433–1440. [PubMed] [Google Scholar]
- 6.Branchereau P, Champagnat J, Denavit-Saubie M. Cholecystokinin-gated currents in neurons of the rat solitary complex in vitro. J Neurophysiol. 1993;70:2584–2595. doi: 10.1152/jn.1993.70.6.2584. [DOI] [PubMed] [Google Scholar]
- 7.Brenner L, Yox DP, Ritter RC. Suppression of sham feeding by intraintestinal nutrients is not correlated with plasma cholecystokinin elevation. Am J Physiol Regul Integr Comp Physiol. 1993;264:R972–R976. doi: 10.1152/ajpregu.1993.264.5.R972. [DOI] [PubMed] [Google Scholar]
- 8.Breukel AI, Wiegant VM, Lopes da Silva FH, Ghijsen WE. Presynaptic modulation of cholecystokinin release by protein kinase C in the rat hippocampus. J Neurochem. 1998;70:341–348. doi: 10.1046/j.1471-4159.1998.70010341.x. [DOI] [PubMed] [Google Scholar]
- 9.Browning KN, Renehan WE, Travagli RA. Electrophysiological and morphological heterogeneity of rat dorsal vagal neurones which project to specific areas of the gastrointestinal tract. J Physiol. 1999;517:521–532. doi: 10.1111/j.1469-7793.1999.0521t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox CL, Huguenard JR, Prince DA. Cholecystokinin depolarizes rat thalamic reticular neurons by suppressing a K+ conductance. J Neurophysiol. 1995;74:990–1000. doi: 10.1152/jn.1995.74.3.990. [DOI] [PubMed] [Google Scholar]
- 11.Crawley JN, Corwin RL. Biological actions of cholecystokinin. Peptides. 1994;15:731–755. doi: 10.1016/0196-9781(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 12.Davison JS, Clarke GD. Mechanical properties and sensitivity to CCK of vagal gastric slowly adapting mechanoreceptors. Am J Physiol Gastrointest Liver Physiol. 1988;255:G55–G61. doi: 10.1152/ajpgi.1988.255.1.G55. [DOI] [PubMed] [Google Scholar]
- 13.Della-Fera MA, Baile CA, Schneider BS, Grinker JA. Cholecystokinin antibody injected in cerebral ventricles stimulates feeding in sheep. Science. 1981;212:687–689. doi: 10.1126/science.7221559. [DOI] [PubMed] [Google Scholar]
- 14.Dun NJ, Wu SY, Lin CW. Excitatory effects of cholecystokinin octapeptide on rat nodose ganglion cells in vitro. Brain Res. 1991;556:161–164. doi: 10.1016/0006-8993(91)90562-a. [DOI] [PubMed] [Google Scholar]
- 15.Gates LK, Ulrich CD, Miller LJ. Multiple kinases phosphorylate the pancreatic cholecystokinin receptor in an agonist-dependent manner. Am J Physiol Gastrointest Liver Physiol. 1993;264:G840–G847. doi: 10.1152/ajpgi.1993.264.5.G840. [DOI] [PubMed] [Google Scholar]
- 16.Glatzle J, Kreis ME, Kawano K, Raybould HE, Zittel TT. Postprandial neuronal activation in the nucleus of the solitary tract is partly mediated by CCK-A receptors. Am J Physiol Regul Integr Comp Physiol. 2001;281:R222–R229. doi: 10.1152/ajpregu.2001.281.1.R222. [DOI] [PubMed] [Google Scholar]
- 17.Guo JJ, Browning KN, Rogers RC, Travagli RA. Catecholaminergic neurons in rat dorsal motor nucleus of vagus project selectively to gastric corpus. Am J Physiol Gastrointest Liver Physiol. 2001;280:G361–G367. doi: 10.1152/ajpgi.2001.280.3.G361. [DOI] [PubMed] [Google Scholar]
- 18.Gutzwiller JP, Drewe J, Ketterer S, Hildebrand P, Krautheim A, Beglinger C. Interaction between CCK and a preload on reduction of food intake is mediated by CCK-A receptors in humans. Am J Physiol Regul Integr Comp Physiol. 2000;279:R189–R195. doi: 10.1152/ajpregu.2000.279.1.R189. [DOI] [PubMed] [Google Scholar]
- 19.Hill DR, Shaw TM, Graham W, Woodruff GN. Autoradiographical detection of cholecystokinin-A receptors in primate brain using 125I-Bolton Hunter CCK-8 and 3H-MK-329. J Neurosci. 1990;10:1070–1081. doi: 10.1523/JNEUROSCI.10-04-01070.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubota Y, Takagi H, Morishima Y, Kaway Y, Smith AD. Relationship between catecholaminergic neurons and cholecystokinin-containing neurons in the caudal part of the dorsomedial medulla oblongata of the rat: light and electron microscopic observations by the mirror technique. Brain Res. 1986;370:343–348. doi: 10.1016/0006-8993(86)90491-9. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Owyang C. Vagal afferent pathway mediates physiological action of cholecystokinin on pancreatic enzyme secretion. J Clin Invest. 1993;92:418–424. doi: 10.1172/JCI116583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liddle RA. Regulation of cholecystokinin synthesis and secretion in rat intestine. J Nutr. 1994;124:1308S–1314S. doi: 10.1093/jn/124.suppl_8.1308S. [DOI] [PubMed] [Google Scholar]
- 23.Lloyd KCK, Raybould HE, Walsh JH. Cholecystokinin inhibits gastric acid secretion through type “A” cholecystokinin receptors and somatostatin in rats. Am J Physiol Gastrointest Liver Physiol. 1992;263:G287–G292. doi: 10.1152/ajpgi.1992.263.3.G287. [DOI] [PubMed] [Google Scholar]
- 24.Mercer LD, Beart PM. Histochemistry in rat brain and spinal cord with an antibody directed at the cholecystokinin A receptor. Neurosci Lett. 1997;225:97–100. doi: 10.1016/s0304-3940(97)00197-3. [DOI] [PubMed] [Google Scholar]
- 25.Moran TH. Cholecystokinin and satiety: current perspectives. Nutrition. 2000;16:858–865. doi: 10.1016/s0899-9007(00)00419-6. [DOI] [PubMed] [Google Scholar]
- 26.Moran TH, Kinzig KP. Gastrointestinal satiety signals. II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286:G183–G188. doi: 10.1152/ajpgi.00434.2003. [DOI] [PubMed] [Google Scholar]
- 27.Moran TH, Ladenheim EE, Schwartz GJ. Within-meal gut feedback signaling. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S39–S41. doi: 10.1038/sj.ijo.0801910. [DOI] [PubMed] [Google Scholar]
- 28.Noble F, Wank SA, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, Roques BP. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol Rev. 1999;51:745–781. [PubMed] [Google Scholar]
- 29.Plata-Salaman CR, Fukuda A, Oomura Y, Minami T. Effects of sulphated cholecystokinin octapeptide (CCK-8) on the dorsal motor nucleus of the vagus. Brain Res Bull. 1988;21:839–842. doi: 10.1016/0361-9230(88)90054-8. [DOI] [PubMed] [Google Scholar]
- 30.Powley TL, Fox EA, Berthoud HR. Retrograde tracer technique for assessment of selective and total subdiaphragmatic vagotomies. Am J Physiol Regul Integr Comp Physiol. 1987;253:R361–R370. doi: 10.1152/ajpregu.1987.253.2.R361. [DOI] [PubMed] [Google Scholar]
- 31.Raybould HE, Holzer H. Dual capsaicin-sensitive afferent pathways mediate inhibition of gastric emptying in rat induced by intestinal carbohydrate. Neurosci Lett. 2000;141:236–238. doi: 10.1016/0304-3940(92)90902-j. [DOI] [PubMed] [Google Scholar]
- 32.Raybould HE, Roberts ME, Dockray GJ. Reflex decreases in intragastric pressure in response to cholecystokinin in rats. Am J Physiol Gastrointest Liver Physiol. 1987;253:G165–G170. doi: 10.1152/ajpgi.1987.253.2.G165. [DOI] [PubMed] [Google Scholar]
- 33.Raybould HE, Tache Y. Cholecystokinin inhibits gastric motility and emptying via a capsaicin-sensitive vagal pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1988;255:G242–G246. doi: 10.1152/ajpgi.1988.255.2.G242. [DOI] [PubMed] [Google Scholar]
- 34.Reidelberger RD. Cholecystokinin and control of food intake. J Nutr. 1994;124:1327S–1333S. doi: 10.1093/jn/124.suppl_8.1327S. [DOI] [PubMed] [Google Scholar]
- 35.Reidelberger RD, Hernandez J, Fritzsch B, Hulce M. Abdominal vagal mediation of the satiety effects of CCK in rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1005–R1012. doi: 10.1152/ajpregu.00646.2003. [DOI] [PubMed] [Google Scholar]
- 36.Rinaman L, Hoffman GE, Dohanics J, Le WW, Stricker EM, Verbalis JG. Cholecystokinin activates catecholaminergic neurons in the caudal medulla that innervate the paraventricular nucleus of the hypothalamus in rats. J Comp Neurol. 1995;360:246–256. doi: 10.1002/cne.903600204. [DOI] [PubMed] [Google Scholar]
- 37.Rinaman L, Hoffman GE, Stricker EM, Verbalis JG. Exogenous cholecystokinin activates cFos expression in medullary but not hypothalamic neurons in neonatal rats. Dev Brain Res. 1994;77:140–145. doi: 10.1016/0165-3806(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 38.Rinaman L, Verbalis JG, Stricker EM, Hoffman GE. Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J Comp Neurol. 1993;338:475–490. doi: 10.1002/cne.903380402. [DOI] [PubMed] [Google Scholar]
- 39.Ritter RC. Gastrointestinal mechanisms of satiation for food. Physiol Behav. 2004;81:249–273. doi: 10.1016/j.physbeh.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Saleh TM, Kombian SB, Zidichouski JA, Pittman QJ. Cholecystokinin and neurotensin inversely modulate excitatory synaptic transmission in the parabrachial nucleus in vitro. Neuroscience. 1997;77:23–35. doi: 10.1016/s0306-4522(96)00463-0. [DOI] [PubMed] [Google Scholar]
- 41.Schick RR, Yaksh TL, Go VL. Intracerebroventricular injections of cholecystokinin octapeptide suppress feeding in rats—pharmacological characterization of this action. Regul Pept. 1986;14:277–291. doi: 10.1016/0167-0115(86)90170-9. [DOI] [PubMed] [Google Scholar]
- 42.Shi M, Jones AR, Ferreira J, Sahibzada N, Gillis RA, Verbalis JG. Glucose does not activate nonadrenergic noncholinergic inhibitory neurons in the rat stomach. Am J Physiol Regul Integr Comp Physiol. 2005;288:R742–R750. doi: 10.1152/ajpregu.00561.2004. [DOI] [PubMed] [Google Scholar]
- 43.Sun K, Ferguson AV. Cholecystokinin activates area postrema neurons in rat brain slices. Am J Physiol Regul Integr Comp Physiol. 1997;272:R1625–R1630. doi: 10.1152/ajpregu.1997.272.5.R1625. [DOI] [PubMed] [Google Scholar]
- 44.Travagli RA, Rogers RC. Receptors and transmission in the brain-gut axis: potential for novel therapies. V. Fast and slow extrinsic modulation of dorsal vagal complex circuits. Am J Physiol Gastrointest Liver Physiol. 2001;281:G595–G601. doi: 10.1152/ajpgi.2001.281.3.G595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods SC. Gastrointestinal satiety signals. I. An overview of gastrointestinal signals that influence food intake. Am J Physiol Gastrointest Liver Physiol. 2004;286:G7–G13. doi: 10.1152/ajpgi.00448.2003. [DOI] [PubMed] [Google Scholar]
- 46.Zheng ZL, Rogers RC, Travagli RA. Selective gastric projections of nitric oxide synthase-containing vagal brainstem neurons. Neuroscience. 1999;90:685–694. doi: 10.1016/s0306-4522(98)00586-7. [DOI] [PubMed] [Google Scholar]